

Abstract
The intestinal epithelium engages in bidirectional transport of fluid and electrolytes to subserve the physiological processes of nutrient digestion and absorption, as well as the elimination of wastes, without excessive losses of bodily fluids that would lead to dehydration. The overall processes of intestinal ion transport, which in turn drive the secretion or absorption of water, are accordingly carefully regulated. We and others have identified the epidermal growth factor receptor (EGFr) as a critical regulator of mammalian intestinal ion transport. In this article, we focus on our studies that have uncovered the intricate signaling mechanisms downstream of EGFr that regulate both chloride secretion and sodium absorption by colonocytes. Emphasis will be placed on the EGFr-associated regulatory pathways that dictate the precise outcome to receptor activation in response to signals that may seem, on their face, to be quite similar if not identical. The concepts to be discussed underlie the ability of the intestinal epithelium to utilize a limited set of signaling effectors to produce a variety of outcomes suitable for varying physiological and pathophysiological demands. Our findings therefore are relevant not only to basic biological principles, but also may ultimately point to new therapeutic targets in intestinal diseases where ion transport is abnormal.
Keywords: Chloride secretion, sodium absorption, colon, epidermal growth factor, PTP1B
Introduction
The intestinal epithelium constantly engages in the transport of water, electrolytes and other solutes to fulfill its physiological functions (Montrose, Keely, & Barrett 2003). Large quantities of gastrointestinal secretions are mixed with nutrients to permit digestion and absorption of substances needed by the body. Conversely, water and electrolytes that are utilized during these processes must later be reclaimed to avoid whole body dehydration and electrolyte derangements. The movement of fluid both into and out of the intestinal lumen is passive, and driven by the active secretion and absorption of electrolytes, as well as absorption of the end products of digestion and other luminal substances, such as bile acids. In the period between meals, moreover, water and electrolytes are transported across the intestinal epithelium to maintain fluidity appropriate for ongoing motility patterns and to flush the lumen free of toxins, microbes and any residues of digestion. During this period when nutrients are not present to drive the uptake of fluid, absorptive mechanisms center primarily around the electroneutral, coupled absorption of sodium and chloride, or, in the colon, by the electrogenic absorption of sodium. Fluid secretion, on the other hand, is driven primarily by the active electrogenic secretion of chloride ions, with a more modest contribution from bicarbonate. Chloride secretion occurs throughout the length of the gastrointestinal tract and during both the post-prandial period as well as the inter-digestive period.
The foregoing should hopefully make it clear that there is a need for close regulation of ion transport by intestinal epithelial cells. Not only is intestinal transport critical for normal physiological functioning of the gut, but there is substantial evidence that dysregulation of intestinal ion transport can lead to diseases, such as diarrheal disorders with a variety of underlying etiologies (Barrett & Keely 2006). Moreover, there is an increasing appreciation that intestinal fluid and electrolyte transport functions not only to determine the fluidity of the bulk luminal contents, but may provide for specific ionic conditions in microenvironments close to the epithelium, as well as contributing globally to the barrier function that permits the intestine to exclude substances and organisms that could be harmful to the host (Bair & Huang 1992;Chu & Montrose 1996;Chu & Montrose 1997;Chu & Montrose 1999;Genz, von Engelhardt, & Busche 1999;McEwan & Lucas 1990;Zamuner et al. 2003). There are accordingly numerous reasons why our laboratory is among many such groups who have sought a molecular-level understanding of the precise mechanisms that regulate intestinal transport.
This article summarizes a presentation made at the Frontiers of Physiology symposium held in Copenhagen, Denmark, in May 2008, to honor the occasion of Professor Ole Petersen's 65th birthday. In both this review and in the talk, we have sought to summarize our studies that have identified the epidermal growth factor (EGF) receptor (EGFr) as a critical regulator of mammalian intestinal ion transport. In particular, we focus here on the intricate signaling mechanisms that regulate both chloride secretion and sodium absorption by colonocytes, with emphasis placed on the factors that dictate the precise outcome to signals that may seem, on their face, to be quite similar if not identical. These concepts therefore underlie the ability of the intestinal epithelium to utilize a limited set of signaling effectors to produce a variety of outcomes suitable for varying physiological and pathophysiological demands. As such, moreover, we believe the subject matter of this review to be apt for the symposium in which the findings were discussed, in that Professor Petersen has devoted the majority of his career to dissecting the complex signals that mediate stimulus-secretion coupling in another gastrointestinal secretory cell type, the pancreatic acinar cell.
The epidermal growth factor receptor and chloride secretion
The epidermal growth factor receptor (EGFr) is a type I receptor tyrosine kinase that is abundantly expressed in the basolateral membranes of intestinal epithelial cells (among many other sites)(Uribe & Barrett 1997). It is well-known for its roles in regulating cell growth, differentiation and migration, in part by triggering changes in gene expression that specify altered cell function (Sibilia et al. 2007). In addition to these “classical” functions, it has likewise been appreciated that EGFr also exerts acute effects on a variety of cell types that are independent of changes in gene transcription and/or translation (Uribe & Barrett 1997).
In this vein, some years ago we showed that the EGFr is involved in the acute regulation of intestinal epithelial chloride secretion, as studied in human colonic epithelial cell lines, such as T84 (Uribe et al. 1996). Not only did EGF, which binds to and activates EGFr (also known as ErbB1), exert a direct inhibitory effect on calcium-dependent chloride secretion, but the receptor seemed also to participate in mediating the net expression of overall secretory processes activated by a variety of chloride secretagogues that act via G-protein coupled receptors (GPCR) (Bertelsen, Barrett, & Keely 2004;Keely, Uribe, & Barrett 1998). In the latter cases, EGFr was “transactivated” via signals that linked GPCR occupancy to EGFr phosphorylation, thereby leading to the recruitment of additional downstream signaling pathways (Keely & Barrett 1999;Keely, Calandrella, & Barrett 2000;Keely, Uribe, & Barrett 1998;McCole et al. 2002). Thus, as shown in Figure 1, the prototypic calcium-dependent chloride secretagogue, carbachol, binds to an M3 muscarinic GPCR to recruit calcium-dependent signals that initially activate chloride secretion. However, subsequently there is a cascade of signals that activate EGFr, both via the soluble tyrosine kinases Pyk2 and Src, and also secondary to the release from epithelial cells of the EGFr ligand, transforming growth factor-α (TGF-α). The effect of these subsequent signaling events, which involve the formation of EGFr homodimers, is to limit, or shut off, secretion. This outcome apparently occurs via the ability of EGFr to recruit the extracellular signal-regulated kinase (Erk) isoforms of mitogen-activated protein kinases, as well as possibly other signals, to inhibit an apical calcium-activated chloride channel. On the other hand, EGF itself can also act to inhibit chloride secretion, without, however, serving as an agonist of this process. Interestingly, the signaling pathways and target(s) that underlie the effect of EGF on secretion are apparently distinct from those used by carbachol, despite a functional outcome that is superficially similar. Thus, binding of EGF to its receptor recruits, in turn, the related EGFr family member, ErbB2 (Keely & Barrett 1999). The homodimers thereby formed appear to signal via a phosphatidylinositol 3-kinase (PI3K)/Akt dependent pathway, rather than Erk, to recruit downstream effectors including protein kinase C-ε (PKC-ε) that inhibit chloride secretion by reducing the activity of a basolateral potassium channel (Chow, Uribe, & Barrett 2000).
Figure 1.
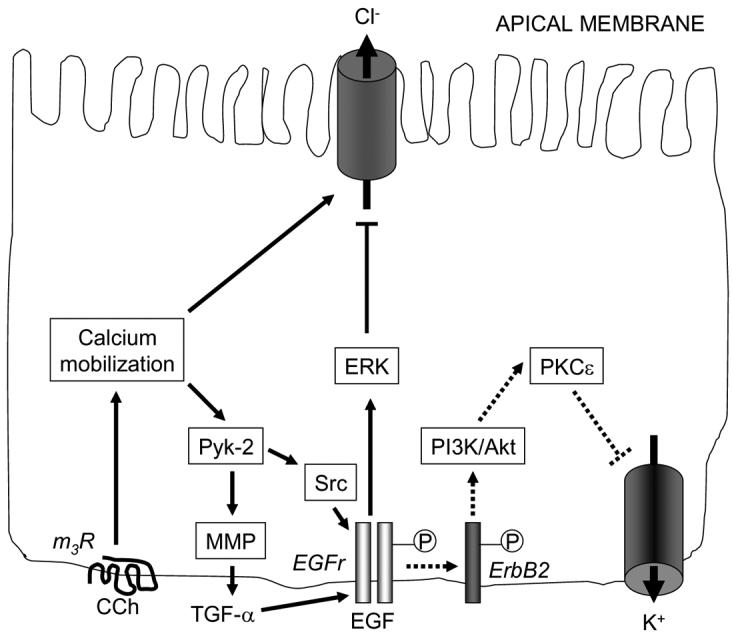
Pathways centering on the receptor for epidermal growth factor (EGFr) that regulate chloride secretion in colonic epithelial cells. Pathways that are activated by binding of EGF to EGFr are shown on the right-hand side and indicated by broken lines; those activated via EGFr transactivation are shown on the left-hand side of the figure, with solid lines. Ligands for receptors coupled to Gq, such as the m3 muscarinic receptor for the agonist, carbachol (CCh), initially cause calcium mobilization that can stimulate chloride secretion. However, they subsequently activate a negative signaling cascade via EGFr to limit further secretion. EGF can also inhibit secretion via its receptor, but via distinct signals that inhibit basolateral potassium channels. Please note that other components of the chloride secretory mechanism (Na+,K+ ATPase, Na+/K+/2Cl− cotransporter) are not shown in the figure for simplicity. MMP, matrix metalloproteinase; TFG-α, transforming growth factor α; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C. For additional details, see text.
Determinants of divergent EGFr signaling in vitro
The foregoing studies left us with an intriguing dilemma. We showed that both transactivation of EGFr, as well as its activation by its cognate ligand (EGF) were able to inhibit chloride secretion, but by making use of different downstream signals and targets to produce this response. Even more intriguingly, at least part of the transactivation response in fact involved activation of EGFr by another ligand for the receptor, TGF-α (McCole, Keely, Coffey & Barrett, 2002). Thus, we were led to hypothesize that EGF and TGF-α either had qualitatively different effects upon binding to the same receptor (which had not been reported hitherto), and/or that the signaling milieu that pertains in cells stimulated with carbachol can modify the consequences of TGF-α binding to EGFr.
In other systems, the various signaling outcomes that can ensue following activation of EGFr are believed to occur, at least in part, because the various tyrosine residues that can be phosphorylated in the cytoplasmic tail of EGFr are able to recruit distinct sets of downstream adaptor and effector proteins. We therefore utilized antibodies specific for the phosphorylated forms of six individual EGFr tyrosine residues to determine the pattern of phosphorylation that could be produced when T84 cells were stimulated with EGF itself, carbachol, or exogenously-added TGF-α (Hynes et al. 2001). These residues included those that are substrates for autophosphorylation (tyrosines 992, 1068, 1086, 1148, and 1173) as well as those that are modified by soluble tyrosine kinases, such as Src (tyrosine 845). T84 cells were treated with the stimulus of interest for various time periods, lysed, immunoprecipitated with antibodies to EGFr, then Western-blotted to reveal the extent to which individual tyrosine residues were phosphorylated.
The results obtained were as follows. While EGF and TGF-α exhibited quantitative differences in the extent to which they evoked phosphorylation of the various residues studied, their overall pattern of receptor phosphorylation was qualitatively similar, with all of the residues studied showing statistically significant increases in their level of phosphorylation in response to both growth factors (McCole et al. 2007). Carbachol, on the other hand, increased phosphorylation of tyrosines 845, 1086, 1148 and 1173, but had no consistent effect on the phosphorylation of either tyrosine 992 or 1068 (McCole, Truong, Bunz, & Barrett 2007). This was particularly intriguing in that carbachol is known to activate EGFr by triggering the cleavage of TGF-α from a membrane-bound precursor form (McCole, Keely, Coffey, & Barrett 2002). Thus, we were left to conclude that carbachol initiates other signaling events in T84 cells, and presumably in native colonocytes, that are capable of redirecting the outcomes of cell exposure to TGF-α (Figure 2).
Figure 2.

Different ligands that activate EGFr via either direct binding or transactivation elicit differing patterns of tyrosine residue phosphorylation in the cytoplasmic tail of the receptor. EGF and TGF-α both induce phosphorylation of six specific residues, albeit to different extents (as indicated by size of the circled letter P). Carbachol activates both the release of TGF-α as well as tyrosine phosphatases such as protein tyrosine phosphatase 1B (PTP1B) that serve to dephosphorylate specific residues.
The fact that fewer EGFr tyrosine residues were phosphorylated in cells exposed to carbachol vs. EGF or TGF-α led us to hypothesize that carbachol might stimulate one or more phosphatases that are active against EGFr. Indeed, broad spectrum tyrosine phosphatase inhibitors “uncovered” an ability of carbachol to phosphorylate both Tyr 992 and 1068 (McCole, Truong, Bunz, & Barrett 2007). Protein tyrosine phosphatase 1B (PTP1B) is one enzyme that has been shown to perform residue-specific cleavage of phosphate groups from the EGFr in other systems (Tonks 2003). In keeping with this, specific knock down of PTP1B in T84 cells using an siRNA reagent revealed an ability of carbachol to phosphorylate Tyr 992 and 1068. More importantly, either pharmacological inhibition or molecular knockdown of PTP1B redirected carbachol-initiated signaling in these colonic epithelial cells (McCole, Truong, Bunz, & Barrett 2007). While carbachol had no significant effect on PI3K-mediated signaling under control conditions, the loss of PTP1B activity was associated with a significant stimulation of the PI3K pathway as assessed by Akt phosphorylation. EGF, on the other hand, was able to stimulate PI3K whether or not PTP1B activity was intact.
Thus, we conclude that the pathways and consequences of EGFr-dependent signaling in colonic epithelial cells, and likely in other cell types, are closely regulated but may diverge depending on the signaling milieu that pertains at the time of cell activation. Specifically, transactivation of the receptor in response to the G-protein coupled ligand, carbachol, results in recruitment of PTP1B and perhaps other phosphatases that remove specific phosphate residues from EGFr, thereby normally precluding PI3-K/Akt dependent signaling (Figure 2). However, if PTP1B is inhibited by pharmacological or molecular means, then PI3-K/Akt signaling is restored.
The physiological significance of this complex signaling cascade and opportunities for differential signaling has yet to be established. However, we can speculate that the ability of carbachol to recruit PTP1B, and thereby reduce potential mitogenic signaling that might otherwise be produced when EGFr is transactivated in response to this ligand, might be important in preventing inappropriate stimulation of cell proliferation in the gastrointestinal tract during daily exposure to acetylcholine and other neurohumoral agonists that are released in response to a meal. On the other hand, PTP1B is over-expressed in the setting of inflammation, implying that EGFr signaling in response to receptor ligands or transactivating stimuli may be altered in diseases such as inflammatory bowel diseases (IBD) (Zabolotny et al. 2008). This may be linked to findings in our in vivo studies of the role of EGFr in regulating intestinal ion transport, which will be addressed below.
Regulation of overall colonic ion transport in vivo
In more recent work, we have sought to extend our observations in cell lines to the setting of intact intestinal tissues, which are likely more relevant to clinical conditions in humans. In particular, we have explored the regulation of intestinal ion transport in vivo, with a view to understanding not only the normal physiological regulation of such processes, but also how derangements of normal transport may underlie diarrheal symptoms in conditions such as ulcerative colitis and Crohn's disease.
As alluded to in the introduction, fluid balance in the normal colon reflects the competing influences of two transport vectors (Montrose, Keely, & Barrett 2003). Fluid absorption, which is performed predominantly by surface epithelial cells, is driven primarily by active electrogenic absorption of sodium ions via the ENaC sodium channel that is expressed particularly in the distal colon, as well as coupled uptake of NaCl and absorption of short chain fatty acids that are produced by bacterial fermentation of undigested carbohydrates (Montrose, Keely, & Barrett 2003). Opposing this fluid absorption, fluid secretion occurs predominantly across crypt epithelial cells, and is driven by the active secretion of chloride ions via the mechanism discussed above. While fluid balance in health tilts markedly in favor of absorption, particularly as one moves more distally in the colon, there is evidence that crypt secretion continues to be expressed, perhaps as the result of local reflexes, to match the fluidity of luminal contents to the need for their movement along the length of the intestine, and perhaps more importantly, to contribute to crypt sterility and protection of the stem cell niche (Humphries & Wright 2008;Muller, Autenrieth, & Peschel 2005;Sidhu & Cooke 1995). In this latter case, fluid secreted from the crypts can be promptly recycled across the surface epithelium to avoid dehydration (Figure 3). Indeed, the emerging evidence that some cases of chronic constipation can effectively be alleviated by treatment with a drug capable of inducing modest levels of intestinal chloride secretion further underscores the importance of an ongoing balance between secretion and absorption even in the most terminal segments of the gastrointestinal tract (JOHANSON & Ueno 2007).
Figure 3.
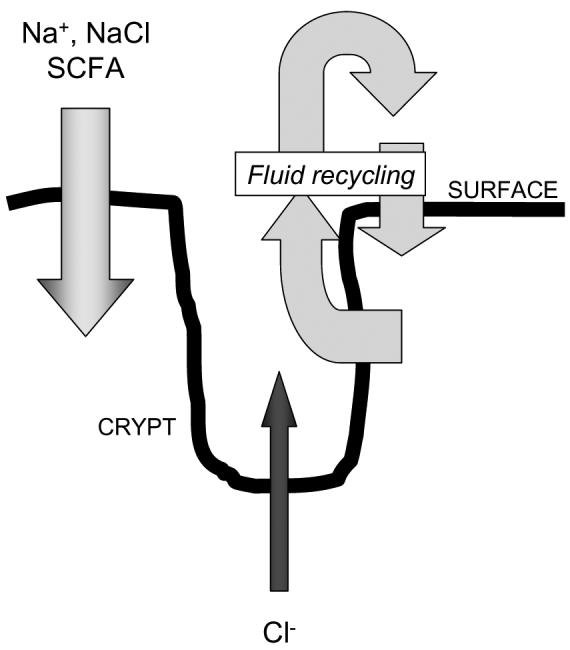
Model for colonic fluid and ion transport in health. Surface cells absorb sodium ions, NaCl and short chain fatty acids (SCFA). Crypt cells secrete chloride ions. The active absorption and secretion of these solutes drives fluid recycling from the crypt lumen followed by reabsorption across the surface epithelium. Absorption exceeds secretion, particularly in the distal colon, so little fluid is lost to the stool.
The foregoing discussion implies that over-expression of chloride secretion may be involved in cases of secretory diarrhea. This is certainly likely to be the case in some infectious disease states, as well as the rare cases of severe diarrhea that are associated with neuroendocrine tumors, such as VIPomas (Barrett & Keely 2006). However, in the particular instance of diarrheal symptoms in IBD, there is little evidence to suggest that chloride secretion is over-expressed, and in fact some evidence to the contrary (Greig & Sandle 2000;Sandle et al. 1990). On the other hand, electrogenic sodium absorption has been described as defective in patients with inflammatory bowel disease, although the precise mechanisms are still the subject of investigation (Greig & Sandle 2000;Greig et al. 2004). This has led us and others to propose a model for inflammatory diarrhea where the distal colon fails to appropriately salvage fluid coming from upstream, leaving it to be lost to the stool (Figure 4). At the same time, diminished chloride secretion from the crypts disrupts the normal flushing function of this site, rendering the crypt lumen vulnerable to the potential accumulation of toxic bacterial products as well, perhaps, to bacterial overgrowth. In turn, this may compromise crypt epithelial barrier function and further exacerbate fluid loss.
Figure 4.
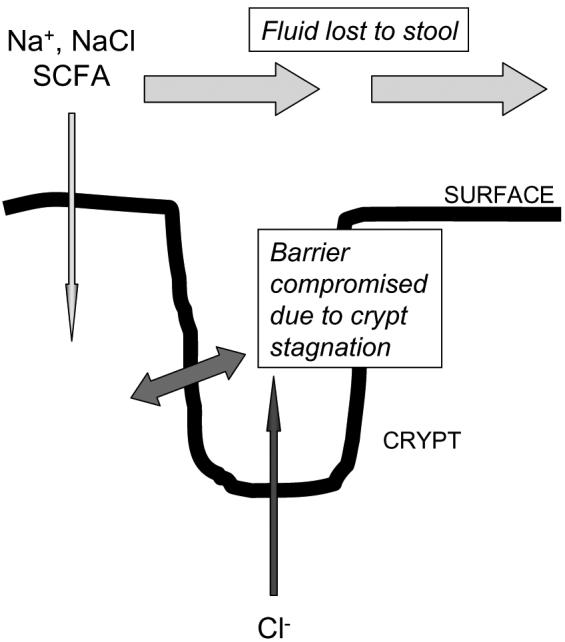
Model for colonic fluid and ion transport in the setting of colitis. Both surface and crypt transport processes are compromised compared to the situation in healthy tissues (compare with Figure 3). The lack of crypt chloride and fluid secretion results in crypt stagnation, the accumulation of toxins, and reduced barrier function in this site. The lack of adequate absorption across surface cells results in a failure to salvage luminal fluid, and the clinical finding of diarrhea.
Given the central role of electrogenic sodium absorption in distal colonic fluid homeostasis, it seems important here to digress slightly into what is known about the regulation of this transport mechanism. In fact, there have been surprisingly few studies of the signaling mechanisms that regulate this transport mechanism specifically in the colon, perhaps as a result of the paucity of suitable cell line models in which such signaling can conveniently be examined. On the other hand, electrogenic sodium absorption has been dissected considerably using epithelial cells of renal origin (Butterworth et al. 2005;Itani, Stokes, & Thomas 2005;Kamynina & Staub 2002;Mies et al. 2007;Rossier et al. 2002;Staub et al. 2000). Assuming that the process of sodium absorption is similar in both organs, some overall insights can be gleaned (Figure 5). First, as in the process of active chloride secretion, electrogenic sodium absorption can be stimulated by agonists that elevate intracellular levels of cAMP (Snyder et al. 2004). cAMP-associated signaling appears to regulate ENaC activity both by direct effects on the channel subunits themselves, and by modifying the activity of an associated ubiquitin ligase, Nedd4-2. Nedd4-2 is ordinarily responsible for ubiquitinating ENaC and inducing its internalization and degradation, including in response (appropriately) to an increase in cytoplasmic sodium concentrations. Thus the apical availability of ENaC is matched to the ability of the epithelium to dispose of excess sodium via the basolaterally-localized Na+,K+-ATPase, thereby also adjusting transepithelial absorption of sodium to meet whole body requirements. In the presence of elevated levels of cAMP, on the other hand, Nedd4-2 is inhibited secondary to protein kinase A-mediated phosphorylation (Snyder, Olson, Kabra, Zhou, & Steines 2004). This has the effect of increasing the residence time of ENaC in the apical membrane, and thereby increasing the rate of sodium absorption (Na+,K+-ATPase activity is not normally rate-limiting) (Kamynina & Staub 2002). Electrogenic sodium absorption can also be regulated by agonists that elevate cytoplasmic calcium concentrations. However, in contrast to cAMP, calcium inhibits ENaC function, perhaps via a direct interaction with the channel complex (Berdiev et al. 2001;Poulsen et al. 2005). This also stands in distinction to the process of chloride secretion, where both cAMP and calcium lead to increases in transport function. In the context of the intact tissue, therefore, agonists that elevate cAMP should have a less pronounced effect on distal colonic luminal fluid accumulation due to simultaneous activation of both chloride secretion and sodium absorption, whereas those acting through calcium (such as acetylcholine and purinergic agonists) should increase the amount of fluid in the lumen by transiently activating secretion while inhibiting absorptive processes.
Figure 5.
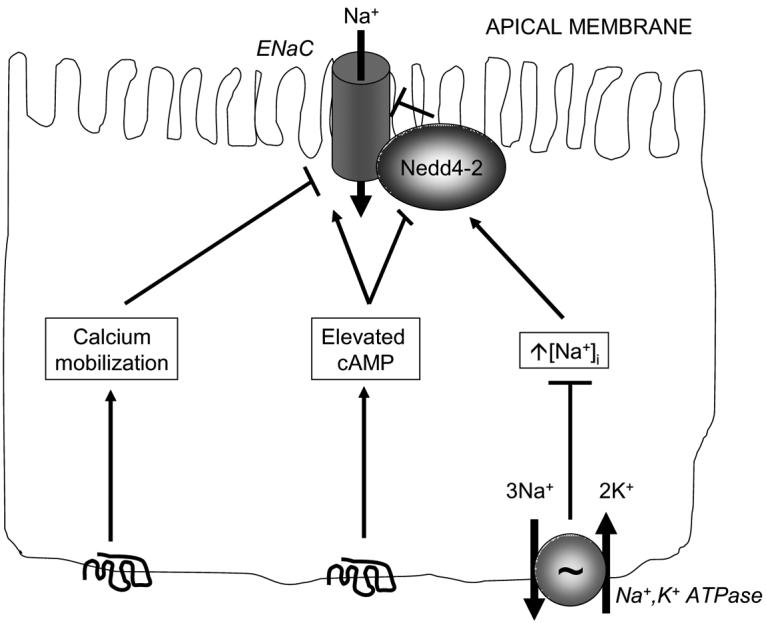
Mechanisms that regulate electrogenic absorption of sodium across epithelial cells. Sodium ions are taken up via the apical epithelial sodium channel (ENaC) and exit the cell via the basolateral Na+, K+ ATPase. The activity of ENaC can be reduced by agonists binding to receptors that stimulate mobilization of intracellular calcium stores. Elevations in intracellular sodium activate the ubiquitin ligase, Nedd4-2, which in turn triggers the internalization and degradation of ENaC. Agonists binding to receptors that elevate intracellular cAMP stimulate ENaC activity both directly, and by inhibiting Nedd4-2 activity. For additional details, see text.
The EGFr and colonic ion transport in vivo
In light of our prior work showing a critical role for EGFr in regulating chloride secretion by colonic epithelial cells in culture, we were interested in determining whether the receptor and/or its ligands are important in modulating overall ion transport in vivo, using as a model segments of distal colon harvested from mice. We first examined whether EGF altered ion transport in normal tissues. As predicted from our work in cell lines, acute addition of EGF inhibited not only carbachol, but also forskolin-induced increases in net ion transport across these tissues. Based on these findings as well as ion substitution studies, we concluded that EGF was able to attenuate both chloride secretion and sodium absorption across normal colonic epithelial cells (McCole et al. 2005).
We next examined the effect of EGF on ion transport in distal colonic tissues derived from animals in which chronic colitis had been induced by repeatedly subjecting the mice to cycles of dextran sulfate sodium (DSS) in their drinking water. This is a widely used model of colitis that mirrors at least some facets of inflammatory bowel disease (Elson et al. 1995). Net ion transport responses to both forskolin and carbachol were markedly reduced in these tissues, and consistent with prior work in both animal models of colitis and samples from patients with inflammatory bowel disease, these diminished responses reflected an inhibition of both chloride and sodium transport, as assessed by ion substitution studies (Asfaha et al. 1999;Greig & Sandle 2000;McCole, Rogler, Varki, & Barrett 2005;McKay & Singh 1997;Zamuner, Warrier, Buret, MacNaughton, & Wallace 2003). However, if EGF was used in these experiments to pre-treat tissues, rather than the inhibition of ion transport that we saw in normal tissues, EGF acutely enhanced net transport, and in the case of forskolin at least, restored it to normal levels (McCole, Rogler, Varki, & Barrett 2005). This response was also observed in another murine model of colitis occurring in animals deficient in the multidrug resistance protein 1a (mdr1a), implying that the effect of inflammation is not restricted to the DSS model (McCole, Rogler, Varki, & Barrett 2005). Such an effect, moreover, could represent a stimulation of chloride secretion, a stimulation of sodium absorption, or both. In chloride-free medium, EGF was still able to enhance ion transport responses evoked by forskolin. However, in the absence of sodium, the potentiating effect of EGF was lost. Coupled with the observation that the ability of EGF to enhance ion transport in inflamed tissues was abolished by the ENaC inhibitor, amiloride, we concluded that EGF acts specifically to enhance electrogenic sodium absorption when applied to inflamed tissues. Indeed, immunohistochemical analyses of colitic tissues revealed that the expression of ENaC is markedly reduced in the surface epithelial cells of inflamed colons versus healthy controls, suggesting that channel availability may be rate-limiting for absorption in the former setting (Greig, Boot-Handford, Mani, & Sandle 2004;McCole, Rogler, Varki, & Barrett 2005). However, our ion transport data would suggest that in spite of reduced ENaC expression, sufficient epithelial ENaC subunits are maintained in these inflamed tissues to facilitate restoration of forskolin-stimulated sodium absorption following acute treatment with EGF. In keeping with this, the ability of EGF to enhance sodium absorption was blocked when tissues were also treated with the PI 3-kinase inhibitor, wortmannin. PI 3-kinase activity has been implicated in preserving ENaC at the apical membrane, at least in renal cells, most likely by a combination of preventing internalization and degradation secondary to Nedd4-2 activity, and/or stimulating the insertion of channels from subapical compartments (Blazer-Yost, Esterman, & Vlahos 2003;Pearce 2003).
In summary, electrogenic transport of both chloride and sodium is reduced in mouse models of colitis (Figure 6). Preliminary observations, as well as the work of others, suggest that these findings can also be extrapolated to the setting of human patients with IBD (Greig & Sandle 2000;Greig, Boot-Handford, Mani, & Sandle 2004). The loss of sodium absorption, in particular, secondary to the inability to salvage fluid in the distal colon, may account for the diarrheal symptoms that are such a disabling feature of these disease states. Further, while EGF inhibits ion transport processes in normal tissues, its effect in the inflamed colon appears to be to restore active sodium absorption (Figure 6). Our findings imply that manipulation of the responsible signaling events might eventually prove useful in treating colitis-associated diarrheal disease. In support of this hypothesis, there is evidence that administration of EGF in enema form, given in conjunction with the orally-administered anti-inflammatory agent, mesalamine, has efficacy as an adjunct therapy in reducing diarrhea, and other parameters of disease, in UC patients (Sinha et al. 2003). However, we have yet to establish whether EGFr-mediated signaling also contributes to sodium transport responses in the intact colon that are mediated by G-protein coupled receptor ligands, such as acetylcholine, in the way that we have mapped out for chloride secretion using cell lines. Likewise, the basis for such seemingly opposite effects of EGF in inflamed vs. normal colon is the subject of ongoing investigation. It is of interest that parallel studies in renal cell lines have revealed both stimulatory and inhibitory effects of EGF on electrogenic sodium absorption mediated by ENaC (Blazer-Yost et al. 1999;Matsumoto et al. 1993;Tong & Stockand 2005). The discordant results appear to depend on the ability of the growth factor to activate ERK vs. PI 3-kinase. We are currently testing the hypothesis that inflammation alters the balance of signaling via these downstream pathways. Preliminary studies suggest that PI 3-kinase dependent signaling is favored, at the relative expense of ERK activation, when colonic epithelial cells are exposed to inflammatory cytokines (Figure 7).
Figure 6.

Differential effects of EGF on colonic ion transport in normal and inflamed tissues.
Figure 7.

Hypothesis as to how conflicting data in the literature showing both positive and negative effects of EGF on electrogenic sodium absorption can be reconciled. We hypothesize that in health (1), EGF predominantly activates ERK-dependent pathways that stimulate Nedd4-2 and thereby limit ENaC activity and sodium absorption. In the setting of inflammation (2), on the other hand, EGF signaling via PI3K-dependent pathways is favored, leading to sequential activation of phosphoinositide-dependent kinase 1 (PDK1), serum and glucocorticoid responsive kinase 1 (SGK1) and the ultimate inhibition of Nedd4-2, thus enhancing ENaC activity.
Conclusions and future perspectives
Based on the foregoing, we have concluded that EGFr, as well as its ligands, play pivotal roles in regulating the ion transport functions of intestinal epithelial cells. Such involvement comprises not only the signaling events that ensue when EGFr, as well as related ErbB receptor family members, are activated directly by their bona fide ligands, but also when the receptor is transactivated in response to signals that are initiated by ligands for G-protein coupled receptors, such as acetylcholine, a key neurohumoral regulator of epithelial function in the intact intestine(Hogenauer et al. 1999). However, the signaling pathways involved, their ultimate targets in terms of cellular transport machinery, and the associated functional outcomes may all depend on the precise cellular context in which EGFr activation is elicited. For example, even in reductionist models, colonic epithelial cells can distinguish between direct and indirect EGFr activation, in part by recruiting intracellular effectors that dictate the precise pattern of tyrosine residues that become stably phosphorylated in the cytoplasmic tail of the receptor, and thereby dictating the repertoire of downstream signaling pathways that the receptor can recruit and activate (McCole, Truong, Bunz, & Barrett 2007). In turn, this signaling diversification leads to variable functional outcomes. Further, in the setting of intact colon, we have developed evidence that the tissue milieu, at least with respect to the presence or absence of inflammation, may also modulate the precise responses to EGFr ligation (McCole, Rogler, Varki, & Barrett 2005).
Many questions remain to be answered in our exploration of the regulation of intestinal epithelial ion transport mechanisms, a set of key processes that contribute both to gastrointestinal health and disease. For example, we are still far from a complete understanding of the mechanisms and regulation of electrogenic sodium absorption in the distal colon, in contrast to the situation in the kidney. It is possible, but not confirmed, that the broad details of regulation may be conserved between these two tissues whose ion transport functions need to be coordinated to sustain whole body electrolyte homeostasis, particularly at times of excessive perturbation (for example, large shifts in sodium intake). But even if sodium absorption follows identical paradigms in the colon and kidney at the epithelial cell level, and in all species, it is likely that marked differences in the extracellular environment between these tissue sites will profoundly influence overall transport function. For example, the colon, but not the kidney, is continuously in a state of at least low-level or so-called “physiological” inflammation, although this can clearly be enhanced in specific disease states. Similarly, the functions of colonic epithelial cells are almost certainly modulated by the fact that they engage constantly in a presumed mutually beneficial crosstalk with a vastly complex intestinal microbiota consisting of hundreds of different species of microorganisms. Third, the function of various segments of the gastrointestinal tract are controlled in a highly coordinated fashion secondary to the influence of the enteric nervous system, which can function in a way that can be largely autonomous of either central nervous system or hormonal input. We also need to learn more about the precise role(s) of EGFr, its ligands, and its activation in regulating chloride secretion in vivo. An important role is implied by reductionist work, but we are not yet at a point where this information can confidently be used to intervene more effectively in disease states where epithelial ion transport is abnormal, and in a way that does not carry the risk of an increased risk of inappropriate cell proliferation and/or malignancy.
New experimental models will most likely be needed to address these questions. For example, our lack of knowledge with respect to the regulation of colonic electrogenic sodium absorption almost certainly reflects, at least in part, the lack of a suitable human intestinal epithelial cell line in which this transport process is robustly and reliably expressed. Our understanding of intestinal chloride secretion was increased exponentially when it became possible to explore this process in a controlled and reductionist fashion. There is hope that a similar situation will soon pertain for sodium absorption given recently described cell lines that express functionally competent levels of ENaC (Zeissig et al. 2006). However, as alluded to above, it will also be critical to examine regulatory mechanisms in the context of native intestinal tissues. Methods to follow signaling events in a cell-specific fashion, and perhaps even to link these to markers of cell transport function, ideally in real time, remain holy grails of research in this area. New optical/confocal methods coupled with genetically-engineered reporters of specific intracellular signals should ultimately assist in this direction.
The long-term goal of our research is to improve our ability to intervene in diseases where intestinal ion transport is abnormal. Significant pathologies can be linked to situations where ion transport responses are both under- and over-expressed. Classical examples include cystic fibrosis and cholera, respectively, and represent experiments of nature in which the basic underlying cellular and molecular defects have been largely defined. However, the gastrointestinal symptoms and overall outcomes of even these diseases are greatly influenced by cell-cell interactions within the complexity of the intact intestine, the enteric nervous system, modifying genetic and environmental influences, and extraintestinal factors (Barrett & Keely 2006). Moving beyond these relatively straightforward conditions, an understanding of the pathogenesis of diarrheal symptoms in patients affected by IBD, or following colonization with a range of enteric pathogens, is incomplete at best. Nevertheless, diarrheal diseases remain a scourge of humanity and can range from minor inconveniences, to disabling and painful manifestations of several disease states, to conditions that can prove fatal in vulnerable populations residing in developing countries. In light of imperfect current therapies for such conditions, worldwide emergence of significant antibiotic resistance among enteric pathogens, and the perpetual problem of supplying clean sources of food and water to all of the world's population even during stable times, to say nothing of times of natural and manmade disasters, it seems likely that the need for a fuller understanding of the basic biology of intestinal transport mechanisms will not abate. Ultimately, we believe that such an understanding should point, eventually, to safer, more targeted, and more effective therapies for both acute and chronic diarrhea.
Acknowledgements
Studies from the authors' laboratory have been supported by grants from the National Institutes of Health (DK28305) and the Crohn's and Colitis Foundation of America. The authors acknowledge the contributions of the following current and former lab members, as well as collaborators, to some of the studies described in this review article: Lone Bertelsen, Michael Bunz, Alfred Chappell, Jimmy Chow, Robert Coffey, Stephen Keely, Gisela Paul, Gerhard Rogler, Michael Scharl, Jane Smitham, Anh Truong, Jorge Uribe, and Nissi Varki. The assistance of Glenda Wheeler with manuscript submission is also gratefully acknowledged.
Footnotes
Conflicts of Interest
There is no conflicts of interest.
References
- Asfaha S, Bell CJ, Wallace JL, MacNaughton WK. Prolonged colonic epithelial hyporesponsiveness after colitis: role of inducible nitric oxide synthase. American Journal of Physiology - Gastrointestinal and Liver Physiology. 1999;276:G703–G710. doi: 10.1152/ajpgi.1999.276.3.G703. [DOI] [PubMed] [Google Scholar]
- Bair CH, Huang JD. Effect of theophylline on the intestinal clearance of drugs in rats. Journal of Pharmacy and Pharmacology. 1992;44:483–486. doi: 10.1111/j.2042-7158.1992.tb03651.x. [DOI] [PubMed] [Google Scholar]
- Barrett KE, Keely SJ. Integrative physiology and pathophysiology of intestinal electrolyte transport. In: Johnson LR, et al., editors. Physiology of the Gastrointestinal Tract. 4 edn Academic Press; San Diego: 2006. pp. 1931–1951. [Google Scholar]
- Berdiev BK, Latorre R, Benos DJ, Ismailov II. Actin Modifies Ca2+ Block of Epithelial Na+ Channels in Planar Lipid Bilayers. Biophysical Journal. 2001;80(no. 5):2176–2186. doi: 10.1016/S0006-3495(01)76190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen LS, Barrett KE, Keely SJ. Gs protein-coupled receptor agonists induce transactivation of the epidermal growth factor receptor in T84 cells: implications for epithelial secretory responses. J.Biol.Chem. 2004;279(no. 8):6271–6279. doi: 10.1074/jbc.M311612200. [DOI] [PubMed] [Google Scholar]
- Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI 3-kinase activity. AJP - Cell Physiology. 2003;284(no. 6):C1645–C1653. doi: 10.1152/ajpcell.00372.2002. [DOI] [PubMed] [Google Scholar]
- Blazer-Yost BL, Helman SI, Lee KD, Vlahos CJ. Phosphoinositide 3-kinase is required for aldosterone-regulated sodium reabsorption. AJP - Cell Physiology. 1999;277(no. 3):C531–C536. doi: 10.1152/ajpcell.1999.277.3.C531. [DOI] [PubMed] [Google Scholar]
- Butterworth MB, Edinger RS, Johnson JP, Frizzell RA. Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. Journal of General Physiology. 2005;125(no. 1):81–101. doi: 10.1085/jgp.200409124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JYC, Uribe JM, Barrett KE. A Role for Protein Kinase Cepsilon in the Inhibitory Effect of Epidermal Growth Factor on Calcium-stimulated Chloride Secretion in Human Colonic Epithelial Cells. Journal of Biological Chemistry. 2000;275(no. 28):21169–21176. doi: 10.1074/jbc.M002160200. [DOI] [PubMed] [Google Scholar]
- Chu S, Montrose MH. Non-ionic diffusion and carrier-mediated transport drive extracellular pH regulation of mouse colonic crypts. Journal of General Physiology. 1996;494:783–793. doi: 10.1113/jphysiol.1996.sp021532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Montrose MH. Transepithelial SCFA fluxes link intracellular and extracellular pH regulation of mouse colonocytes. Comparative Biochemistry and Physiology - A: Comparative Physiology. 1997;118:403–405. doi: 10.1016/s0300-9629(96)00329-5. [DOI] [PubMed] [Google Scholar]
- Chu S, Montrose MH. The glow of the colonic pH microclimate kindled by short-chain fatty acids, chloride and bicarbonate. Journal of Physiology. 1999;517:315. doi: 10.1111/j.1469-7793.1999.0315t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–1367. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- Genz AK, von Engelhardt W, Busche R. Maintenance and regulation of the pH microclimate at the luminal surface of the distal colon of guinea pig. J Physiol. 1999;517:507–519. doi: 10.1111/j.1469-7793.1999.0507t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig E, Sandle GI. Diarrhea in ulcerative colitis. The role of altered colonic sodium transport. Ann.N.Y.Acad.Sci. 2000;915:327–332. doi: 10.1111/j.1749-6632.2000.tb05260.x. [DOI] [PubMed] [Google Scholar]
- Greig ER, Boot-Handford RP, Mani V, Sandle GI. Decreased expression of apical Na+ channels and basolateral Na+, K+-ATPase in ulcerative colitis. J Pathol. 2004;204(no. 1):84–92. doi: 10.1002/path.1613. [DOI] [PubMed] [Google Scholar]
- Hogenauer C, Aichbichler BW, Porter JL, Fordtran JS. Effect of atropine and octreotide on normal active chloride secretion by the human jejunum in vivo. Gastroenterology. 1999;116:A880. [Google Scholar]
- Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer. 2008;8(no. 6):415–424. doi: 10.1038/nrc2392. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Horsch K, Olayioye MA, Badache A. The ErbB receptor tyrosine family as signal integrators. Endocr.Relat Cancer. 2001;8(no. 3):151–159. doi: 10.1677/erc.0.0080151. [DOI] [PubMed] [Google Scholar]
- Itani OA, Stokes JB, Thomas CP. Nedd4-2 isoforms differentially associate with ENaC and regulate its activity. AJP - Renal Physiology. 2005;289(no. 2):F334–F346. doi: 10.1152/ajprenal.00394.2004. [DOI] [PubMed] [Google Scholar]
- JOHANSON JF, Ueno R. Lubiprostone, a locally acting chloride channel activator, in adult patients with chronic constipation: a double-blind, placebo-controlled, dose-ranging study to evaluate efficacy and safety. Alimentary Pharmacology & Therapeutics. 2007;25(no. 11):1351–1361. doi: 10.1111/j.1365-2036.2007.03320.x. [DOI] [PubMed] [Google Scholar]
- Kamynina E, Staub O. Concerted action of ENaC, Nedd4-2, and Sgk1 in transepithelial Na(+) transport. Am.J Physiol Renal Physiol. 2002;283(no. 3):F377–F387. doi: 10.1152/ajprenal.00143.2002. [DOI] [PubMed] [Google Scholar]
- Keely SJ, Barrett KE. ErbB2 and ErbB3 receptors mediate inhibition of calcium-dependent chloride secretion in colonic epithelial cells. Journal of Biological Chemistry. 1999;274:33449–33454. doi: 10.1074/jbc.274.47.33449. [DOI] [PubMed] [Google Scholar]
- Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated transactivation of epidermal growth factor receptor and MAP kinase in T{−84} cells is mediated by intracellular Ca{+2+}, PYK-2, and p60{+src} Journal of Biological Chemistry. 2000;275:12619–12625. doi: 10.1074/jbc.275.17.12619. [DOI] [PubMed] [Google Scholar]
- Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and MAP kinase in T{−84} cells: implications for carbachol-stimulated chloride secretion. Journal of Biological Chemistry. 1998;273:27111–27117. doi: 10.1074/jbc.273.42.27111. [DOI] [PubMed] [Google Scholar]
- Matsumoto PS, Ohara A, Duchatelle P, Eaton DC. Tyrosine kinase regulates epithelial sodium transport in A6 cells. American Journal of Physiology. 1993;264:C246–C250. doi: 10.1152/ajpcell.1993.264.1.C246. [DOI] [PubMed] [Google Scholar]
- McCole DF, Keely SJ, Coffey RJ, Barrett KE. Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-à. Journal of Biological Chemistry. 2002;277:42603–42612. doi: 10.1074/jbc.M206487200. [DOI] [PubMed] [Google Scholar]
- McCole DF, Rogler G, Varki N, Barrett KE. Epidermal growth factor partially restores colonic ion transport responses in mouse models of chronic colitis. Gastroenterology. 2005;129:591–608. doi: 10.1016/j.gastro.2005.06.004. [DOI] [PubMed] [Google Scholar]
- McCole DF, Truong A, Bunz M, Barrett KE. Consequences of direct versus indirect activation of epidermal growth factor receptor in intestinal epithelial cells are dictated by protein-tyrosine phosphatase 1B. J Biol Chem. 2007;282(no. 18):13303–13315. doi: 10.1074/jbc.M700424200. [DOI] [PubMed] [Google Scholar]
- McEwan GTA, Lucas ML. The effect of E. coli STa enterotoxin on the absorption of weakly dissociable drugs from rat proximal jejunum in vivo. Br J Pharmacol. 1990;101:937–943. doi: 10.1111/j.1476-5381.1990.tb14184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay DM, Singh PK. Superantigen activation of immune cells evokes epithelial (T84) transport and barrier abnormalities via IFN-gamma and TNF alpha: inhibition of increased permeability, but not diminished secretory responses by TGF-beta2. The Journal of Immunology. 1997;159(no. 5):2382–2390. [PubMed] [Google Scholar]
- Mies F, Spriet C, Heliot L, Sariban-Sohraby S. Epithelial Na+ channel stimulation by n-3 fatty acids requires proximity to a membrane-bound A-kinase-anchoring protein complexed with protein kinase A and phosphodiesterase. J Biol Chem. 2007;282(no. 25):18339–18347. doi: 10.1074/jbc.M611160200. [DOI] [PubMed] [Google Scholar]
- Montrose MH, Keely SJ, Barrett KE. Electrolyte secretion and absorption: small intestine and colon. In: Yamada T, editor. Textbook of Gastroenterology. Lippincott Williams and Wilkins; Philadelphia: 2003. pp. 308–339. [Google Scholar]
- Muller CA, Autenrieth IB, Peschel A. Intestinal epithelial barrier and mucosal immunity. Cellular and Molecular Life Sciences (CMLS) 2005;62(no. 12):1297–1307. doi: 10.1007/s00018-005-5034-2. [DOI] [PubMed] [Google Scholar]
- Pearce D. SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem. 2003;13(no. 1):13–20. doi: 10.1159/000070245. [DOI] [PubMed] [Google Scholar]
- Poulsen AN, Klausen TL, Pedersen PS, Willumsen NJ, Frederiksen O. Regulation of ion transport via apical purinergic receptors in intact rabbit airway epithelium. Pflugers Arch. 2005;450(no. 4):227–235. doi: 10.1007/s00424-005-1388-4. [DOI] [PubMed] [Google Scholar]
- Rossier BC, Pradervand S, Schild L, Hummler E. EPITHELIAL SODIUM CHANNEL AND THE CONTROL OF SODIUM BALANCE: Interaction Between Genetic and Environmental Factors. Annual Review of Physiology. 2002;64(no. 1):877–897. doi: 10.1146/annurev.physiol.64.082101.143243. [DOI] [PubMed] [Google Scholar]
- Sandle GI, Higgs N, Crowe P, Marsh MN, Ventkatesan S, Peters TJ. Cellular basis for defective electrolyte transport in inflamed human colon. Gastroenterology. 1990;99:97–105. doi: 10.1016/0016-5085(90)91235-x. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Kroismayer R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007;75:770–787. doi: 10.1111/j.1432-0436.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- Sidhu M, Cooke HJ. Role for 5-HT and ACh in submucosal reflexes mediating colonic secretion. American Journal of Physiology. 1995;269:G346–G351. doi: 10.1152/ajpgi.1995.269.3.G346. [DOI] [PubMed] [Google Scholar]
- Sinha A, Nightingale J, West KP, Berlanga-Acosta J, Playford RJ. Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N.Engl.J Med. 2003;349(no. 4):350–357. doi: 10.1056/NEJMoa013136. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol.Chem. 2004;279(no. 44):45753–45758. doi: 10.1074/jbc.M407858200. [DOI] [PubMed] [Google Scholar]
- Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger JD, Schild L, Rotin D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000;57(no. 3):809–815. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- Tong Q, Stockand JD. Receptor tyrosine kinases mediate epithelial Na+ channel inhibition by epidermal growth factor. AJP - Renal Physiology. 2005;288(no. 1):F150–F161. doi: 10.1152/ajprenal.00261.2004. [DOI] [PubMed] [Google Scholar]
- Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Letters. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- Uribe JM, Barrett KE. Non-mitogenic actions of growth factors: an integrated view of their role in intestinal physiology and pathophysiology. Gastroenterology. 1997;112:255–268. [PubMed] [Google Scholar]
- Uribe JM, Gelbmann CM, Traynor-Kaplan AE, Barrett KE. Epidermal growth factor inhibits calcium-dependent chloride secretion in T{−84} human colonic epithelial cells. American Journal of Physiology. 1996;271:C914–C922. doi: 10.1152/ajpcell.1996.271.3.C914. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Kim Y-B, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J.Biol.Chem. 2008;283:14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamuner SR, Warrier N, Buret AG, MacNaughton WK, Wallace JL. Cyclooxygenase 2 mediates post-inflammatory colonic secretory and barrier dysfunction. Gut. 2003;52(no. 12):1714–1720. doi: 10.1136/gut.52.12.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeissig S, Fromm A, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Restoration of ENaC expression by glucocorticoid receptor transfection in human HT-29/B6 colon cells. Biochem Biophys.Res Commun. 2006;344(no. 4):1065–1070. doi: 10.1016/j.bbrc.2006.04.012. [DOI] [PubMed] [Google Scholar]