

Abstract
Extracellular superoxide dismutase (SOD3), a secretory copper-containing antioxidant enzyme, plays an important role in various oxidative stress-dependent cardiovascular diseases. Although cofactor copper is required for SOD3 activity, it remains unknown whether it can regulate SOD3 transcription. We previously demonstrated that SOD3 activity requires the copper chaperone Antioxidant-1 (Atox1) involved in copper delivery to SOD3 at the trans-Golgi network (TGN). Here we show that copper treatment in mouse fibroblasts significantly increases mRNA and protein levels of SOD3, but not SOD1, which is abolished in Atox1-deficient cells. Copper promotes Atox1 translocation to the nucleus. Promoter deletion analysis identifies copper- and Atox1-response element (RE) at the SOD3 promoter. Gel shift and ChIP assays reveal that Atox1 directly binds to the Atox1-RE in a copper-dependent manner in vitro and in vivo. Adenovirus-mediated re-expression in Atox1-/- cells with nucleus-targeted Atox1 (Atox1-NLS), but not TGN-targeted Atox1 (Atox1-TGN), increases SOD3 transcription without affecting SOD3 activity. Importantly, re-expression of both Atox1-NLS and Atox1-TGN together, but not either alone, in Atox1-/- cells increases SOD3 activity. SOD3 transcription is positively regulated by copper through transcription factor function of Atox1, while full activity of SOD3 requires both copper chaperone and transcription factor function of Atox1. Thus, Atox1 is a potential therapeutic target for oxidant stress-dependent cardiovascular disease.
Keywords: Antioxidant-1, SOD3, Copper, Transcription Factor, Copper Chaperone
Introduction
SOD3 is the major extracellular antioxidant enzyme against O2•-. It is synthesized and secreted by vascular smooth muscle cells and fibroblasts, thereby preventing oxidative inactivation of nitric oxide released from endothelial cells and superoxide- or peroxynitrite-mediated degradation of extracellular matrix proteins [1-3]. Thus, SOD3 plays an important role in various oxidative stress-dependent pathophysiologies including hypertension, ischemia-reperfusion injury and lung injury [1-3]. Indeed, the R213G polymorphism in the SOD3 gene has been linked to an increase in cardiovascular risk [4]. Furthermore, mice deficient in SOD3 reveal an essential role of SOD3 in angiotensin II-induced hypertension, ischemia-induced cardiac remodeling and angiogenesis [5-8]. Accumulating evidence reveals that SOD3 activity and expression are altered by various stimulants and pathological states at multiple levels such as transcriptional and post-translational levels [9]. Little is known about the mechanism by which SOD3 transcription is regulated.
Both SOD3 and SOD1 activity requires a catalytic copper to scavenge O2•- by reduction and reoxidation of the copper ion at the active site of the enzyme [10, 11]. Under physiological conditions, the level of intracellular free copper is extraordinarily restricted [12]. Thus, soluble copper carrier proteins termed “copper chaperones” are required to directly transfer copper to specific cellular targets. SOD1 has been shown to obtain catalytic copper ion through interaction with the cytosolic copper carrier protein CCS, a copper chaperone for SOD1, thereby increasing its activity [13]. In contrast, we have previously demonstrated that SOD3 activity requires the copper chaperone, Antioxidant-1 (Atox1) which delivers copper to SOD3 via interacting with the copper transporter Menkes ATPase at the trans-Golgi network (TGN) [11, 14]. Unexpectedly, our preliminary study shows that the cofactor copper upregulates SOD3 mRNA expression without affecting SOD1 mRNA. However, underlying mechanisms are unknown.
Atox1 is originally isolated from yeast, and has been reported to protect SOD1-deficient yeast from oxidative damage [15]. This oxidative protection is dependent on both conserved copper binding residues in the N-terminal domain and conserved lysine residues in the C-terminal domain, whereas the copper chaperone function of Atox1 is dependent on copper binding residues, but not on its lysine residues [16]. Most recently, we demonstrated that Atox1 functions as a copper-dependent transcription factor to increase cell proliferation by upregulating cyclin D1 transcription, whose effect is dependent on both copper binding and conserved lysine residues which contains nuclear-localization signal [17]. Furthermore, we found that basal SOD3 mRNA expression is decreased in Atox1-/- fibroblasts [11].
We performed the present study to test the hypothesis that Atox1 functions as a copper-dependent transcription factor to regulate SOD3 transcription in addition to its role as a copper chaperone. Here we demonstrate that copper increases SOD3 expression without affecting SOD1 expression, which is abolished in Atox1-/- fibroblasts, and that copper stimulates nuclear translocation of endogenous Atox1. We also show that Atox1 binds directly to the GAAAGA sequence (Atox1-RE) in the SOD3 promoter in a copper-dependent manner. Functional studies with transfection of nuclear-targeted and TGN-targeted Atox1 reveal that SOD3 expression is regulated by transcription factor function of Atox1, while SOD3 full activity is dependent on both transcription factor and copper chaperone functions of Atox1. These findings suggest that Atox1 functions to sense copper to increase SOD3 transcription and activity, which may be important for physiological function of copper by protecting cells from oxidative stress.
Materials and Methods
Materials and Cell Culture Used
Atox1-/- and wild-type immortalized mouse embryonic fibroblast cells (MEFs) were cultured in DMEM with 10% fetal bovine serum as previously described.[17] Experiments were performed in medium containing 1% serum with no additives. All other cells were grown in DMEM with 10% FBS. Before stimulation, cells were serum-starved for 24 hours. All reagents were purchased from Sigma Chemical Co, except when specified (see the online data supplement).
Plasmid constructs
The promoter-reporter constructs consist of 5’ regions of the SOD3 promoter inserted into the luciferase reporter vector pGL3-Basic (pGL3-SOD3 -2500/104, -1293/104, -208/104) as previously described [18]. Other expression promoter-reporter constructs (-633/104, -513/104, -393/104, -303/104) were generated by PCR and subcloned into the pGL3-Basic vector (Promega, Madison, WI, U.S.A.).
For production of recombinant Atox1 protein, its coding region was ligated into the pGEX-4T-1 expression vector (Amersham) and used to transform Escherichia coli strain BL21 (DE3) as previously described [17].
Generation of nuclear targeted Atox1 (Flag-Atox1-NLS) constructs, trans-Golgi network (TGN)-targeted Atox1 (Flag-Atox1-TGN) constructs, and plasmids for creation of adenovirus are described in the online data supplement.
Western blot analysis
Western blot analysis was performed as previously described [11, 17, 19]. For Atox1 detection, samples were separated by 16.5% Tricine-SDS/PAGE (BioRad). Equal loading of proteins was confirmed by Ponceau or Coomassie blue staining.
Quantitative Real-time PCR
Real-time PCR to quantify SOD3 mRNA was performed as previously described [11].
Transient transfection and Reporter assay
Transfection assays were carried out using Polyfect reagent (Qiagen, CA) according to the manufacturer’s protocols. The pGL3-SOD3 constructs were transfected into Atox1-/- or Atox1+/+ mouse embryonic fibroblast cells along with pRL-CMV (Promega) according to the manufacturer’s protocol. Forty-eight hours after transfection, luciferase activity was measured using a luminometer. Transfection efficiency was normalized on the basis of Renilla luciferase activity.
Electrophoretic Mobility Shift Assays (EMSAs)
DNA-protein binding assays were carried out with nuclear extract from mouse embryonic fibroblast cells as previously described (see the online data supplement for the details) [17].
Chromatin immunoprecipitation assay (ChIP)
Identical numbers of mouse embryonic fibroblast cells were allowed to adhere onto tissue-cultured plates in basal media. ChIP assays were performed as previously described (see the online data supplement for the details) [17].
Adenoviral transfection
Adenoviruses expressing Flag-Atox1-WT and Flag-Atox1 mutants were generated by standard techniques (Vector Biolabs, Philadelphia, PA). Adenovirus expressing the bacterial β-galactosidase gene (Lac Z) was used as a control. Cells were incubated with various multiplicity of infection (MOI) of indicated Adenovirus in serum-free medium for 5 hours, followed by incubation in 10% FBS containing culture medium overnight. After an overnight infection, the media were replaced with fresh 0.5 % FBS without virus for 72 hours before experiments. Adenovirus-transfected cells were then used to examine activity and expression of SOD3 and ceruloplasmin.
Immunofluorescence Studies
Cells were fixed in 4% paraformaldehyde, permeabilized in 0.05% Triton X-100, and rinsed sequentially in PBS, 50mM NH4Cl, and PBS as previously described.[17] Immunofluorescence analysis was performed as described in the online data supplement.
SOD and ceruloplasmin (Cp) activity assays
SOD activity was assayed by monitoring inhibition of the rate of xanthine/xanthine oxidase–mediated reduction of cytochrome c [11], and Cp activity was measured by p-phenylenediamine (PPD) oxidation as previously described[20]. SOD3 and Cp secreted in the culture medium and SOD1 in cell lysates were collected at 72-hour after addition of copper. Con A-Sepharose chromatography (Pharmacia Biotech, Piscataway, NJ) was used to isolate SOD3 and Cp from conditioned media of Atox1-/- and Atox1+/+ cells as previously described [11, 17].
Statistical analysis
All data are expressed as mean ± SEM. Comparisons were made by one-way ANOVA followed by the Turkey-Kramer post hoc test. Values of p<0.05 were considered statistically significant.
Results
Expression of SOD3, but not SOD1, is upregulated by copper at transcription level
To confirm our preliminary data, we examined the effects of copper on expression of the copper-containing enzymes SOD3 and SOD1 in mouse embryonic fibroblasts (MEFs). As shown in Fig. 1A, copper markedly increased protein expression of SOD3, but not SOD1, in a dose-dependent manner, with a maximum at 10 μM which is its physiological concentration [17, 21]. Note that membrane-impermeable specific copper chelator bathocuproine disulfonic acid (BCS)[22] significantly decreased SOD3 expression (Fig. 1A). This response was specific to copper, because neither iron nor zinc had any effect on SOD3 expression (Supplemental Fig. IA). Polyethylene glycol (PEG)-SOD or PEG-catalase had no effect on copper-induced SOD3 expression (Supplemental Fig. IB), suggesting that reactive oxygen species are not involved in this response.
Fig. 1. Copper stimulates expression of SOD3, but not SOD1, at the transcription level in mouse fibroblasts.

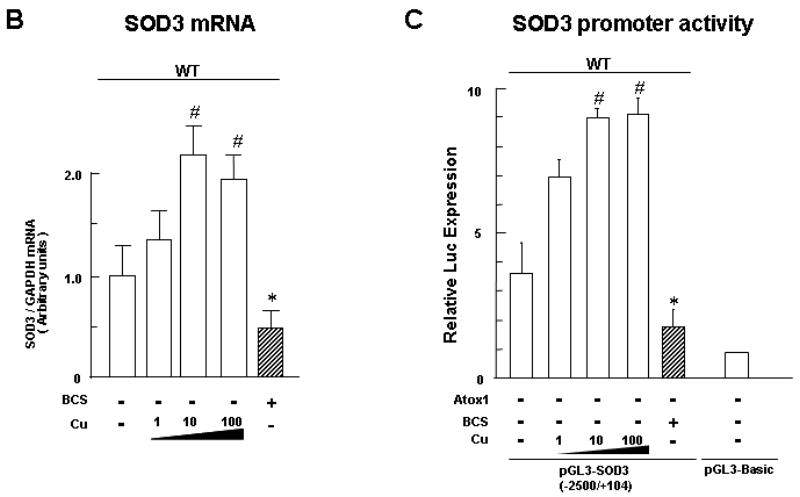
A, Effect of copper on protein expression of secreted SOD3 (left) and SOD1 (right) in mouse embryonic fibroblasts (MEFs) treated either with the copper chelator BCS (200 μM) or CuCl2 at the dose indicated for 72 h (n=4, *p<0.01 vs. non-treated cells). The SOD3 secreted in the culture medium was collected, and concentrated by Concanavalin-A sepharose chromatography as previously described.[11] Protein expression of SOD3 in concentrated cultured medium and of SOD1 in cell lysates was examined as previously described.[11] Equal protein loading was confirmed by Ponceau staining or Coomassie blue staining. B, Quantitative real-time PCR analysis showing SOD3 mRNA levels in MEFs treated either with the copper chelator BCS (200 μM) or CuCl2 at the dose indicated for 12h (n=4, *p<0.01; #p<0.001 vs. non-treated cells). C, Effect of copper on transactivation of the SOD3 promoter in MEFs (n=3, each in quadruplicate, *p<0.01; #p<0.001 vs. non-treated WT cells). Cells were transiently transfected with SOD3 promoter luciferase reporter constructs, and treated with either the copper chelator BCS (200 μM) or CuCl2 at the dose indicated.[11] Forty-eight hours after transfection, the luciferase activity was assayed and normalized to the Renilla luciferase activity produced by the co-transfected control plasmid pRL-CMV.
We next examined whether copper upregulates SOD3 expression at the mRNA level using real-time quantitative RT-PCR analysis (Fig. 1B). Copper markedly increased SOD3 mRNA level in a dose-dependent manner with maximum at 10 μM in MEFs. In contrast, copper chelator BCS decreased SOD3 mRNA levels. To determine whether copper increases SOD3 gene at the transcriptional level, we performed luciferase reporter assays (Fig. 1C). Copper treatment in MEFs markedly increased the SOD3 promoter activity in a dose-dependent manner with maximum at 10 μM. By contrast, zinc treatment had no effect (data not shown). These results suggest that copper transcriptionally upregulates the SOD3 gene.
Copper-induced increase in SOD3 transcription requires Atox1
We next examined whether copper-induced SOD3 transcription is mediated through Atox1. Fig. 2A showed that the copper-induced increase in protein expression of SOD3, but not that of SOD1, was abolished in Atox1-/- MEFs. Note that SOD3 protein expression in the baseline was markedly decreased in Atox1-/- cells. Furthermore, inhibition of the copper-induced increase in SOD3 protein levels in Atox1-/- MEFs was associated with inhibition of its mRNA levels (Fig. 2B). These results suggest that Atox1 is required for the copper-induced increase in SOD3 transcription.
Fig. 2. Copper-induced increase in SOD3 transcription is dependent on Atox1.

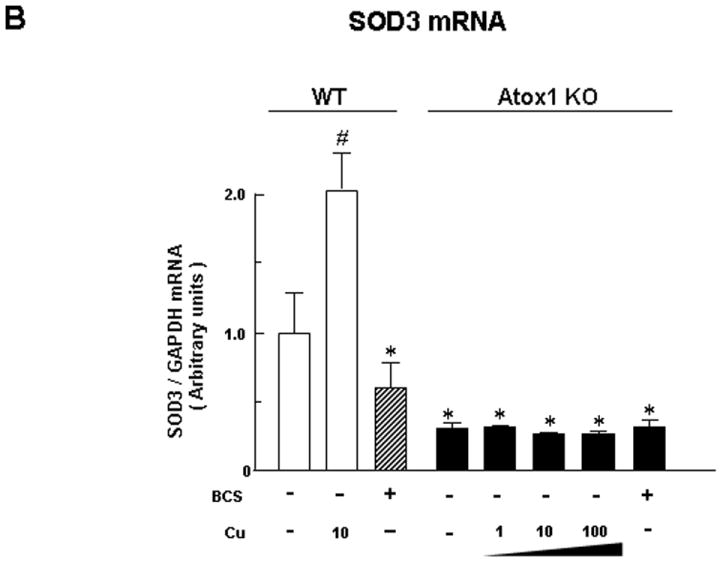
A, Effect of copper on protein expression of secreted SOD3 in cultured medium and SOD1 in cell lysates in wild-type (WT) and Atox1-/- (Atox1-KO) MEFs (n=4, *p<0.01 vs. non-treated cells). B, Quantitative real-time PCR analysis showing SOD3 mRNA levels in wild-type and Atox1-/-MEFs (n=4, *p<0.01; #p<0.001 vs. non-treated cells).
Atox1 translocates to the nucleus, and binds to and activates the SOD3 promoter in a copper-dependent manner
To determine if Atox1 functions as a transcription factor for SOD3, we examined the subcellular localization of endogenous Atox1 in mouse fibroblasts using specific Atox1 antibody. Copper treatment markedly increased Atox1 staining in the nucleus (Supplemental Fig. II). Luciferase reporter assays demonstrated that the copper-induced increase in SOD3 promoter activity was abrogated in Atox1-/- cells (Fig. 3A). The specific effect of Atox1 on copper-induced SOD3 transcription was further confirmed by the observation that transfection of WT Atox1 in Atox1-/- cells rescued the copper-induced response of SOD3 (Fig. 3A). A 5’ deletion analysis of the SOD3 promoter identified the copper-responsive region in -393/-303 of the SOD3 promoter (Fig. 3B). We confirmed that all of the 5’ deletion constructs of the SOD3 promoter had no response to copper in Atox1-/- cells (Fig. 3C). Furthermore, the Atox1 responsive region regulating SOD3 promoter activity resides within the copper responsive element between -393 and -303 (Fig. 3D).
Fig. 3. Identification of copper/Atox1 responsive elements at a proximal 90-bp SOD3 promoter element.

A, Effect of copper on transactivation of the SOD3 gene promoter in wild-type and Atox1-/- MEFs. Cells were transiently transfected with SOD3 promoter luciferase reporter constructs in the presence or absence of pcDNA/Atox1. Luciferase activity was assayed as described in Fig. 1C (n=4, each experiment in quadruplicate, *p<0.01; #p<0.001 vs. BCS-treated WT cells, or BCS-treated Atox1-/- cells transfected with pcDNA/Atox1). B, C, and D, WT and Atox1-KO MEFs were transiently transfected with 5’ deletion constructs of SOD3 in the presence of either CuCl2 (10 μM)(+Cu) or BCS (200 μM)(-Cu) (n=4, each experiment in quadruplicate, *p<0.01 vs. BCS-treated WT (B), or CuCl2-treated Atox1-/- cells (D)).
To determine if nuclear proteins could specifically bind to this region, we performed EMSA in WT cells (Fig. 4A). We found that specific DNA-protein binding complexes were produced as a major slower migrating band only by a biotinylated SOD3 promoter fragment -333/-304 (Fig. 4A), but not by -393/-364 and -373/-344 fragments (data not shown). The DNA-protein complex formation was competed by 50-fold molar excess of unlabeled -333/-304 (Fig. 4A, lanes 4 and 5). To map the -333 to -304 binding area in greater detail, mutations which spanned the region with sequential nucleotide substitutions were created (Fig. 4B). The major DNA-protein complexes were competed by 100-fold molar excess of unlabeled mutants, except for the unlabeled mutants in the region of -314 to -304 (Fig. 4B) and the region of -312 to -307 (data not shown). These findings suggest that nuclear proteins bind to the region -312/-307 in the SOD3 promoter.
Fig. 4. Atox1 binds to the region of -312 to -307 in the SOD3 promoter in a copper-dependent manner in vitro and in vivo.
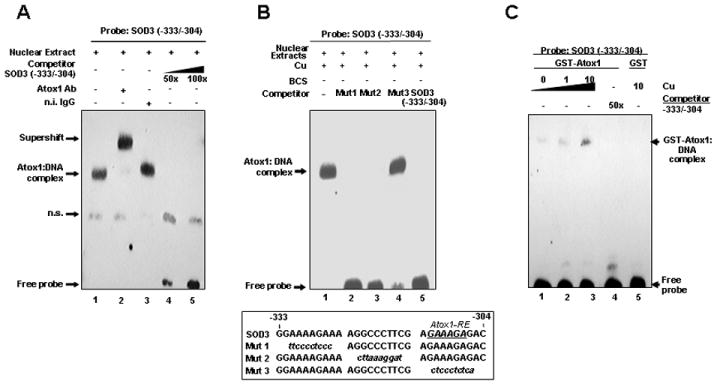

A, B, and C. EMSAs were performed as previously described.[17] A and B, Nuclear extracts from MEFs were incubated with biotinylated SOD3 promoter fragment with indicated treatments. C, purified GST or GST-Atox1[17] was incubated with the DNA probe with indicated treatments. D, ChIP assay showing association of Atox1 with SOD3 promoter in a copper dependent manner in vivo. Results are representative of three independent experiments. E, The region -312 to -307 is required for copper-induced activation of SOD3 promoter (n=4, each in quadruplicate, #p<0.001 vs. BCS-treated cells).
To confirm that Atox1 binds to the region -312 to -307 in the SOD3 promoter, we performed supershift assays on nuclear extracts of WT MEFs. An antibody specific for Atox1, but not preimmune rabbit serum, supershifted DNA-protein complexes, suggesting that the major slower migrating band represents binding of Atox1 to the region -312 to -307 (Fig. 4A, lanes 1, 2, and 3). To test if Atox1 directly binds to the SOD3 promoter, we performed EMSA using recombinant GST-Atox1. The SOD3 promoter segment (-333/-304) directly associated with GST-Atox1, but not with GST alone. This association was dependent on copper (Fig. 4C, lanes 1, 2, and 3), which was competed by 50-fold molar excess of unlabeled SOD3 (-333/-304) (Fig. 4C, lane 4).
To test whether endogenous Atox1 binds to the SOD3 promoter in vivo, we performed chromatin immunoprecipitation (ChIP) assays using Atox1 antibodies to precipitate Atox1 in WT cells treated with copper or with the copper chelator BCS. The promoter region of SOD3, but not those of SOD1 that lack Atox1 binding sites, was immunoprecipitated from cell extracts from Atox1 antibodies, but not by normal IgG, in a copper-dependent manner (Fig. 4D). Finally, mutation of the GAAAGA sequence (-312/-307) in the SOD3 promoter abolished the copper-induced activation of the SOD3 promoter (Fig. 4E), suggesting that this sequence acts as a core element for Atox1 binding to target DNA. These findings indicate that Atox1 binds to the SOD3 promoter at -312 to -307 that is essential for copper-dependent expression of SOD3.
Both transcription factor function and copper chaperone function of Atox1 are essential for full activation of SOD3
Atox1 also functions as a copper chaperone that delivers copper to the secretory compartment which includes trans-Golgi network (TGN) [23]. To segregate the copper-dependent transcription factor and the copper chaperone function of Atox1, we examined the effect of re-expression of WT-, TGN- or nuclear-targeted Atox1 (Flag-Atox1-WT, Flag-Atox1-TGN or Flag-Atox1-NLS, respectively) in Atox1-/- MEFs on SOD3 expression and activity (Fig. 5). We confirmed that each Atox1 protein was expressed with the intended subcellular localization (Fig. 5A), and that transduction efficiency of each adenovirus was similar as measured by percentage of Flag positive cells using immunofluorescence analysis (data not shown). Transfection of Atox1-WT and Atox1-NLS, but not Atox1-TGN, in Atox1-/-cells markedly increased SOD3 promoter activity (Fig. 5B) and protein expression (Fig. 5C). By contrast, either Atox1-NLS or Atox1-TGN alone had no effect on SOD3 activity, while both Atox1-NLS and Atox1-TGN re-expression in Atox1-/- cells increased SOD3 activity (Fig. 5D). Under this condition, transfection of Atox1-TGN, but not Atox1-NLS, increased specific activity of SOD3, as determined by the ratio of activity to relative amount of protein as previously described (Fig. 5E) [11]. These results suggest that full activation of SOD3 requires both the transcription factor and the copper chaperone functions of Atox1.
Fig. 5. Both transcription factor function and copper chaperone function of Atox1 are essential for full activation of SOD3.

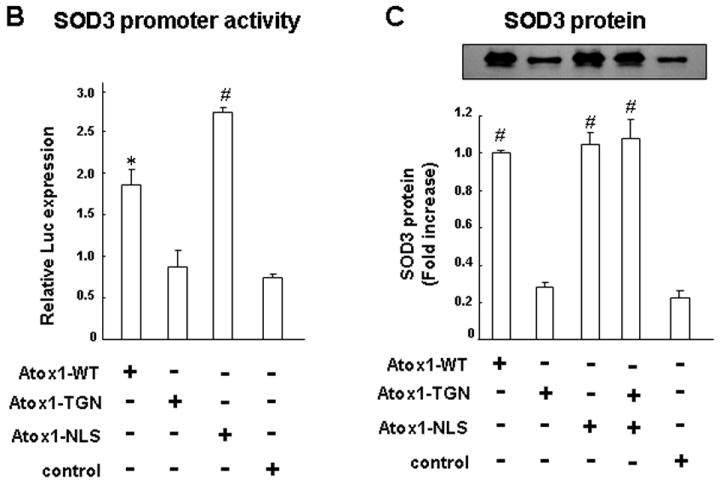

A, Subcellular localization of Atox1-WT, Atox1-TGN, and Atox1-NLS fusion proteins. Atox1-/- mouse fibroblasts were transfected with Adenovirus expressing Flag-Atox1-WT or Flag-Atox1 mutants, immunolabeled with antibodies for Flag tag (green) and TGN-marker, γ-adaptin (red) or nuclear marker, DAPI (blue). Upper panel; WT Atox1 was found in both nucleus and cytoplasm. Atox1-NLS (middle) and Atox1-TGN (lower) were mainly localized at nucleus and TGN, respectively. (B, C, D, E) Effect of re-expression of Atox1-WT, Atox1-TGN, or Atox1-NLS on transactivation of the SOD3 promoter (B), protein expression (C), activity (D), and specific activity (E) of SOD3 in Atox1-/- MEFs. SOD3 promoter activity was examined as described in Fig. 1C (n=4, each in quadruplicate, *p<0.01; #p<0.001 vs. control-treated cells). For protein expression, activity, and specific activity of SOD3, cells were transfected with Adenovirus expressing Flag-Atox1-WT, Flag-Atox1 mutants, or LacZ (control) in the presence of CuCl2 (10 μM) (n=4, #p<0.001 vs. control-treated cells). Three days after transfection, conditioned media were collected, and protein levels and enzymatic activity of SOD3 were assayed as described above. Specific activity of SOD3 was determined by the ratio of activity to relative amount of protein as previously described.[11]
To confirm the specific effect of transcription factor function of Atox1 on SOD3, we examined effects of Atox1-WT or Atox1-TGN or Atox1-NLS on expression and activity of ceruloplasmin that obtains copper at TGN [24]. Either Atox1-WT or Atox1-NLS alone had no effect on expression of ceruloplasmin [24], while Atox1-TGN, but not Atox1-NLS, alone increased specific activity of this secretory enzyme (Supplement Fig. III). This result suggests that the transcription factor function of Atox1 is specific to SOD3 but not to ceruloplasmin, and that the copper chaperone function of Atox1 is required for ceruloplasmin activity.
Discussion
The present study provides novel evidence that cofactor copper increases not only catalytic activity of SOD3 but also SOD3 transcription without affecting SOD1 gene expression via an Atox1 dependent mechanism. We found that: 1) copper-induced increase in SOD3 gene expression is abolished in Atox1-/- cells; 2) copper promotes Atox1 binding to the GAAAGA sequence (Atox1-RE) in the SOD3 promoter, which is consistent with the recently identified Atox1 responsive element in the cyclin D1 promoter; 3) full SOD3 activity is rescued by transfection of both nuclear- and TGN-targeted Atox1 in Atox1-/- cells, but not either Atox1 construct alone. Thus, not only the copper chaperone function but also the transcription factor function of Atox1 plays an essential role in full expression of SOD3 activity (Fig.6).
Fig. 6. Proposed model for the dual role of Atox1 as a copper-dependent transcription factor and a copper chaperone in regulating full activity of SOD3.
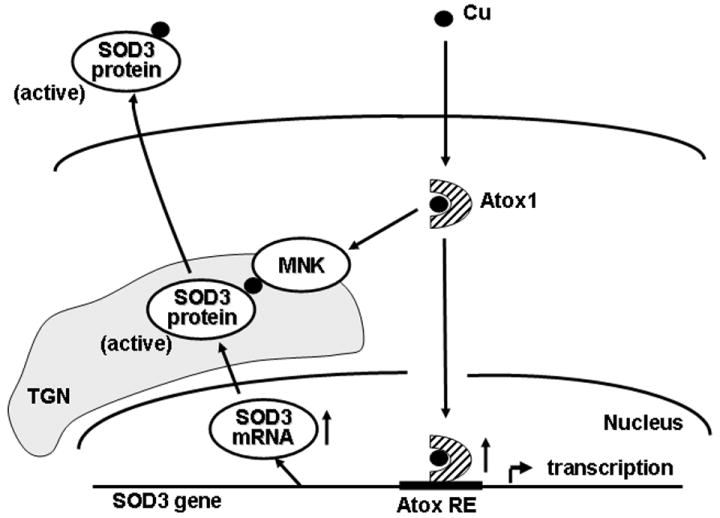
Nuclear Atox1 regulates SOD3 transcription by binding to the Atox1-RE in the SOD3 promoter, while cytosolic Atox1 at the TGN functions as a copper chaperone by delivering copper to SOD3 via interaction with Menkes ATPase (MNK). Full activation of SOD3 requires both functions of Atox1. However, it remains unclear whether they occur in parallel or sequentially.
Copper has been recognized to be essential for the catalytic activity of SOD3 [11]. In the present study, we demonstrate that copper treatment significantly increases mRNA and protein levels of SOD3, but not SOD1, which is abolished in Atox1-deficient cells. Our result is in contrast with a previous report in yeast showing that copper upregulates SOD1 through copper-dependent transcription factor ACE1 [25] whose orthologs have not been identified in the mammalian genome. PEG-catalase and PEG-SOD have no effect on copper-induced SOD3 expression (Supplemental Fig. IB), suggesting that ROS are not involved in these responses. Copper promotes nuclear translocation of Atox1. Promoter deletion analysis, EMSAs, and ChIP assays reveal that copper induces the binding of Atox1 to the Atox1-responsive element (RE) of the SOD3 promoter, which is indispensable for the copper-dependent SOD3 transcription. It is conceivable that copper-induced conformational change of Atox1 [26, 27] may facilitate Atox1 binding to negatively charged DNA. These results suggest that Atox1 functions as a copper-dependent transcription factor to transmit the copper-dependent signal to upregulate SOD3 transcription. Importantly, transcription factor function of Atox1 is specific to SOD3 but not to another secretory copper enzyme ceruloplasmin, although copper chaperone function of Atox1 is required for ceruloplasmin activity. Note that the Atox1 responsive region of SOD3 (5’-GAAAGA-3’) is consistent with the Atox1-RE in the cyclin D1 promoter which we identified previously [17], indicating that this sequence acts as a core element for the copper and the Atox1-RE. Interestingly, both ROS and copper regulate the entry into the S phase by regulating cyclin D1 expression [28, 29]. Given that SOD3 is a major copper-dependent antioxidant enzyme that regulates extracellular levels of O2•- and that Atox1 functions as a copper-dependent transcription factor for SOD3 and cyclin D1, it is likely that Atox1 serves as a sensor of intracellular copper concentrations to regulate cell proliferation by controlling SOD3 and cyclin D1 expression.
Atox1 has been proposed to transfer copper to Menkes ATPase through direct interaction with its N-terminal cytoplasmic domains at TGN [27]. In the present study, we found that transfection of TGN-targeted Atox1 (Atox1-TGN) in Atox1-/- cells rescues specific activity of either SOD3 or ceruloplasmin which obtains copper through a TGN-dependent secretory pathway [14, 30]. Under this condition, Atox1-TGN transfection into Atox1-/- cells restores enzymatic activity of SOD3 only in the presence of Atox1-NLS. These findings suggest that both copper chaperone and transcription factor functions of Atox1 are required for full activation of SOD3. It remains unclear whether these two functions of Atox1 occur in parallel or sequentially (Fig. 6). It is conceivable that copper may at first bind to Atox1 to promote transcription factor function in the nucleus to increase SOD3 gene expression, and then target Atox1 to the cytoplasmic face of the TGN to deliver copper to the transporter Menkes ATPase where SOD3 obtains copper to increase its activity [14]. Future studies are required to clarify the mechanism by which copper is transported from the extracellular space into the cells to deliver copper to Atox1.
The functional significance for the copper- and Atox1-dependent SOD3 expression and activity remains unclear. First, upregulation of the SOD3 gene induced by Cu-Atox1 will prevent inactivation of bioavailable nitric oxide (NO) involved in endothelial function due to the rapid reaction of NO and O2•-, as well as production of highly toxic oxidants such as peroxynitrite and hydroxyl radical by either reaction of NO• and O2•- or by metal-mediated Harber-Weiss reactions [31, 32]. Second, Cu-Atox1 will transfer sufficient levels of intracellular copper to target the SOD3 enzyme to ensure the formation of active holoenzyme. This mechanism could be important, because it will limit the amount of non-copper bound, inactive SOD3 which may have deleterious effects as reported for non-copper bound SOD1 [33]. Third, Cu-Atox1 may contribute to vascular pathology by increasing extracellular levels of H2O2 due to dismutation of O2•- by SOD3. Fourth, Cu-Atox1 may contribute to intracellular copper buffering in a similar fashion to SOD1 [34]. In various lower organisms such as insects, plants, yeast, and fungi, transcription factors activated by copper regulate the expression of genes encoding proteins involved in copper sequestration and/or protection against copper toxicity [35]. Whether other genes linked to copper homeostasis are transcriptionally activated or repressed by Cu-Atox1 is the subject of future study.
Copper deficiency is proposed as a risk factor for coronary heart disease [36]. Several prospective epidemiological studies have shown that high serum copper levels are associated with an increased future risk of coronary heart disease in Dutch men [37], and in the Finnish [38] and US population [39]. Moreover, in experimental animal models, it has been suggested that optimal dietary copper intake in the cholesterol-fed rabbit is linked to a reduced susceptibility to aortic atherosclerosis, while copper deficiency and an excessive daily copper supplement are associated with increased atherogenesis [40]. Thus, tight control of intracellular copper homeostasis by regulating copper containing enzymes through Atox1 and Menkes ATPase seems to be essential to prevent oxidant stress-dependent cardiovascular diseases by regulating SOD3. This notion is supported by our recent study that angiotensin II-induced hypertension,O2•- production and endothelial dysfunction are significantly increased in Menkes ATPase mutant mice [41]. The functional role of Atox1 in vivo using Atox1 knockout mice is current under investigation.
In summary, we demonstrate that SOD3 transcription and activity are regulated by copper through transcription factor function of Atox1, and by both copper chaperone and transcription factor function of Atox1, respectively. This novel finding should provide insight into not only SOD3 but also Atox1 as potential therapeutic targets for treatment of oxidant stress-dependent cardiovascular disease such as atherosclerosis and hypertension.
Supplementary Material
Acknowledgments
This research was supported by NIH R01 HL70187 (T.F.), P01 HL58000 (T.F.), and AHA Grant-In-Aid 0455242B (T.F.); NIH R01 HL 077524 (MU-F), and AHA Grant-in-Aid 0555308B and 0755805Z (MU-F). The authors thank Dr. Lula Hilenski, Emory Universtiy, for editorial assistance.
Footnotes
Disclosures None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res. 2002;55:239–249. doi: 10.1016/s0008-6363(02)00328-0. [DOI] [PubMed] [Google Scholar]
- 2.Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236–256. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 3.Qin Z, Reszka KJ, Fukai T, Weintraub NL. Extracellular superoxide dismutase (ecSOD) in vascular biology: an update on exogenous gene transfer and endogenous regulators of ecSOD. Transl Res. 2008;151:68–78. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: the Copenhagen City Heart Study. Circulation. 2004;109:59–65. doi: 10.1161/01.CIR.0000105720.28086.6C. [DOI] [PubMed] [Google Scholar]
- 5.Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension. 2006;48:473–481. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 6.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. 2003;93:622–629. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 7.Kim HW, Lin A, Guldberg RE, Ushio-Fukai M, Fukai T. Essential role of extracellular SOD in reparative neovascularization induced by hindlimb ischemia. Circ Res. 2007;101:409–419. doi: 10.1161/CIRCRESAHA.107.153791. [DOI] [PubMed] [Google Scholar]
- 8.van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, Duncker DJ, Chen Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic Biol Med. 2008;44:1305–1313. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 10.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 11.Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG, Fukai T. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ Res. 2005;96:723–729. doi: 10.1161/01.RES.0000162001.57896.66. [DOI] [PubMed] [Google Scholar]
- 12.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. see comments. [DOI] [PubMed] [Google Scholar]
- 13.Culotta VC, Yang M, O’Halloran TV. Activation of superoxide dismutases: putting the metal to the pedal. Biochim Biophys Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J. 2006;20:334–336. doi: 10.1096/fj.05-4564fje. [DOI] [PubMed] [Google Scholar]
- 15.Lin SJ, Culotta VC. The ATX1 gene of Saccharomyces cerevisiae encodes a small metal homeostasis factor that protects cells against reactive oxygen toxicity. Proc Natl Acad Sci U S A. 1995;92:3784–3788. doi: 10.1073/pnas.92.9.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hung IH, Casareno RL, Labesse G, Mathews FS, Gitlin JD. HAH1 is a copper-binding protein with distinct amino acid residues mediating copper homeostasis and antioxidant defense. J Biol Chem. 1998;273:1749–1754. doi: 10.1074/jbc.273.3.1749. [DOI] [PubMed] [Google Scholar]
- 17.Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, Fukai T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. 2008;283:9157–9167. doi: 10.1074/jbc.M709463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zelko IN, Folz RJ. Myeloid zinc finger (MZF)-like, Kruppel-like and Ets families of transcription factors determine the cell-specific expression of mouse extracellular superoxide dismutase. Biochem J. 2003;369:375–386. doi: 10.1042/BJ20021431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu FF, Olden K. The expression of ceruloplasmin, an angiogenic glycoprotein, by mouse embryonic fibroblasts. Biochem Biophys Res Commun. 1985;126:15–24. doi: 10.1016/0006-291x(85)90565-0. [DOI] [PubMed] [Google Scholar]
- 20.Sunderman FW, Jr, Nomoto S. Measurement of human serum ceruloplasmin by its p-phenylenediamine oxidase activity. Clin Chem. 1970;16:903–910. [PubMed] [Google Scholar]
- 21.Hainaut P, Rolley N, Davies M, Milner J. Modulation by copper of p53 conformation and sequence-specific DNA binding: role for Cu(II)/Cu(I) redox mechanism. Oncogene. 1995;10:27–32. [PubMed] [Google Scholar]
- 22.Mohindru A, Fisher JM, Rabinovitz M. Bathocuproine sulphonate: a tissue culture-compatible indicator of copper-mediated toxicity. Nature. 1983;303:64–65. doi: 10.1038/303064a0. [DOI] [PubMed] [Google Scholar]
- 23.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 24.Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- 25.Gralla EB, Thiele DJ, Silar P, Valentine JS. ACE1, a copper-dependent transcription factor, activates expression of the yeast copper, zinc superoxide dismutase gene. Proc Natl Acad Sci U S A. 1991;88:8558–8562. doi: 10.1073/pnas.88.19.8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenzweig AC. Copper delivery by metallochaperone proteins. Acc Chem Res. 2001;34:119–128. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 27.Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC. Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat Struct Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 28.Burch PM, Heintz NH. Redox regulation of cell-cycle re-entry: cyclin D1 as a primary target for the mitogenic effects of reactive oxygen and nitrogen species. Antioxid Redox Signal. 2005;7:741–751. doi: 10.1089/ars.2005.7.741. [DOI] [PubMed] [Google Scholar]
- 29.Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, Fukai T. Novel role of antioxidant-1 (atox1) as a copper dependent transcription factor involved in cell proliferation. J Biol Chem. 2008 doi: 10.1074/jbc.M709463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan DS, Dancis A, Klausner RD. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem. 1997;272:25787–25793. doi: 10.1074/jbc.272.41.25787. [DOI] [PubMed] [Google Scholar]
- 31.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutteridge JM. Superoxide dismutase inhibits the superoxide-driven Fenton reaction at two different levels. Implications for a wider protective role. FEBS Lett. 1985;185:19–23. doi: 10.1016/0014-5793(85)80732-8. [DOI] [PubMed] [Google Scholar]
- 33.Banci L, Bertini I, Durazo A, Girotto S, Gralla EB, Martinelli M, Valentine JS, Vieru M, Whitelegge JP. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc Natl Acad Sci U S A. 2007;104:11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Culotta VC, Joh HD, Lin SJ, Slekar KH, Strain J. A physiological role for Saccharomyces cerevisiae copper/zinc superoxide dismutase in copper buffering. J Biol Chem. 1995;270:29991–29997. doi: 10.1074/jbc.270.50.29991. [DOI] [PubMed] [Google Scholar]
- 35.Winge DR, Graden JA, Posewitz MC, Martins LJ, Jensen LT, Simon JR. Sensors that mediate copper-specific activation and repression of gene expression. J Biol Inorg Chem. 1997;2:2–10. [Google Scholar]
- 36.Klevay LM. Cardiovascular disease from copper deficiency--a history. J Nutr. 2000;130:489S–492S. doi: 10.1093/jn/130.2.489S. [DOI] [PubMed] [Google Scholar]
- 37.Kok FJ, Van Duijn CM, Hofman A, Van der Voet GB, De Wolff FA, Paays CH, Valkenburg HA. Serum copper and zinc and the risk of death from cancer and cardiovascular disease. Am J Epidemiol. 1988;128:352–359. doi: 10.1093/oxfordjournals.aje.a114975. [DOI] [PubMed] [Google Scholar]
- 38.Salonen JT, Salonen R, Korpela H, Suntioinen S, Tuomilehto J. Serum copper and the risk of acute myocardial infarction: a prospective population study in men in eastern Finland. Am J Epidemiol. 1991;134:268–276. doi: 10.1093/oxfordjournals.aje.a116080. [DOI] [PubMed] [Google Scholar]
- 39.Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151:1182–1188. doi: 10.1093/oxfordjournals.aje.a010168. [DOI] [PubMed] [Google Scholar]
- 40.Lamb DJ, Avades TY, Ferns GA. Biphasic modulation of atherosclerosis induced by graded dietary copper supplementation in the cholesterol-fed rabbit. Int J Exp Pathol. 2001;82:287–294. doi: 10.1046/j.1365-2613.2001.00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin Z, Gongora MC, Ozumi K, Itoh S, Akram K, Ushio-Fukai M, Harrison DG, Fukai T. Role of Menkes ATPase in Angiotensin II-Induced Hypertension: A Key Modulator for Extracellular SOD Function. Hypertension. 2008 doi: 10.1161/HYPERTENSIONAHA.108.116467. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.