

Abstract
The mechanisms that trigger the phototoxic response to 2-chlorophenothiazine derivatives are still unknown. To better understand the relationship between the molecular structure of halogenated phenothiazines and their phototoxic activity, their photophysics and photochemistry were studied in several alcohols. The photodestruction quantum yields were determined under anaerobic conditions using monochromatic light (313 nm). Absorption- and emission-spectroscopy, 1H- and 13C-NMR, and GC-MS were used to characterize the photoproducts and reference compounds. An electron transfer mechanism had been previously proposed by Bunce and coworkers (J. Med. Chem. 22, 202–204) to explain the large difference between the photodestruction quantum yield of 2-chlorpromazine (φ = 0.46) and 2-chlorphenothiazine (φ = 0.20). According to these authors, the alkylamino chain transfers an electron to the phenothiazine moiety. Our results demonstrate that this mechanism is incorrect, because the photodestruction quantum yields of all chlorinated derivatives of this study are the same under the same conditions of solvent and irradiation wavelength. The quantum yield has no dependence on the 10-substituent, but it depends on the solvent. The percentage of each photoproduct, on the other hand, strongly depends on that substituent, but not very much on the solvent. Finally, it is demonstrated that the phototoxic effect of chlorinated phenothiazines is not related to the photodechlorination, although both processes share the same transient.
INTRODUCTION
Chlorpromazine (CPZ 2b, Fig. 1) is a major Tricyclic antidepressant drug (TCA). The studies on the photochemistry of this and other related TCAs were initially stimulated by the observation that it produces skin rashes and ocular changes in patients being treated with large doses. The mechanism responsible for this promazine-induced phototoxicity is still unknown, although several proposals have been made to account for the phototoxicity of CPZ (1–2). Kochevar and coworkers (3) attributed this response to the formation of dimers and higher multimers of the drug produced by pre-irradiation of CPZ. Nevertheless, the dimers and polymers of CPZ cannot form in concentrations high enough to be toxic. The in vivo therapeutic concentration of CPZ is only 0.03–3.0 µM, which is far less than the critical concentration required for dimerization (4).
Figure 1.

Structure of the phenothiazine derivatives and related compounds [1 R1 = H; 2 R1 = (CH2)3-N(CH3)2, 3 R1 = (CH2)3-CH(CH3)2]
Other mechanisms consider that the biochemical damages are produced by free radicals (5–6), ground state complexation (7–8), or the photoaddition of CPZ to ds-DNA (9–13). Based on the observation that the photobinding of CPZ in vivo can be induced even with longwave UVA light, Ljunggren and Moller (14) suggested that these adverse photobiological effects could also be caused by the CPZ-metabolites. Therefore, the attention was once focused on chlorpromazine sulfoxide (CPZSO), the major metabolite of CPZ (15). According to Rosenthal et. al. (16) and Davies et. al. (5), the sulfoxide can also be produced by the attack of singlet oxygen to the ground state of CPZ. These photooxidation reactions of CPZ were exclusively observed in aqueous solutions, but never in organic solvents. Photolysis of the sulfoxide derivative in aqueous solution further resulted in a species capable of oxidizing ascorbate, cysteine, gluthatione, NADH, and azide by single electron transfer (15). In addition, this species can abstract hydrogen atoms from ethanol and dimethyl sulfoxide. Since the oxidation does not require the presence of dissolved oxygen, the oxidizing species was proposed to be the hydroxyl free radical arising from the homolytic cleavage of the S-O bond of the sulfoxide. Nevertheless, Schoonderwoerd (17) ruled out the metabolic product of CPZ to be responsible for the drug phototoxicity. They based their conclusion on the bioavailability of CPZ and its oxidation product (CPZSO) in the skin and the absorption spectrum in the UVA spectral region. They also concluded that, in fact, sulfoxidation of CPZ results in less photobinding.
The ground state complexes between CPZ and DNA cannot be responsible for the phototoxicity either, because no complex formation was detected at physiological conditions (18). The involvement of the covalent binding of CPZ to DNA in the phototoxic side-effect has also been questioned. Rosenthal (16) found no toxic effects on E. Coli attributed to CPZ-sensitization. Our previous results showed that the triplet quantum yield of most TCAs strongly depends on the substituent at the 2-position and the solvent (19–21). It was further demonstrated that the triplet state of halogenated phenothiazine derivatives is efficiently quenched by a hydrogen transfer process and that the most phototoxic derivatives have a high triplet quantum yield and a short lifetime. Therefore, they concluded that the triplet intermediate is somehow involved in the phototoxic mechanism of the promazines (20).
Studies on the photochemistry of the phenothiazine family have produced a series of reports with different and, most of the time, contradictory results. This fact is mainly due to the spectrum of experimental conditions and initial drug concentration used in each study. Felmeister and coworkers (22) studied the photodecomposition of CPZ-HCl under aerobic and anaerobic conditions at 253.5 nm. For the kinetic studies and photodestruction quantum yield determination, they used microirradiation to excite its π→π* transition. The characterization of the photoproducts, on the other hand, was done in a concentration range of 10−2 to 10−3 M using a Hanovia ultraviolet lamp and removable glass filters transmitting from 360 to 370 nm. These wavelengths correspond to the excitation of the n→π* transition. They observed that the absorption spectra taken during the photolysis under aerobic conditions, changed in a different way than those taken under anaerobic conditions, implicating the formation of different products. Among the photoproducts, they characterized an alcohol derivative using acetylation reactions and IR spectroscopy. The photoproducts formed in the bulk reaction appeared to be more hydrophilic than CPZ and CPZSO. They reported photodestruction quantum yield values in aerobic and anaerobic conditions at 253.5 nm of 0.18 and 0.14, respectively. The major problem in these sets of experiments is that the characterized photoproducts at 360 nm are not necessarily the same ones quantified with 253.5 nm.
Davies and coworkers (5) found no spectral changes on the >300 nm irradiation of CPZ-HCl in iPrOH under aerobic conditions. Singlet oxygen is evidently incapable of oxidizing CPZ, confirming the previous observation of Iwaoka and Kondo (12). Under anaerobic conditions, on the other hand, they reported a photodestruction quantum yield of 0.12 and detected the formation of PZ and PZOCH(CH3)2. Based on these results, they proposed a direct homolysis of the triplet state of CPZ to produce the free radicals. The formation of these radicals was confirmed by irradiating an air-free CPZ solution in methyl methacrylate, which undergoes polymerization. For the determination of the quantum yield, they used a 3 mM solution and a Pyrex cell with a pathlength of 3.3 cm. According to this setup, an absorption gradient is produced within the first centimeters of the solution, rendering the quantum yield values uncertain.
Rosenthal and coworkers (16) used selectively labeled methanol to elucidate the mechanism of the photoreaction of CPZ-HCl under anaerobic conditions at wavelength longer than 300 nm. No hydrogen scrambling occurred along the reaction pathway between methyl and hydroxyl hydrogen as could be expected for the suggested all-radical mechanism proposed by Davies (5). They found that the isotopic composition of the methoxy group in the alkoxy-derivative is identical to that of the original methyl group in the methyl alcohol. They concluded that the methyl group of the solvent is the exclusive source of the hydrogen atom that replaces the chlorine. Therefore, for the formation of the methoxy-phenothiazine derivative, they proposed an ionic photonucleophilic substitution of the chlorine. They further found that the photolysis of CPH 2b under the same conditions produces 90% PH and ~2% MOPH. Similarly, the direct photolysis of CPZ-HCl resulted in almost total conversion to PZ and MOPZ, in addition to minor amounts of other decomposition products. The photodestruction quantum yield and the chemical yield of the CPZ photoproducts were not reported, but they observed that the N-alkyl side chain is not a critical requirement for this reaction. The concentration of CPZ-HCl in this reaction was 14 mM and the pathlength of the cylindrical Pyrex cell was not mentioned.
Until now, no direct evidence has been presented to confirm or rule out the participation of the TCAs neutral radicals in their phototoxicity. For the dehalogenation of 2-chlorinated promazines from the triplet state (3CPZ*), two parallel mechanisms have been proposed: homolytic cleavage of the C-Cl bond and nucleophilic attack of the solvent (5, 23–24). Nevertheless, data on the quantification of this process is scarce and most of the values show very poor reproducibility. Davies and coworkers found that the irradiation of the hydrochloride salt of CPZ (CPZ-HCl) in deoxygenated propan-2-ol solution yielded free chloride ion and a concomitant equimolar amount of hydronium ion with a quantum yield of 0.12 (5). Therefore, these authors proposed a direct homolysis of 3CPZ* affording radicals, which - in turn - abstract hydrogen atoms from the solvent. Bunce et. al. proposed that electron transfer is the major reason for the photochemical instability of CPZ and reported a quantum yield of 0.46 for the dehalogenation of chlorpromazine free base in degassed 4:1-acetonitrile-water mixtures (23). They also measured the quantum yield of the parent compound 2-chlorophenothiazine (CPH 1b) in the same solvent and reported a value of 0.20. Based on these findings, they concluded that the N-alkyl substituent is not a necessary requirement for the photodehalogenation, but may accelerate the process of chloride removal by the intramolecular electron transfer mechanism. Moore and coworkers reported a quantum yield value of 0.65 for this dehalogenation process in three different degassed solvents (propan-2-ol, methanol and water), but they did not specify if the drug was protonated or in the free form (24).
In this work, we report a systematic study of the photophysical properties and the quantum yields for the dehalogenation of CPH 1b and CPZ-HCl in methanol, ethanol, 1-propanol, 2 -propanol and t-butanol. The photolysis of the novel 2-chloro-10-(4-methyl)-pentyl phenothiazine (CMPPH, 3b) was also performed in selected alcohols to assert the contribution of the N-alkyl substituent to the dehalogenation process. A general photodestruction mechanism is proposed to account for the measured quantum yields, the characterized photoproducts, and the phototoxicity of these TCAs.
MATERIALS AND METHODS
Materials and chemicals
Phenothiazine (PH, 1a), chlorphenothiazine (CPH, 1b), the hydrochloride salts of promazine (PZ, 2a) and chlorpromazine (CPZ, 2b), anhydrous ethyl alcohol, anhydrous 2-propanol, anhydrous 1-propanol and anhydrous tert-butanol were purchased from Sigma-Aldrich (Illinois, USA). The hydrochloride salts of 2-methoxypromazine (MOPZ, 2c) and 2-trifluoromethylpromazine were a gift from the NIH-National Cancer Institute (Drug Synthesis & Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment). CPZ and MOPZ were purified by addition of NaOH to an aqueous solution of the protonated drug and then extracting with diethyl ether. All other compounds were used as received. Other HPLC-grade solvents were obtained from Fisher Scientific (Cayey, PR). High purity helium and nitrogen were purchased from Air Products (Humacao, PR).
Synthesis of MPPH (3a), CMPPH (3b), and MMPPH (3c)
Compounds 3a and 3b were synthesized by a method based on literature procedures with some minor modifications (25–26). Briefly, a solution of DMSO (25 mL) containing 0.0051 mol of the corresponding phenothiazine (1a or 1b) and 0.0051 mol of potassium hydroxide was stirred at room temperature, while adding 0.084 mL (0.0056 mol) of 1-bromo-4-methylpentane. After 4 h, 30 mL of water were added and the product was extracted by washing the solution several times with methylene chloride, saving the organic phase. This organic phase was then washed with water and brine, and dried over magnesium sulfate. The solvent was removed by rotary evaporation and the oily product was then purified with silica gel column chromatography with a hexane/ethyl acetate mobile phase. MPPH 3a was obtained with 47 % yield: 1H-NMR = 7.20-7.12 (m, 4H), 6.99-6.90 (m, 4H), 3.83-3.80 (t, J = 6.8 Hz, 2H), 1.69-1.62 (m, 2H), 1.51-1.44 (m, 1H), 1.27-1.22 (m, 2H), 0.80-0.78 (d, J = 6.4 Hz, 6H); 13C-NMR = 144.8, 127.8, 127.5, 127.0, 123.6, 122.3, 115.7, 46.6, 35.34, 27.0, 24.0, 22.4; and MS = 284(26), 283(100), 213(18), 212(84), 199(19), 198(33), 181(11), 180(19); CMPPH 3b was obtained with a 51% yield: 1H-NMR 400 MHz in CD3SOCD3: 7.23-7.13 (m, 3H), 7.03-6.94 (m, 4H), 3.86-3.82 (t, J = 6.9 Hz, J = 13.8 Hz, 2H), 1.53-1.44 (m, 1H), 1.28-1.22 (quartet, J = 6.9 Hz, J = 15.20 Hz, 2H), 0.80 (d, J = 6.6 Hz, 6H);13C-NMR = 146.8, 144.5, 132.9, 128.6, 127.7, 123.9, 123.2, 122.5, 116.8, 116.2, 47.1, 35.7, 27.5, 24.4, 22.9; and MS = 319(26), 318(14), 317(82), 248(36), 247(16), 246(100), 234(24), 233(25), 232(44). MMPPH 3c was obtained by a bulk photolysis of 3a in methanol. The product was obtained by solvent evaporation and separation with the same column chromatography. After purification, MMPPH 3c is obtained with a 16% yield: 1H-NMR = 7.21-7.16 (ddd, J = 8.2 Hz, J = 7.4 Hz, J = 1.4 Hz, 1H), 7.14-7.12 (dd, J = 7.6 Hz, J = 1.2 Hz, 1H), 7.04 -6.99 (m, 2H), 6.95-6.91 (ddd, J = 1.2 Hz, J = 0.80 Hz, J = 0.80, 1H), 6.59-6.54 (q, 2H), 3.86-3.82 (t, J = 7.0), 3.74 (s, 3H), 1.69-1.66 (q, 2H), 1.53-1.49 (m,1H), 1.30-1.24 (quartet, 2H), 0.82-0.81 (d, J = 6.4 Hz, 6H). 313(100), 243(21), 242(56), 229(35), 228(39); 13C-NMR = 160.0, 146.8, 145.1, 127.9, 127.8, 127.5, 124.8, 122.9, 116.4, 114.9, 107.9, 103.4, 55.8, 47.2, 35.9, 27.5, 24.6, 22.9; and MS = 313(100), 243(21), 242(56), 229(35), 228(39).
Absorption Spectroscopy, Gas Chromatography, NMR and Mass Spectroscopy
Absorption spectra were taken with a HP 8453 UV-Vis photodiode array spectrophotometer (California, USA). The chromatograms were taken with an Agilent GC 6850 gas chromatograph (California, USA) with a capillary column model Restek 176832 (Stationary phase=35% diphenyl-65% dimethylpolysiloxane, Nominal length=30 m). The oven conditions were set to: Initial Temp=200 °C, Final Temp= 300 °C and Ramp= 10 °C/min. The detector conditions were set to: Detector=FID, Temp=350 °C, Hydrogen Flow=40 mL/min, and Air Flow=450 mL/min. The inlet conditions were: Mode=Split, Initial Temp=280 °C, Split Flow=10 mL/min and Gas Type= Helium. The proton- and carbon-NMR spectra were taken with an Advance 400 NMR spectrometer (Texas, USA) using the 5 mm Bruker BioSpin BBO probe (Boston, USA). Deuterated dimethyl sulfoxide was used for all solutions. For the mass spectra, the separation of the products was done with a Thermo Finnigan Trace GC/Polaris Q chromatograph with a capillary column model Restek 12623 (Stationary phase RTX-5MS: Crossbond® 5% diphenyl / 95% dimethyl polysiloxane, Nominal length =30 m). The oven conditions were set to: Initial Temp=90 °C, Final Temp=250 °C, and Ramp=10 °C/min. The detector conditions were set to: Ion Source=200 °C, Transfer Line=275 °C, Scan Mode=Full Scan (Range 50–650), Electron Impact=70 eV, and Mass Selector=Ion Trap. The inlet conditions were: Mode= Split, Temp= 200 °C, Split Flow=26 mL/min, Split Ratio=17, Gas Type=Helium, and Constant Flow= 1.5 mL/min.
Photodestruction Quantum Yields
The photolysis light source was a Sylvania 200 W high pressure Hg–Xe lamp and the 313 nm line was isolated with a 1/8 m Spectral Physics grating monochromator (Cincinnati, USA). The lamp intensity was determined before and after each set of photolysis with the Packard and Hatchard method using the potassium ferrioxalate actinometer (27). All photoreactions were carried out in a quartz cuvette (1 ×1 ×4 cm3) for up to 10–80 % conversion of the starting material and using the same cell orientation. 3 mL of multiple solutions of ~0.22 mM of the hydrochlorinated TCA or its free base in each alcohol, previously saturated with helium or dry nitrogen (~15 min), were irradiated with 313 nm for different times at room temperature. The photoreaction was controlled with an electronic shutter managed by a Labview 7.5 based program (Texas, USA). After photolysis, 45 µL of a 20.00 mM alcohol-solution of 2-trifluoromethylpromazine (TFMPZ) were added as internal standard for the determination of the conversion percent and the yields of the photodestruction. Then, 2 µL of this mixture were injected at least three times in a 6850 gas chromatograph to determine the quantity of remaining TCA. Calibration curves of amount ratio vs area ratio of each phenothiazine derivative or the corresponding photoproduct were prepared using a concentration range of 0.05 mM - 0.30 mM and a constant concentration of 0.15 mM of TFMPZ. All solutions used for calibration were injected three times and the average of the integrated area was used for the curve. An absorption spectrum was taken for all solutions before and after irradiation. Since the photodestruction of the TCA is a zeroth order reaction for small irradiation times, its quantum yields were determined from the linear part of the[TCA] vs time plot using the following equation:
| (1) |
where k is the photodestruction rate constant (slope of the plot in M/s), V is the reaction volume (3 mL), Io is the lamp intensity at 313 nm, [TCA]o is the initial drug concentration, and α is the destructed fraction of the TCA. The division by 2 in the absorption term is included to account for the gradient produced in the Iabs term, i.e. the absorbed intensity is taken as the average of the initial and final absorption. In the same manner, the formation quantum yield of PZ and other photoproducts were determined with the equation:
| (2) |
where [PZ]f is the amount of PZ formed after irradiation and the factor 0.5 is introduced also to average the absorption of PZ and taking [PZ]o = O. The determination of [PZ]f for small irradiation times is difficult, since there is almost no product formed and sometimes the regression gives non-zero intercepts. For these cases, corrections were done by forcing a zero intercept and keeping the same value of k.
Characterization of the Photoproducts
The photoproducts were identified and characterized with GC-MS using standards. All of them have a very similar mass spectrum, since they have a very similar MS fragmentation pattern (data not shown). Typical values of m/z (%) are the following: PH 1a: 166(27), 167(70), 168(11), 199(100), 200(15); CPH 1b: 198(63), 199(42), 234(22), 235(49); MOPZ 2c : 299(50), 297(49), 218(100), 217(50), 185(35); EOPZ 2d: 328(100), 243(48), 86(30), 58(72); POPZ 2e: 342(100), 257(45), 86(28), 58(61); iPOPZ 2f : 299(50), 297(49), 218(100), 217(50); and TBOPZ 2g: 356(100), 300(27), 215(27), 86(27), 58(50).
Theoretical Calculations
All geometry optimizations were initially done at the semiempirical level with the Polak-Ribiere conjugated gradient protocol with 1×10−5 convergence limit and 0.01 kcal/(Å mol) rms limit, as previously described (28). Density functional theory (DFT) with B3LYP/6–31 G(d) was used for the optimization refinement and the calculation of the dissociation energies of solvents and TCA molecules, according to the equations (3) and (4):
| (3) |
| (4) |
where ΔHf (X-Y) is the formation enthalpy of the species XY and D(X-Y) is the dissociation enthalpy of the X-Y bond. For some radicals, including ΔHf(H•) = 52.1 kcal/mol and ΔHf(Cl•) = 28.95 kcal/mol, the experimental values are taken from the literature (29–32). For systems with known D(R-H), the D(R1-R2) is calculated using the combination of all reactions given in Eq. (3), according to:
| (5) |
Evaluation of steps in the photodestruction mechanism
The number of steps or “independent reactions” (s) involved in the photodestruction of the 2-chlorinated phenothiazine derivatives is required for the proper formulation of a mechanism. As previously mentioned, Rosenthal (16) and Grant and Greene (6) proposed a mechanism, in which the first step is the homolytic cleavage of the C-Cl bond in the triplet excited state. Then the promazinyl radical abstracts a hydrogen from the solvent to yield the parent compound. The second proposed reaction is the nucleophilic attack of the solvent to the 3TCA* to produce the alkoxy-derivative. Therefore, the mechanism of the photolysis of chlorinated phenothiazines should consist of two reactions.
The evaluation of s in the TCA photodestruction was done according to the method described by Mäuser (33). Briefly, for r parallel reaction-steps involving n components A of the general form given in Eq. (6), the total change in the concentration of the ith component (Δ[A]i) is given by Eq. (7), where xk is the degree of advancement of the kth reaction and νki is the coefficient of the ith component in the kth reaction.
| (6) |
| (7) |
If for this system, h absorbencies Eλ (t)(λ = 1, h) are measured at m different times tj (j = 1, m), the matrix for the difference in absorption at a particular wavelength (ΔEλ) can be rearranged to give Eq. (8)
| (8) |
In this set of equations, d is pathlength of the cell and ε is the extinction coefficient of Ai at a particular wavelength. If at a given time t, there is any relationship between the xk (with constant coefficients ak) or the qλk (with constant coefficients bk), i.e. if some of the reactions are not independent from one another and there are only s independent steps (i.e. the rank of the matrix is s < r), then all linear relationships are equal to zero for k=1,r (Eq. 9) and Eq. (8) yields Eq. (10).
| (9) |
| (10) |
According to this equation, it is always possible to determine the value of s by measuring absorbencies, without knowledge of the individual absorption coefficients. For an isosbestic point, for instance, this equation equals zero for a given wavelength at all times. Nevertheless, isosbestic points only give limited information about the uniformity of a chemical reaction. More reliable expressions can be made using absorbency difference diagrams (ED-diagrams) (33). The total change in absorbency for two different wavelengths with time is given by Eq. (11), which yields Eq. (12) for s = 1.
| (11) |
| (12) |
The ED-diagram results in a straight line passing through zero with the slope Q11/Q21. The ED-diagrams must be examined at as many wavelength combinations as possible, because an ΔE1 vs ΔE2 plot can be linear by chance (34–35). In this case, Eq. (11) requires three different wavelengths (λ = 1, 2, 3), equation (11) is given in the same way for the extra ΔE3(t) and the corresponding Kronecker-Capelli′s determinant is equal to zero (Eq 13).
| (13) |
In this case, D23ΔE1 + D13 ΔE2 + D12ΔE3 = O with D23 = Q21Q32 − Q31Q22, D13 = Q11Q32 − Q31Q12 and D12 = Q11Q22 − Q12Q21. These equations result in a 3-dimensional space spanned by the absorbency differences ΔE1, ΔE2 and ΔE3. They are presented graphically in a two dimensional plot by rearranging them into equation (14):
| (14) |
where ρ=−D23/D12 and σ=−D13/D12. Plots of ΔE3/ΔE1 vs ΔE2/ΔE1 are called “extinction difference quotient diagram” or EDQ-plots (33). This type of diagrams become linear if either the mechanism consists of only two steps or, if in more than two linear independent partial reactions, the rank of the matrix Q reduces to two by chance. For a clear discrimination between these cases, as many combinations as possible have to be chosen in a large wavelength range extended far into the short wavelength region.
RESULTS AND DISCUSSION
Photophysics of CPH 1b
The absorption spectra of the free base phenothiazines present two main bands in the 250–265 nm and 300–320 nm wavelength ranges (Table 1 and Fig 2) (20). The first band is attributed to a π→π* transition and the other belongs mainly to an n→π* transition involving the sulfur lone-electron pairs (36). The absorption spectra of PH 1a and CPH 1b show some significant differences (Fig. 2), including the blue shift of ~5 nm in the absorption maximum of PH relative to CPH (Table 1). This shift and the corresponding higher oscillator strength is introduced by the chlorine atom. CPH has larger absorption extinction coefficients than PH at wavelengths > 310 nm and smaller values for wavelengths < 310 nm, producing an isosbestic point at 310 nm. Therefore, the resulting absorption spectrum of a mixture of CPH and PH has a maximum wavelength at 321 nm. This behavior should describe the spectral changes of photolyzed solutions of CPH, if its only photoproduct is PH.
Table 1.
Photophysical properties of the phenothiazines in different alcohols under anaerobic conditions, including the irradiation wavelength (313 nm). The emission maxima were measured in methanol.
| Phenoth | λmax (nm) [ε(M−1cm−1) × 10−4] | λmax (nm) [f] | λemm (nm) | |
|---|---|---|---|---|
| Methanol | Ethanol | Theoretical | ||
| 254 [5.1], 313 [0.439], | 319 [0.508], 313 [0.501] | 208 [0.08], 253 [0.32], | 442 | |
| PH 1a | 318 [0.48] | 262 [1.08], 323 [0.11] | ||
| 256 [4.86], 313 | 257 [4.55], 313 [402], | 206 [0.46], 216 [0.16], | 450 | |
| CPH 1b | [0.484], | 325 [0.467] | 262 [1.12], 321 [0.23] | |
| 323 [0.430] | ||||
Figure 2.

LEFT: Normalized absorption spectra of methanol solutions of PH 1a (•••), CPH 1b (□) and their equimolar mixture (- - -). RIGHT: Absorption curves for the photolysis of CPH in methanol under anaerobic conditions, Irradiation wavelength = 313 nm; Lamp Intensity = 4.70 × 10−10E/s, Time intervals for the irradiation = 200 s with t(a) = o s.
Photochemistry of CPH 1b
The major photoproduct found for CPH (1b) in all alcohols was PH (1a). In methanol and ethanol, a small amount of the corresponding alkoxide (PH-OR) and other unidentified products was detected for long irradiation times (t>720 s), as illustrated in Eq. (15) for R1 = H.
 |
(15) |
The absorption spectra of irradiated solutions of CPH changes according to the type and amount of products formed. In methanol, for instance, the absorption increases for wavelengths < 320 nm, indicating that the photoproducts have larger molar absorption coefficients at these wavelengths (Fig 2). Although the general characteristics at wavelengths > 330 nm are very similar to those expected, the formation of the isosbestic point is observed at 328 nm and not at 310 nm. Besides, for long irradiation times, the absorption maximum at 308 nm does not match with the maximum of the mixture at 321 nm. These new characteristics are most probably due to the formation of several other photoproducts with different absorption profiles.
For the determination of the quantum yield of the CPH photodestruction and the PH photoformation, the concentration of each compound was determined as function of irradiation time using the integration capabilities of the GC. For very long irradiation times (t > 720 s), single exponential behavior was observed for both processes, which is characteristic of first order reactions (Fig. 3). It also indicates that there might be, among other processes, filter effects and secondary reactions. For short irradiation times (t < 720 sec), on the other hand, there is a linear concentration/time dependence, indicating a zeroth order photoreaction. The quantum yields were calculated using the linear part of the corresponding plot, which is maintained for up to 45% CPH photodestruction. Table 2 summarizes the kinetic values for the photoreactions in methanol and ethanol, which corroborate that the major photoproduct of this reaction is PH. The fact that the photodestruction quantum yield of CPH is slightly larger than unity, definitely indicates that other processes might be going on in this reaction and/or the amount of absorbed light is underestimated in equation (1). This is further confirmed by the smaller value of φPH, compared to φCPH. The quantum yield for the formation of the alkoxide and all other unidentified photoproducts can be calculated to be φOther = 0.34 in methanol and only 0.11 in ethanol. To verify this statement, the mass balance of the photoreaction was determined in terms of the recovered mass percent. It was found that the larger the irradiation time, the bigger the amount of unrecovered material. For long irradiation times, a limiting chemical yield for PH of 68% was obtained for methanol, in disagreement of the yield measured by Rosenthal of 90% (16).
Figure 3.

Photokinetics of CPH 1b in methanol under anaerobic conditions and irradiating with 313 nm light. The solid lines represent the linear regression for 0–720 sec. The photodestruction of CPH (□) produces mostly PH (●). The linear regressions yield: [CPH]t = 0.219 – 1.39 × 10−4t [r2=0.9982], and [PH]t = 0.0003 + 9.15 × 10−5t [r2= 0.9979]
Table 2.
Isosbestic point of the PH-CPH mixtures before and after photolysis, number of independent reactions (s), kinetic constant (k) and quantum yield (φ) for the production of PH and destruction of CPH.
| Solvent | Isosbestic Point (nm) | S | k (mM/s) × 105 | φ | |||
|---|---|---|---|---|---|---|---|
| before | after | PH | CPH | PH | CPH | ||
| MeOH | 310 | 328 | 1 | 9.15 | 1.39 | 0.70 ± 0.03 | 1.04 ± 0.03 |
| EtOH | 330 | 320 | 1 | 12.4 | 14.7 | 1.03 ± 0.09 | 1.14 ± 0.04 |
The number of independent reactions was asserted with the ED and EDQ diagrams. For both methanol and ethanol, excellent linear ED-plots were obtained with zero intercept and r2 > 0.999 (Fig. 4, Left). This indicates that, since there is only one main photoproduct, the photodecomposition of CPH occurs through a single reaction, yielding the reduction product PH (s=1, Table 2). This was further corroborated with the EDQ plots (not shown), which have r2 values smaller than 0.83, especially for wavelengths in the 300 – 310 nm range and irradiation times >720s.
Figure 4.

LEFT: ED-plots for the photolysis of CPH in methanol for the following wavelengths combinations (nm): ΔE280 vs ΔE290 (●); ΔE280 vs ΔE300 (□); ΔE280 vs ΔE360 (□); ΔE290 vs ΔE300 (□); ΔE290 vs ΔE360 (□); and ΔE300 vs ΔE360 (□). RIGHT: ED-plots for the photolysis of CPZ-HCl in ethanol for the following wavelengths combinations (nm):ΔE280 vs ΔE290 (●); ΔE280 vs ΔE310 (□); ΔE280 vs ΔE320 (□); ΔE290 vs ΔE310 (Δ); ΔE290 vs ΔE320 (□), and ΔE310 vs ΔE320 ( □)
Photophysics of CPZ (2b) and CMPPH (3b)
The absorption and emission properties of CPZ 2b and similar compounds have been previously reported (19,20) (Table 3). The emission spectra of all 10-alkylated phenothiazines consist of a broad band with maximum between 440 and 470 nm in all solvents. They also present Stoke’s shift larger than 104 cm−1, which is a considerable magnitude. The emission maxima are more solvent dependant than the corresponding absorption maxima. No differences were observed in the absorption properties of CMPPH 3b. Compared to the parent PZ 2a, chlorinated derivatives have small fluorescence quantum yields, especially in methanol. All promazines have φf values in the order of 10−2 – 10−3. Therefore, other deactivation mechanisms for the S1 state must be competing favorably with the fluorescence process for these molecules. The 10-alkylamino chain was found to be very flexible in solution, given to all molecules several thermally accessible stable conformations (37–38). The emission properties of promazines 2 are also very sensitive to the solvent and the 2-substituent, but not to the alkylamino chain (20, 39). The fluorescence lifetime (τf) shows the same substituent and solvent dependency. The 10-alkyl chain has no effect on the lifetime values, as shown by the τf values reported by these authors. The small lifetime values (<1.0 ns) reported for the chlorinated derivatives are also attributed to the chlorine atom, which can enhance the spin-orbit coupling in the S1→So non-radiative or the ISC processes (40).
Table 3.
Absorption, emission and triplet-state properties of the promazine derivatives measured in methanol
| Phenothiazine derivative | λmax (nm) [ε(M−1cm−1) × 10−4] | λmax (nm) , Stoke’s Shift (cm−1), φf × 103 and τf (ns) | λmax (nm), εT × 10−4 (M−1cm−1), φT, τT (µs) |
|---|---|---|---|
| PZ 2a | 255 [3.3 ± 0.2], | 444 | 460 |
| 307 [0.42 ± 0.03] | 10,050 | 2.65 | |
| 4.5 | 0.41 | ||
| 1.75 † | 61 | ||
| CPZ 2b | 258 [3.66 ± 0.01], | 449 | 460 |
| 311 [0.46 ± 0.01]‡ | 9,474 | 1.95 | |
| 0.95 | 0.90 | ||
| 0.89 † | 2.2 § | ||
| CMPPH 3b | 258 [3.41 ± 0.03], | -- | -- |
| 313 [0.44 ± 0.02] |
Values from reference (19).
Corresponding values in ethanol are: 257 [3.26 ± 0.04], 308 [0.409 ± 0.001], 313 [0.400 ± 0.001]; 1-propanol: 257 [3.83 ± 0.06], 309 [0.472 ± 0.001], 313 [0.461 ± 0.001]; 2-propanol: 257 [3.37 ± 0.03], 309 [0.417 ± 0.002], 313[0.408 ± 0.002]; and t-butanol: 257 [3.21 ± 0.05], 310 [0.398 ± 0.001], 313 [0.391 ± 0.001].
Values from reference (20).
The laser flash transient absorption spectrum of nitrogen saturated solutions of 10-alkylated phenothiazines 2 at high laser intensities generally consists of an intense band with a maximum between 460–480 nm, one near 530 nm and another very broad one extending into the red region of the spectrum (Table 3) (19,20,21). A self-quenching process of their triplet state was reported by Barra and coworkers (41). Self-quenching rate constants in the order of 107–108 M−1s−1 were obtained for several derivatives, in excellent agreement with those previously reported for the non substituted phenothiazines (41). The triplet state molar absorption coefficients are of the order of 1.5 – 7.8 ×104 M−1cm−1 (19,20,39). The intersystem crossing quantum yields (φT) are in the range of 0.2 – 0.9 and show some solvent dependence. For the chlorinated derivatives, for instance, φT cannot be measured in aqueous solutions, because their triplet state is rapidly quenched by a proton transfer process (19). In methanol, on the other hand, CPZ-triplet forms with an impressive quantum yield of 0.90. The triplet lifetimes is a very solvent/substituent-sensitive property too, but the 10-substitution has the least effect. Davies reported triplet lifetime values for PZ-HCl and CPZ-HCl in isopropanol of 22.8 and 3.2 µs, respectively (5). The 2-substitution, on the other hand, induces a larger variation in the triplet lifetime values, as noted for the promazines in methanol (19,20).
Photochemistry of CPZ 2b and CMPPH 3b
The photolysis of CPZ-HCl 2 was carried out in methanol, ethanol, 1-propanol, 2-propanol and t-butanol. The photolysis of CMPPH 3b was studied only in MeOH and EtOH. In all these solvents the ground state molar absorption spectra of CPZ-HCl show two bands with maxima around 258 and 312 nm (Table 4). The spectra of photolyzed CPZ-HCl in methanol, ethanol and isopropanol present an isosbestic point in the 304–312 nm range, although the isosbestic points of the corresponding non-photolyzed mixtures with PZ and the alkoxy derivative are not the same. In methanol, the isosbestic point is blue-shifted with irradiation time. In tert-butanol, no isosbestic point is observed, the only product is tertbutoxypromazine, and the photoreaction is slower than in the other solvents. For CMPPH 3b , the isosbestic points are observed at 309 nm and 308 nm in MeOH and EtOH, respectively, and both shift by 9 nm on irradiation. To be certain about the participation of the triplet excited state in the dehalogenation process of CPZ, some of these photoreactions were performed in the presence of 0.35 mM the triplet quencher 1,3 cyclohexadiene [ET = 52.6 kcal/mol (42)], keeping all experimental conditions constant. In this case, no photodestruction was observed, confirming that the photodehalogenation occurs from the 3CPZ*.
Table 4.
Isosbestic point of the PZ-CPZ mixtures before and after photolysis, number of independent reactions (S), kinetic constant (k), percentage of photo-conversion of CPZ and formation of PZ, and quantum yield (φ) for the production of PZ and destruction of CPZ.
| Solvent | Isosb. Point (nm) | S | k (mM/s) × 105 | % | φ | ||||
|---|---|---|---|---|---|---|---|---|---|
| Before | After | PZ | CPZ | CPZ | PZ | PZ | CPZ | ||
| MeOH | 306 | 298–302† | 2 | 7.0 | 1.62 | 82 | 37 | 0.39 ± 0.01 | 0.93 ± 0.03‡ |
| EtOH | 315 | 306§ | 1¶ | 12.0 | 16.3 | 89 | 66 | 1.02 ± 0.06 | 1.15 ± 0.05□ |
| 1-PrOH | 304 | 303 | 1 | 13.4 | 16.1 | 84 | 83 | 0.85 ± 0.05 | 1.01 ± 0.06 |
| 2-PrOH | 312 | 306 | 1 | 16.4 | 16.4 | 91 | 89 | 1.1 ± 0.1 | 0.99 ± 0.04¤ |
| t-BuOH | 312 | -- | 2 | -- | 11.0 | 62 | 0 | -- | 0.75 ± 0.03 |
Formation of a well-defined isosbestic point was not observed. For CMPPH 3b the isosbestic point is observed at 309 nm.
For CMPPH 3b, φ= 0.88 ± 0.07.
For CMPPH 3b, the isosbestic point is observed at 308 nm.
A value of 2 is obtained for long irradiation times.
For CMPPH 3b, φ= 1.01 ± 0.02.
φ = 0.12 [Reference (5)].
The photolysis of CPZ-HCl in methanol produces PZ 1a and MOPZ 1c in a 1:1 atio [Eq. 6 with R1 = (CH2)3N(CH3)2], as previously identified by Grant and coworkers (6). This product distribution differs significantly from that corresponding to the photochemistry of CPH 1b in the same solvent, since only less than 2% of converts to the methoxy derivative and other compounds. The photodestruction quantum yield of 0.93 is in excellent agreement with the triplet quantum yield of 0.90 reported by our group (19). Since MOPZ is produced in the same amount PZ, φMOPZ = 0.39 and, using φCPZ = φPZ + φMOPZ + φOther (Table 4), a quantum yield of 0.15 is obtained for the formation of all other photoproducts. For CMPPH 3b, the main photoproduct in MeOH and EtOH is the reduction product (MPPH, 3a). The corresponding destruction quantum yields are slightly smaller than those for CPZ (Table 4), but the quantum yield for the formation of MPPH is higher than those for PZ. From the expected alkoxy derivatives, only the methoxy one (MMPPH, 3c) formed with a quantum yield high enough to allow its characterization (φ = 0.41). Obviously, the nitrogen at the alkyl amino chain does not affect the photodestruction quantum yield of these TCAs, but somehow influences the distribution of the photoproducts.
The destruction percentage of CPZ-HCl in MeOH and EtOH was not constant for a constant irradiation time interval of 120 s. The unrecovered material percent was larger than that observed for other alcohols and increased with irradiation time. This is mainly due to the fact that, in these two solvents, PZ is not the only photoproduct. This was confirmed by the ED-EDQ analysis, which gave s=2 for MeOH (data not shown). In EtOH, linear ED-plots with zero intercepts and s=1 were obtained for irradiation times where the time-concentration curve is linear. For longer times, on the other hand, the linearity is lost and an s=2 is also obtained (Fig 4, Right). Exactly the same behavior was observed for CMPPH in both solvents, although the linearity of the ED-plots is EtOH is lost at very long irradiation times (t >900 s). In the case of 1-PrOH and 2-PrOH, the destruction percentage of CPZ is constant up to 720 s and 420 s, respectively. Under this time restriction, an s=1 is found for both propanol isomers. After that, the percentage increases with irradiation time, indicating that secondary processes are taking place. For t-BuOH the calculated percentage of destruction is constant for all irradiation intervals of 120 s for up to 960 s and no PZ was detected. Therefore, an s=1 value was obtained from the Mäuser analysis.
Davies and coworkers reported a photodestruction quantum yield of only 0.12 for CPZ-HCl in isopropanol under anaerobic conditions (5). They further reported the formation of PZ as the major photoproduct and iPOPZ as a minor product. The quantum yield determined in this work is in better agreement with the triplet quantum yield of this drug. The large difference between the destruction quantum yields is mainly due to differences in the experimental conditions. For instance, Davies et. al. used 3.0 mM CPZ-HCl solutions, which have a large absorption at wavelengths >300 nm. This obviously introduces primary and secondary filter effects. Moreover, they measured the formation of HCl assuming that the only photoprocess is the dehalogenation with no alkoxy-derivative formation. In this work, on the other hand, the destruction of the drug was measured using optically diluted solutions (Abs < 1.0 at 313 nm) without any assumption regarding the mechanism. This approach allows a better quantification of the destruction process.
The mechanism proposed for the photodehalogenation in Fig. 5 accounts very well for all experimental results on the photochemistry of CPH (1b), CPZ (2b), and CMPPH (3b) in terms of the photoproducts, their photodestruction quantum yields, and the effect of the solvent on the product distribution. Table 5 shows that the amount of decomposed CPZ converted to PZ increases with the length of the alcohol alky chain. The difference between the percentage of CPZ-destruction and the PZ-formation rapidly drops to zero, indicating that the mechanism of formation of PZ is definitely affected by the R-group of the alcohol. This is explained in terms of the stability of the alcohol radicals and the OH-BDE (See next section). t-Butanol produces no PZ, since it has no alpha hydrogen to be abstracted.
Figure 5.

Mechanism proposed for the photodehalogenation of chlorinated phenothiazines in alcohols.
Table 5.
Bond dissociation energy of the solvents used for the photolysis of halogenated phenothiazines and the TCA-chlorine bond (E[H•] = −0.500273 hartree; E[Cl•] = −460.136242 hartree; 1 Hartree = 627.5095 kcal/mol).
| R-X | E (UBLYP) Hartrees | BDE (kcal/mol) | ||
|---|---|---|---|---|
| R-X | R• | This work | Exp.† | |
| CH3O–H | −115.714405 | −115.050462 | 102.7 | 102 |
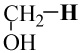 |
−115.714405 | −115.062993 | 94.8 | 92 |
| CH3CH2O–H | −155.034287 | −154.370492 | 102.6 | 103 |
 |
−155.034287 | −154.375316 | 99.6 | - |
| CH3CH2CH2O–H | −194.348026 | −193.685044 | 102.1 | 103 |
 |
−194.348026 | −193.688468 | 100.0 | -- |
 |
−194.353452 | −193.688795 | 103.2 | 103 |
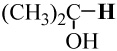 |
−194.353452 | −193.697975 | 97.4 | 94 |
| (CH3)3CO—H | −233.670958 | −233.006172 | 103.2 | 102 |
| −233.670958 | −232.997898‡ | 108.4 | -- | |
| PH-Cl | −1375.239118 | −914.955531 | 92.5 | -- |
| 3(PH-Cl)* | −1375.140165 | −914.955531 | 30.4 | -- |
| PZ-Cl | −1627.126232 | −1166.842675 | 92.4 | -- |
| 3(PZ-Cl)* | −1627.027190 | −1166.842675 | 30.3 | -- |
Values from references (28–31).
Calculated for the beta hydrogen.
Quantum Chemical calculations
The photodestruction process of CPZ is effective and PZ is the main product, only if both the promazyl and the solvent radicals are relative stable (Eq. 3) and/or the corresponding BDEs are relative low. In other words, for the formation of the alkoxy derivative, the R2O-radical formation should be kinetically favored over the promazyl radical formation (43). Table 5 contains the BDEs of the TCA-chlorine and the solvent-H bonds determined with DFT. The ground and triplet excited state BDEs for both TCA-chlorine bonds are, respectively, ~92 and ~30 kcal/mol in all alcohols. This indicates that the TCA-chlorine bond cleavages with the same thermodynamic feasibility efficiency for both compounds, especially in the excited state. These values further show that, within the experimental and theoretical errors, the BDE for the phenothiazine-chlorine bond is not affected by the alkylamino chain. Therefore, the efficiency of the photodestruction of CPH 1b, CPZ 2b, CMPPH 3b, and any 2-chlorinated phenothiazine derivative should be almost the same in all alcohols. This is experimentally observed for each separate alcohols included in this study, i.e. all TCAs have about the same destruction quantum yield in the same solvent (Table 2 and Table 4). Nevertheless, the BDE values of the TCAs cannot be used to explain the differences in the TCA photodehalogenation quantum yields in different solvents, nor the electron transfer mechanism previously proposed by Bunce (23) for this process. The solvent-H BDEs, on the other hand, clearly explain why it is easier for the alpha hydrogen to be abstracted than the hydroxyl hydrogen, especially for alcohols with long and branched alkyl chains. The corresponding energy for this R2O-H bond also increases with the alkyl chain, making the formation of the alkoxy derivative less competitive. The BDE of the α- hydrogen also increases with the chain length, but this increase is compensated by the stability of the corresponding radical. In summary, the formation of the alkoxy product is very inefficient and can only compete with the reduction one if the solvent radicals are not stable, as is the case of methanol.
CONCLUSIONS
To solve the controversy regarding the so many different values reported for the photodestruction quantum yield of halogenated phenothiazines, a methodology was developed in this work to determine the quantum yields using a monochromatic 313 nm light for irradiation of optically diluted solutions and a GC “total quantification” procedure for the determination of the quantum yield (44). All these parameters are very important because: (a) the light intensity and the photoprocess quantum yield strongly depend on the selected wavelength; (b) by using an excitation wavelength range, several transitions can be excited at once; (c) by using an excitation wavelength range, there is no easy way to determine the absorbed light intensity; (d) irradiation of concentrated solutions induces filter effects; and (e) the total drug photodestruction can be measured without assuming a specific mechanism or a specific product distribution.
The results of this approach indicate that: (a) according to the values of the s parameter, the photodestruction of 2-chlorinated TCAs consists of only one reduction reaction. The effectiveness of this reaction is determined by the BDE and the radical stability of the participating partner. Under special conditions of prolonged irradiation and/or small alcohols the formation of the corresponding alkoxide can be observed; (b) the larger the alkyl chain of the alcohol, the lesser the amount of alkoxide photoproduct formed, in agreement with the calculated differences in the solvent BDEs and the mechanism of the photonucleophilic substitution proposed by Grant and Green (6). The biggest effect is introduced by tBuOH, in which the photodestruction quantum yields is small and no reduction product was obtained; (c) the photodestruction quantum yields of the TCAs depend more on the solvent than on the alkylamino chain at the 10-position. Therefore, the electron transfer mechanism proposed by Bunce (23) is not correct; and (d) the phototoxicity of the phenothiazine-type drugs is not directly determined by their dehalogenation, since their constant photodestruction quantum yield cannot so far account for their differences in toxicity strength. Nevertheless, these species might somehow induce this side-effect through a mechanism involving membrane components in vivo (45).
Acknowledgments
This work has been supported in part by NIH-MBRS Grant SO6GM08216 to UPR-Humacao. We specially thank Melvin De Jesus and Jorge Castillo for the support with the NMR spectroscopy and the GC-MS Chromatography.
REFERENCES
- 1.Kochevar IE. Phototoxicity mechanism: Chlorpromazine photosensitized damage to DNA and cell membranes. J. Invest. Dermatol. 1981;76:59–64. doi: 10.1111/1523-1747.ep12479244. [DOI] [PubMed] [Google Scholar]
- 2.Kochevar IE. Mechanisms of drug photosensitation. Photochem. Photobiol. 1987;45:891–895. doi: 10.1111/j.1751-1097.1987.tb07899.x. [DOI] [PubMed] [Google Scholar]
- 3.Kochevar IE, Hom J. Photoproducts of chlorpromazine that cause red blood cell lysis. Photochem. Photobiol. 1983;37:163–168. doi: 10.1111/j.1751-1097.1983.tb04452.x. [DOI] [PubMed] [Google Scholar]
- 4.Curry SH. Plasma protein binding of chlorpromazine. J. Pharm. Pharmacol. 1970;22:193–197. doi: 10.1111/j.2042-7158.1970.tb08496.x. [DOI] [PubMed] [Google Scholar]
- 5.Davis AK, Navaratnam S, Phillips GO. Photochemistry Of Chlorpromazine (2-chloro-N-(3dimethylamino Propyl) Phenothiazine) In Propan-2-ol Solution. J. Chem. Soc. Perkin Trans. II. 1976;25:25–29. [Google Scholar]
- 6.Grant FW, Green J. Phototoxicity and photonucleophilic aromatic substitution in chlorpromazine. Toxic. Appl. Pharmacol. 1972;23 doi: 10.1016/0041-008x(72)90205-0. 71Error! Main Document Only.-74. [DOI] [PubMed] [Google Scholar]
- 7.Epstein S. Chlorpromazine photosensitivity, phototoxic and photoallergic reactions. Arch. Dermatol. 1968;98:354–363. doi: 10.1001/archderm.98.4.354. [DOI] [PubMed] [Google Scholar]
- 8.Kahn G, PDB In vitro studies on longwave ultraviolet light dependent reactions of the skin photosensitizer chlorpromazine with nucleic acids. Purines and pyrimidines. J. Invest. Dermatol. 1970;55:47–53. doi: 10.1111/1523-1747.ep12290532. [DOI] [PubMed] [Google Scholar]
- 9.Ciulla TA, Epling GA, Kochevar IE. Photoaddition of chlorpromazine to guanosine-5'-monophosphate. Photochem. Photobiol. 1986;43:607–613. doi: 10.1111/j.1751-1097.1986.tb05635.x. [DOI] [PubMed] [Google Scholar]
- 10.Fujita H, Yanagisawa F, Endo A, Suzuki K. Photoreactivity of Chlorpromazine with Native DNA in an Aqueous solution. J. Rad. Research. 1980;21:279–287. doi: 10.1269/jrr.21.279. [DOI] [PubMed] [Google Scholar]
- 11.Fujita H, Endo A, Suzuki K. Inactivation of bacteriophage lambda by near ultraviolet irradiation in the presence of chlorpromazine. Photochem. Photobiol. 1981;33:215–222. doi: 10.1111/j.1751-1097.1981.tb05327.x. [DOI] [PubMed] [Google Scholar]
- 12.Iwaoka T, Kondo M. Mechanistic studies on the photooxidation of chlorpromazine in water and ethanol. Bull. Chem. Soc. Japan. 1974;47:980–986. [Google Scholar]
- 13.Kochevar IE, Chung F, Jeffrey AM. Photoaddition of Chlorpromazine to DNA. Chem. Biol. Interactions. 1984;51:273–284. doi: 10.1016/0009-2797(84)90153-4. [DOI] [PubMed] [Google Scholar]
- 14.Ljungreen B, Moeller H. Phenothiazine phototoxicity: an experimental study on chlorpromazine and its metabolites. J. Invest. Dermatol. 1977;68:313–317. doi: 10.1111/1523-1747.ep12494582. [DOI] [PubMed] [Google Scholar]
- 15.Buettner GR, Motten AG, Hall RD, Chignell CF. Free radical production by chlorpromazine sulfoxide. An ESR spin trapping and flash photolysis study. Photochem. Photobiol. 1986;44:5–10. doi: 10.1111/j.1751-1097.1986.tb03556.x. [DOI] [PubMed] [Google Scholar]
- 16.Rosenthal I, BenHur E, Prager A, Riklis E. Photochemical reactions of chlorpromazine: chemical and biochemical implications. Photochem. Photobiol. 1978;28:591–594. doi: 10.1111/j.1751-1097.1978.tb06975.x. [DOI] [PubMed] [Google Scholar]
- 17.Schoonderwoerd S, Beijerbergen G, Henegouwen V, Leijendijk J. Photobinding of chlorpromazine and its sulfoxide in vitro and in vivo. Photochem. Photobiol. 1988;48:621–626. doi: 10.1111/j.1751-1097.1988.tb02872.x. [DOI] [PubMed] [Google Scholar]
- 18.Kochevar IE, Garcia C, Geacintov NE. Photoaddition to DNA by Nonintercalated Chlorpromazine Molecules. Photochem. Photobiol. 1998;68(5):692–697. [PubMed] [Google Scholar]
- 19.García C, Smith GA, Grant M, Kochevar IE, Redmond RW. Mechanism and Solvent Dependence for the Photoionization of Promazine and Chlorpromazine. J. Am. Chem. Soc. 1995;117:10871–10877. [Google Scholar]
- 20.García C, Oyola R, Piñero LE, Arce R, Silva J, Sánchez V. Substitution and Solvent Effect on the Photophysical Properties of Several Series of 10alkylated Phenothiazine Derivatives. J. Phys. Chem. A. 2005;109:3360–3371. doi: 10.1021/jp044530j. [DOI] [PubMed] [Google Scholar]
- 21.Oyola R. Design, Construction and use of a Nanosecond Laser Transient Absorption Spectrokinetic System for the study of the Photochemical and Photophysical Properties of Relevant Biological Molecules: dTpA, dApT and Tricyclic antidepressive drugs Ph. D. Thesis. Rio Piedras: University of Puerto Rico; 2001. [Google Scholar]
- 22.Felmeister A, Discher CA. Photodegradation of chlorpromazine hydrochloride. J. Pharm. Sci. 1964;53:756–762. doi: 10.1002/jps.2600530712. [DOI] [PubMed] [Google Scholar]
- 23.Bunce NJ, Kumar Y, Ravanal L. Phototoxicity of chlorpromazine. J. Med. Chem. 1979;22:202–204. doi: 10.1021/jm00188a016. [DOI] [PubMed] [Google Scholar]
- 24.Moore D, Tamat S. Photosensitization by drugs: photolysis of some chlorine-containing drugs. J. Pharm. Pharmacol. 1980;32:172–177. doi: 10.1111/j.2042-7158.1980.tb12884.x. [DOI] [PubMed] [Google Scholar]
- 25.Kricka LJ, Ledwith A. Reactions of condensed N-Heteroaromatic Molecules. Part I. Alkylation by thallium(I)ethoxide. J. Chem. Soc. Perkin Trans. I. 1972:2292–2293. [Google Scholar]
- 26.Wang Z, McGimpsey WG. One and two laser photochemistry of iminodibenzyl. J. Phys. Chem. 1993;97:9668–9672. [Google Scholar]
- 27.Kuhn HJ, Braslavsky SE, Schmidt R. Chemical Actinometry. Pure & Appl. Chem. 1989;61(2):187–210. [Google Scholar]
- 28.García C, Oyola R, Piñero LE, Cruz N, Alejando F, Arce R, Nieves I. Photophysical, Electrochemical and Theoretical study of Protriptyline in several solvents. J. Phys. Chem. B. 2002;106:9794–9801. [Google Scholar]
- 29.Benson SW. Thermochemical Kinetics: Methods for the estimation of thermochemical data and rate parameter. New York, USA: John Wiley & Sons; 1976. [Google Scholar]
- 30.McMillen DF, Golden DM. Hydrocarbon bond dissociation energies. Ann. Rev. Phys. Chem. 1982;33:493–532. [Google Scholar]
- 31.NISTWebBook. NIST Standard Reference Database Number 69. 2005 Http://webbook.nist.gov/chemistry;
- 32.Wedenejew VW, Gurwitsch LW, Kondratjew WH, Medwedew WA, Frankenwitsch EL. Energien chemischer bindungen, Ionisationspotentiale und Elektronenaffinitae VEB Deutcher Verlag Fuer Grundstoffindustrie. Leipzig: 1971. [Google Scholar]
- 33.Mäuser H. Formale Kinetic: Experimentelle Methoden der Physik und der Chemie. Bertelsmann Universitätsverlag. 1974;1:297–363. [Google Scholar]
- 34.Daniil D, Gauglitz G, Meier H. Kinetic investigation of the photolysis of 2-oxo-2,5-dihydro-1,3,4-oxadiazoles. Photochem. Photobiol. 1977;26:225–229. [Google Scholar]
- 35.Mäuser H, Gauglitz G. Photokinetiks: Theoretical fundamentals and applications. In: Compton RG, Hancock G, editors. Comprehensive chemical kinetics. Vol. 36. New York: Elsevier Science Publishers; 1998. [Google Scholar]
- 36.Aaron J, Maafi M, Kersebet C, Párkányi C, Shafik AM, Motohashi N. A solvatochromic study of new benzo [a] phenothiazines for the determination of dipole moments and specific solute solvent interactions in the first excited singlet state. J. Photochem. Photobiol. A:Chem. 1996;101:127–136. [Google Scholar]
- 37.Casarotto MG, Craick DJ. Ring flexibility within tricyclic antidepressant drugs. J. Pharm. Pharmac. Sci. 2001;90:713–721. doi: 10.1002/jps.1027. [DOI] [PubMed] [Google Scholar]
- 38.Casarotto MG, Craik DJ, Munro SL. 13C NMR studies of the solution molecular dynamics of Tricyclic antidepressants. Magn. Reson. Chem. 2005;28:533–540. [Google Scholar]
- 39.Elisei F, Latterini L, Aloisi GG, Mazzucato U, Viola G, Miolo G, Vedaldi D, Dall’Acqua F. Excited state properties and in vitro phototoxicity studies of three phenothiazine derivatives. Photochem. Photobiol. 2002;75(1):11–21. doi: 10.1562/0031-8655(2002)075<0011:espaiv>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Turro NJ, Khudyakov IV, Willigen Hv. Photoionization of Phenothiazine: EPR Detection of Reactions of the Polarized Solvated Electron. J. Am. Chem. Soc. 1995;117(49):12273–12280. [Google Scholar]
- 41.Barra M, Calabrese GS, Allen MT, Redmond RW, Sinta R, Lamola AA, Small RD, Scaiano J. Photophysical and photochemical studies of phenothiazine and some derivatives: Explanatory studies of novel photosensitizers for photoresist technology. Chem. of Materials. 1991;3:610–616. [Google Scholar]
- 42.Toth M. Triplet-triplet absorption of cyclohexadiene-1,3 and ergosterol-Sensitized total ground state depletion of dienes by triplet energy transfer from the ruby laser excited 2fluorenylphenylketone. Chem. Phys. Letter. 1980;46:437–443. [Google Scholar]
- 43.Wright JS, Johnson ER, DiLabio GA. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001;123:1173–1183. doi: 10.1021/ja002455u. [DOI] [PubMed] [Google Scholar]
- 44.Piñero LE. Synthesis, characterization, photophysics, and photochemistry of 2chlorinated phenothiazine derivatives in several solvents Master Thesis. University of Puerto Rico; 2006. [Google Scholar]
- 45.Chignell CF. Application of physicochemical and analytic techniques to the study of drug interactions with biological systems. CRC Critical Rev. In Toxicology. 1972:413465. [Google Scholar]