

Abstract
In the current study, we determined the functional significance of sodium-dependent/-independent glucose transporters at the neurovasculature during oxygen glucose deprivation (OGD). Confluent brain endothelial cells cocultured with astrocytes were exposed to varying degrees of in vitro stroke conditions. Glucose transporter (GLUT) 1 and sodium glucose cotransporter (SGLT) activity were investigated by luminal membrane uptake and transport studies using [3H]d-glucose and also by [14C]α-methyl d-glucopyranoside (AMG), a specific, nonmetabolized substrate of SGLT. In vivo middle cerebral artery occlusion experiments were tested to determine whether blood-brain barrier (BBB) SGLT activity was induced during ischemia. Increases in luminal d-glucose and AMG uptake and transport were observed with in vitro stroke conditions. Specific inhibitor experiments suggest a combined role for both SGLT and GLUT1 at the BBB during OGD. A time-dependent increase in the uptake of AMG was also seen in mice exposed to permanent focal ischemia, and this increase was sensitive to the SGLT inhibitor, phlorizin. Infarct and edema ratio during ischemia were significantly decreased by the inhibition of this transporter. These results show that both GLUT1 and SGLT play a role at the BBB in the blood-to-brain transport of glucose during ischemic conditions, and inhibition of SGLT during stroke has the potential to improve stroke outcome. Pharmacological modulation of this novel BBB transporter could prove to be a brain vascular target in stroke.
The central nervous system is protected by three main physiological cell barriers, which consist of the arachnoid epithelium, the choroid plexus epithelium, and the brain endothelium, which form the blood central nervous system interface. The brain itself is protected by brain endothelial cells that restrict the passage of many substances into and out of the brain, forming a selective blood-brain barrier (BBB). For example, various transporters are expressed at the BBB on both the luminal (blood facing) and the abluminal (brain facing) surfaces of the neurovascular barrier (Kumagai et al., 1995) such as the glucose transporter, Na,K-ATPase, Na,K,2Cl-cotransporter, and iron-bound transferrin receptor-mediated transporter. These all play a vital role in the transport of nutrients and ions and endogenous substances into and out of the brain. Transporter expression changes during disease states and overexpression or underexpression of some transporters occur either on the luminal or abluminal sides. For example, increased luminal glucose transporter (GLUT) 1 density occurs with hypoglycemia (Simpson et al., 1999), increased density of the Na,K,2Cl-cotransporter on the luminal (O'Donnell et al., 2004) and abluminal side (Abbruscato et al., 2004) occurs with stroke conditions, and decreased activity of Na,K-ATPase on the abluminal side occurs with oxygen glucose deprivation (OGD) (Kawai et al., 1996; Abbruscato et al., 2004). It is apparent that the neurovascular unit does not simply function as a static barrier yet has the ability to adapt during pathological states such as ischemia by its ability to transport ions and nutrients into and out of the brain.
Glucose is a major energy substrate for mammalian brain metabolism, and a continuous supply of glucose is required for neuronal function. Under conditions of hypoxia, optimum glucose levels are needed to maintain low reactive oxygen species levels and high cell viability in primary cultured neurons (Shi and Liu, 2006). The major transporter through which glucose gains access through BBB is the 55-kDa form of the facilitative glucose transporter protein GLUT1, which is independent of insulin (Harik et al., 1994). GLUT1 is known to be modulated by many pathophysiological conditions, such as Alzheimer's disease, where decreased density of GLUT1 is observed (Kalaria and Harik, 1989). An increase in brain glucose transporter capillary density was observed in chronic hypoxia (Harik et al., 1995), hypoglycemia (Kumagai et al., 1995), and ischemia (Harik et al., 1994). It is apparent that the BBB can increase or decrease nutrient transport depending on the pathophysiological state of the central nervous system.
Sodium glucose cotransporter (SGLT) is another glucose transporter that contributes to nutrient transport. SGLT was originally characterized in kidney proximal tubule epithelial cells and is known to be expressed more on the apical surface of the kidney and on the brush border membrane of the intestine (Wright, 2001). SGLT1, which transports 2Na+/glucose (Mackenzie et al., 1998) is expressed in the intestine epithelial cells, and both SGLT1 and SGLT2 (which transport 1Na+/glucose; Mackenzie et al., 1996) are expressed in kidney epithelial cells. In addition, SGLT1, the 70- to 75-kDa high-affinity isoform, was shown to be expressed in neurons and up-regulated during conditions, such as metabolic stress, when there was a decrease in d-glucose content (Poppe et al., 1997). SGLT, like GLUT1, has also been shown to be present on the brain artery endothelial cells, and its importance was suggested to be in the maintenance of glucose levels in the arteries during the conditions of stress such as hypoglycemia (Nishizaki and Matsuoka, 1998). Immunodetection of SGLT1 was demonstrated at the BBB and was shown to be up-regulated after brain ischemia and reperfusion (Elfeber et al., 2004a). Recently, the mRNA encoding for SGLT2 was also shown to be present and enriched in isolated rat brain microvessels (Enerson and Drewes, 2006). However, a functional role for SGLT has not been established in the brain. In the present study using an in vitro model of varying degrees of OGD to mimic brain ischemia at the BBB (Abbruscato and Davis, 1999) and an in vivo model of middle cerebral artery occlusion, we demonstrated a functional role for SGLT in the blood-to-brain movement of glucose.
Materials and Methods
Materials. [3H]d-Glucose (specific activity of 21.4 Ci/mmol), [3H]mannitol (specific activity of 15–30 Ci/mmol), and [14C]α-methyl d-glucopyranoside (specific activity of 250–350 mCi/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). [14C]Sucrose (specific activity of 625 mCi/mmol) was purchased from American Radiolabeled Chemicals (St. Louis, MO). Transwell Cell Culture assembly with polyester membrane inserts (0.4-μm pore size, 12-mm diameter) were obtained from Corning Life Sciences (Acton, MA). The SGLT inhibitor phlorizin was purchased from Sigma-Aldrich (St. Louis, MO), and the GLUT1 inhibitor phloretin was purchased from Calbiochem (San Diego, CA)
Cell Culture. Primary bovine brain microvascular endothelial cells (BBMECs) were isolated from fresh bovine brains as described previously (Abbruscato and Davis, 1999). In vitro BBMECs have been used extensively to model the BBB and mimic in vivo BBB transport and enzymatic activity. These cells are a pure culture of endothelial cells, free of astrocytes, neurons, and pericytes, and exhibit high transendothelial electrical resistance and polarity of expression of transporters (Abbruscato and Davis, 1999; Abbruscato et al., 2002, 2004). First passage cells were seeded at a cell density of 50,000 cells/cm2 on 12-well transwell plate inserts (0.4-μm pore size) coated with collagen and fibronectin. Factors released by astrocytes have been shown to mimic the in vivo conditions (Brillault et al., 2002); therefore, on day 10 of BBMEC culturing, C6 astroglioma cells (American Type Culture Collection, Manassas, VA) were seeded on the bottom well of BBMEC transwell inserts at a cell density of 40,000 cells/cm2 and were cultured to confluence for 48 h. On day 12, BBMECs were ready for later experiments. All experiments described in these studies were reproduced with at least three separate isolates.
Hypoxia, Hypoxia/Hypoglycemia, Hypoxia/Aglycemia. Confluent monolayer of BBMECs cocultured with astrocytes in the bottom well was exposed to varying degrees of oxygen and glucose deprivation. The concentration of glucose for normoxic conditions was 5.5 mM. Hypoglycemia was induced by decreasing the glucose levels to 50% in the media, and aglycemic conditions were induced by adding RPMI 1640 medium. Hypoxia was induced as described previously (Abbruscato and Davis, 1999; Abbruscato et al., 2004) by placing the cells in a custom-made hypoxic polymer glove box (Coy Laboratory Products Inc., Grass Lakes, MI), which was infused with 95% N2 and 5% CO2, and the temperature was maintained at 37°C. The concentration of oxygen in the atmosphere was maintained at 0%, and the PO2 in the media was below 25 mm Hg. Phlorizin, a phenolic glucoside, a competitive inhibitor of SGLT (Eskandari et al., 2005), and phloretin, a competitive inhibitor of GLUT1 (Betz et al., 1975), were used. A concentration of 50 μM (Nishizaki et al., 1995; Nishizaki and Matsuoka, 1998) was used for both the inhibitors. Bumetanide at a concentration of 20 μM was used to inhibit the Na,K,2Cl-cotransporter (Abbruscato et al., 2004).
Glucose Uptake Studies. After hypoxia (H), hypoxia/hypoglycemia (H/Hg), and hypoxia/aglycemia (H/A) exposure, [3H]d-glucose or [14C]AMG was added to the luminal side, and the plates were rotated for 15 min and then washed three times with ice-cold 0.1 M Tris buffer. BBMECs were solubilized with 1% Triton X-100, and the radioactivity present was determined by counting the samples in a Beckman LS 6500 liquid scintillation counter (Beckman Coulter, Fullerton, CA). Protein content was calculated by using the detergent-compatible BCA assay (Pierce Chemical, Rockford, IL) (Abbruscato et al., 2004). For studies using inhibitors, phlorizin or phloretin or a combination of both the inhibitors was added to the luminal side. Phloretin- and phlorizin-sensitive uptakes were considered GLUT1 and SGLT activity, respectively. For sodium-dependent uptake studies, sodium was replaced by mannitol to maintain consistent osmolarity.
Western Blotting Analysis. The protein was isolated from BBMECs using TRI Reagent (Molecular Research Center, Inc. Cincinnati, OH) at 0.4 ml/100 cm2 culture surface. Protein concentration was determined by using the Pierce BCA assay kit, and total cell membrane was denatured in Laemmli buffer, with β-mercaptoethanol, at 95°C for 5 min, resolved in SDS-polyacrylamide gel electrophoresis (20 μg protein/lane), transferred to polyvinylidene difluoride membrane (GE Healthcare, Chalfont St. Giles, UK), and the polyvinylidene difluoride membrane was incubated with the primary rabbit polyclonal human SGLT1 antibody (hSGLT1) 1:1000 overnight at 4°C or a primary rabbit polyclonal β-actin antibody (1:4000) for 2 h, followed by secondary antibody (1:5000) (goat anti-rabbit IgG-horse radish peroxidase) 2 h at room temperature, and the protein band was detected by enhanced chemiluminescence detecting reagents, Western Lightening (GE Healthcare) (Sabolić et al., 2006).
Transport Studies. Transport studies were performed to determine the role of GLUT1 and SGLT on the blood-to-brain transport of glucose. Confluent BBMECs with astrocytes in the bottom well were exposed to H/A conditions for 12 h. [3H]d-Glucose, [14C]AMG, and [14C]sucrose transport studies were done as described previously (Weber et al., 1993; Abbruscato et al., 1996; Abbruscato and Davis, 1999). For studies using inhibitors, phlorizin or phloretin were added to both the luminal and abluminal sides to inhibit the transporters on both sides. For studies using the Na,K,2Cl-cotransporter inhibitor, bumetanide was added to the abluminal side to inhibit the transporter on abluminal side. The results were expressed as a permeability coefficient with the sucrose (the internal control for paracellular leakiness) space subtracted from the [3H]d-glucose or [14C]AMG values. Apical-to-basolateral permeation of [14C]sucrose and [3H]glucose were calculated by:
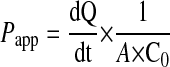 |
(Dehouck et al., 1992), where dQ/dt is the flux of radiolabeled compounds (disintegrations per minute per second), A is the surface area available for transport, and C0 is the original donor concentration (disintegrations per minute per milliliter) of the radioactive compounds.
Middle Cerebral Artery Occlusion. All studies were approved by the Institutional Animal Care and Use Committee of the Texas Tech University Health Sciences Center and were conducted in accordance with the Guide for Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996). Permanent middle cerebral artery occlusion (MCAO) was performed on CD-1 mice (20–22 g), which were anesthetized with 4% isoflurane by inhalation and maintained at 1% isoflurane in a nitrous oxide/oxygen 70:30 mixture. An incision was made in the skin, and the left occipital and superior thyroid arteries, branches of the external carotid artery, and the pterygopalatine, a branch of the internal carotid artery, were cauterized. A suture of 21-mm 5-0 nylon monofilament was introduced through the left external carotid artery, internal carotid artery, and finally to the middle cerebral artery (Mdzinarishvili et al., 2005). A successful occlusion was verified by the sudden drop of the blood flow by 10 to 15% of the basal flow as monitored by the laser Doppler flowmetry. After successful occlusion, the monofilament was secured in place with ligature, and the skin incision was closed by surgical clips. The permanent MCAO was maintained for a period of 1, 3, 6, or 12 h. After MCAO, mice were anesthetized, and 2 μCi of [14C]AMG or [14C]sucrose (the internal control for paracellular leakiness) was injected through the femoral artery. After 30 min, 0.9% saline was infused through the heart for 1 min before decapitation to wash the brain's vascular space free of blood so that any radiolabeled substrate trapped in the vasculature would be removed. The perfusion was considered successful when the brain blanched completely, and then tissues were processed as described previously (Weber et al., 1991). For studies using the inhibitor, the mice were pretreated with an intraperitoneal dose of 200 mg/kg body mass phlorizin (dissolved in 1,3-propanediol) 1 h before injecting AMG (Bormans et al., 2003; Elfeber et al., 2004b).
AMG Influx Rate Constant. For the measurement of AMG uptake during 6 h of permanent focal ischemia, mice received an intravenous bolus injection of [14C]AMG, and blood was collected at different time intervals. [3H]Mannitol, a plasma volume marker, was injected 2 min before the sacrifice. A terminal blood sample was collected at the end of 30 min, and the mice were deeply anesthetized and decapitated. The brain was removed immediately, blotted dry, and weighed. Both the blood and tissue samples were placed in a scintillation vial, and the samples were solubilized with 1 ml of tissue solubilizer and were then counted on a dual-labeled scintillation counter. The influx rate constant, Kin (Kawai et al., 1998), was calculated as:
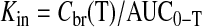 |
(1) |
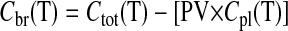 |
(2) |
Cbr (T) is the brain tissue concentration accumulated at time T, AUC0-T (area under the curve) is the integral of the plasma concentrations from time = 0 to time = T, PV is the plasma volume, and Cpl (T) is the final tracer plasma concentration. For studies using the inhibitor, the mice were pretreated with an intraperitoneal dose of 200 mg/kg body mass phlorizin (dissolved in 1,3-propanediol) 1 h before injecting AMG (Bormans et al., 2003; Elfeber et al., 2004b).
Determination of Infarct and Edema Ratio. After 6 h of MCA occlusion, mice were deeply anesthetized and decapitated, and the brain was cut into 1-mm-thick coronal block slices. The brain slices were immersed in 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC) in normal saline at 37°C for 20 min and then fixed in 4% paraformaldehyde at 4°C. TTC stains viable brain tissue dark red based on intact mitochondrial function, whereas infarcted tissue areas remain unstained (white). The size of infarct was calculated using an image analysis system (Image J 1.30 and Scion Image version Beta 4.0.2; National Institutes of Health, Bethesda, MD). Infarct and edema ratios of hemispheric areas were calculated as described previously (Mdzinarishvili et al., 2005). Brain infarct was calculated as the ratio of infarct to the total brain ratio, and brain edema (brain swelling) was quantified by comparing the area of the ipsilateral (ischemic) hemisphere with the contralateral (nonischemic) hemisphere. For studies with the SGLT inhibitor, phlorizin (200 mg/kg) (or vehicle, 1,3 propanediol) was given intraperitoneally 1 h after the induction of focal ischemia, and the animals were sacrificed at the 6th h.
Statistical Methods. All the data were expressed as mean ± S.D., and values were compared by Student's t test or one-way analysis of variance. This test was followed by Newman-Keuls multirange post hoc comparison of the means. Difference in p values less than 0.05 were considered statistically significant.
Results
Time Course Selection. To determine BBMEC sensitivity to H and H/A condition, we performed studies at four different time points (1, 3, 6, and 12 h) and measured d-glucose uptake. Six hours of H/A and 12 h of H and H/A showed significant differences in the glucose uptake compared with the control (p < 0.05; Fig. 1). We observed a maximum uptake of the glucose at 12 h of H/A. Glucose uptake peaks at 12 h; therefore, a time point of 12 h was chosen in the later glucose in vitro uptake and transport studies.
Fig. 1.

Time-dependent studies of luminal glucose uptake into BBMECs cocultured with astrocytes. A, hypoxia; B, hypoxia with aglycemia. Data represent mean ± S.D., n = 5. *, p < 0.05 compared with the control using one-way analysis of variation and Newman-Keuls multiple comparison.
Glucose and AMG Uptake Studies. Luminal [3H]d-glucose uptake studies were performed under control, H, H/Hg, and H/A conditions in the presence or absence of either the GLUT1 inhibitor phloretin (50 μM) or the SGLT inhibitor phlorizin (50 μM). The effect of the inhibitors was dose-dependent at the BBB (data not shown). Under control conditions, with the inhibition of GLUT1, there was a significant decrease in the uptake of glucose, but with SGLT inhibition, there was no change. With the inhibition of SGLT and GLUT1 independently during H, H/Hg, and H/A conditions, there was a significant decrease in the uptake (p < 0.05; Fig. 2, A and B). The combination of both phlorizin and phloretin during H/A significantly reduced glucose uptake compared with each inhibitor by itself (p < 0.05; Fig. 2, A and B). Control values averaged 1.4 ± 0.178 × 10-5 nmol/mg/min for d-glucose uptake. [3H]d-Glucose uptake was also confirmed to be sodium dependent under H/A conditions and not sodium dependent under control conditions (p < 0.05; Fig. 2C). Furthermore, luminal [14C]AMG (nonmetabolizable substrate) uptake studies were carried out in the presence or absence of phlorizin. We observed that under control conditions, there was no significant change in the [14C]AMG uptake in the presence of phlorizin. However, in H, H/Hg, and H/A conditions, phlorizin caused a significant decrease in AMG uptake (p < 0.05; Fig. 3B). The viable cell count of BBMECs after OGD, in the presence or absence of inhibitors, remained constant throughout the experiment, as confirmed by trypan blue exclusion (data not shown).
Fig. 2.

Effect of H, H/Hg, and H/A on luminal glucose uptake into BBMECs with [3H]d-glucose in the presence or absence of phloretin (A), the presence or absence of phlorizin (B), and Na+ dependence of BBMEC on [3H]d-glucose uptake with H/A (C). Data represent mean ± S.D., n = 5. *, p < 0.05 compared with the control using one-way analysis of variation and Newman-Keuls multiple comparison. PHT, phloretin; PHZ, phlorizin.
Fig. 3.

A, SGLT1 Immunoreactivity with H, H/Hg, and H/A conditions. B, effect of H, H/Hg, and H/A on luminal [14C]AMG uptake into BBMECs in the presence or absence of phlorizin. Data represent mean ± S.D., n = 5. *, p < 0.05 compared with the control using one-way analysis of variation and Newman-Keuls multiple comparison. PHZ, phlorizin.
Immunoreactivity for SGLT1. Western blot analysis on the BBMEC total cell lysate confirmed immunoreactivity at 75 kDa in control, H, H/Hg, and H/A conditions, which is consistent for SGLT1 (Sabolić et al., 2006) (Fig. 3A). In all experiments, we observed increased immunoreactivity for SGLT1 in conditions of oxygen and glucose deprivation.
Glucose and AMG Transport Studies. Luminal-to-abluminal transport studies were performed across the confluent BBMEC monolayer (Fig. 4). Control values for [3H]d-glucose permeability averaged 280 × 10-6 ± 79.7 × 10-6 mm/s, which compares well with literature values of 235 × 10-6 mm/s for apparent permeability done in a rat in vivo model (Gjedde, 1981). Control values for [3H]d-glucose were significantly greater than [14C]sucrose values, which averaged 346 × 10-7 ± 48.8 × 10-7 mm/s. [3H]d-Glucose transport in control conditions was sensitive to phloretin but not phlorizin, suggesting the absence of SGLT-mediated blood-to-brain glucose transport during control conditions. There was a significant increase in total glucose transport with H/A treatment, and this increase in glucose transport was sensitive to the presence of phlorizin or phloretin, suggesting a role of both GLUT1 and SGLT in blood-to-brain transport of glucose (p < 0.05; Fig. 4A). The magnitude of inhibition by phloretin was not different between control and H/A. Furthermore, [14C]AMG transport studies were carried out in the presence or absence of phlorizin. Control values of [14C]AMG averaged 262 × 10-7 ± 81.8 × 10-7 mm/s, which was not significantly different from [14C]sucrose values. There was a significant increase in [14C]AMG transport with H/A, and this was inhibited with phlorizin (p < 0.05; Fig. 4B). [3H]d-Glucose apical-to-basolateral transport was sensitive to the Na,K,2Cl-cotransporter inhibitor bumetanide in H/A conditions, suggesting the reliance of abluminal SGLT on the Na,K,2Cl-cotransporter during OGD conditions (p < 0.05; Fig. 5).
Fig. 4.

A, transport of [3H]d-glucose across BBMECs monolayer during H/A in the presence or absence of SGLT inhibitor phlorizin or GLUT1 inhibitor phloretin. B, transport of [14C]AMG across BBMEC monolayer during H/A in the presence or absence of the SGLT inhibitor, phlorizin. Data represent mean ± S.D., n = 5 to 6. *, p < 0.05 compared with the control using one-way analysis of variation and Newman-Keuls multiple comparison. PHT, phloretin; PHZ, phlorizin.
Fig. 5.

Modulation of BBB SGLT transport by bumetanide during H/A. Data represent mean ± S.D., n = 4 to 5. *, p < 0.05 compared with the control using one-way analysis of variation and Newman-Keuls multiple comparison.
Blood-to-Brain Transport and Influx Rate Constant of AMG. Blood-to-brain transport of [14C]AMG was measured in mice exposed to permanent focal ischemia for different time points of 1, 3, 6, and 12 h. Control value averaged to 0.14 ± 0.03% total [14C]AMG injected/g tissue. [14C]AMG uptake values in sham-operated animals were found to be no different from control animals. A significant increase in the uptake of [14C]AMG was observed with all the time points tested for the total brain, 6 and 12 h for the ipsilateral hemisphere and 12 h for the contralateral hemisphere compared with the control conditions (p < 0.05; Fig. 6A). Blood-to-brain transport of [14C]sucrose, an impermeable marker, was also measured in mice exposed to permanent focal ischemia for 12 h. Control values averaged 0.132 ± 0.016, and the 12-h stroke value averaged to 0.122 ± 0.04% total sucrose injected/g tissue.
Fig. 6.

A, transport of [14C]AMG into mouse brain exposed to permanent focal ischemia for 1, 3, 6, or 12 h. Control values averaged to 0.14 ± 0.03% total [14C]AMG injected/g tissue. B, influx rate constant (Kin) of [14C]AMG into mouse brain exposed to permanent focal ischemia for 6 h. Data represent mean ± S.D., n = 4 to 5. *, p < 0.05 compared with the control using one-way analysis of variation and Newman-Keuls multiple comparison.
Further studies to measure the influx rate constant (Kin) of AMG were carried out at a time point of 6 h in the presence of a vascular marker, mannitol (formula weight = 182.17), similar in size to AMG (formula weight = 194). A significant increase in the value of Kin of [14C]AMG (corrected for vascular space with [3H]mannitol)was observed at 6 h of focal ischemia, and this increase was significantly decreased with phlorizin (p < 0.05; Fig. 6B). The vehicle had no effect on AMG distribution.
Brain Infarct and Edema Ratio. The infarct and edema ratios measured in mouse brain subjected to focal ischemia were significantly reduced in animals treated with phlorizin in comparison with vehicle controls (p < 0.05; Fig. 7). The vehicle alone had no effect on the infarct and edema ratio.
Fig. 7.

Effect of phlorizin on infarct ratio (infarct area/brain slice area) and edema ratio (ipsilateral/contralateral hemisphere slice area) after 6 h of MCAO. Visual inspection shows that there is a striking attenuation of the ischemic area in the slices treated with phlorizin. Data represent mean ± S.E.M. of five independent determinations containing five to six slices each. *, p < 0.05 compared with the control using the Student's t test.
Discussion
Glucose is the primary energy source for the brain and in stroke conditions, where there is a need for more glucose because of rapid oxygen depletion; regulation of brain glucose transport becomes an important factor. Glucose is known to be transported at the BBB by GLUT1, a saturable transporter that is independent of energy or ion substrates, but an additional sodium-dependent glucose transporter was also suggested to play a role in blood-to-brain movement of glucose during pathophysiological conditions.
The significance and the novelty of this study is the functional characterization of SGLT at the BBB, which was previously reported to be present mostly in the kidney and intestines (Wright, 1993, 2001). Although studies have demonstrated SGLT1 immunoreactivity at the BBB (Elfeber et al., 2004a) and its up-regulation with ischemia, our studies are the first to show a functional role for SGLT at the BBB, with varying degrees of in vitro and in vivo ischemic conditions. In our studies, we used confluent BBMECs cocultured with C6 astrocytes to best mimic the association of endothelial cells with the astrocytes at the BBB (Brillault et al., 2002). We investigated glucose uptake into the BBMECs with H, H/Hg, and H/A conditions, which was used to model varying degrees of in vitro stroke conditions (Abbruscato and Davis, 1999). Luminal uptake experiments suggest the presence of GLUT1 and absence of SGLT activity in control conditions but suggest a role for both these transporters during H, H/Hg, and H/A conditions. This was further confirmed by luminal AMG (nonmetabolizable substrate) uptake studies and positive immunoreactivity of SGLT1 (Fig. 3). In addition, we determined that the combination of phlorizin and phloretin during H/A significantly reduced glucose uptake compared with each inhibitor by itself (Fig. 2, A and B), thus showing the specificity of each inhibitor for their respective transporter. Further studies investigated the effects of H/A on d-glucose and [14C]AMG transport, and our results strongly suggest the involvement of SGLT in the blood-to-brain transport of glucose during OGD, an in vitro condition that mimics neurovascular ischemia (Abbruscato et al., 2004) (Fig. 4).
Blood-to-brain transport across the neurovascular unit requires movement across both the luminal and abluminal surface of the brain endothelial cell. SGLT is also known to have the ability to function in reverse mode when the intracellular sodium concentration is increased, and it can transport glucose from the interior of the cell into the extracellular fluid (Eskandari et al., 2005). The presence of a higher affinity sodium-dependent transporter on the abluminal surface of the BBB was suggested in isolated plasma membranes from bovine microvascular endothelial cells (Lee et al., 1997), although these experiments were not confirmed using in vivo models. The Na,K,2Cl-cotransporter, which transports 1Na+, 1K+, and 2Cl- ions, pumps sodium from the brain extracellular fluid into the cell and is up-regulated during OGD conditions at the BBB on both luminal and abluminal sides (Abbruscato et al., 2004; O'Donnell et al., 2004). This BBB Na,K,2Cl-cotransporter may provide a sodium gradient to fuel sodium-dependent brain glucose transport during H/A by SGLT from the inside of brain endothelial cells to the brain extracellular fluid. Therefore, to study the possible dependence of SGLT on the Na,K,2Cl-cotransporter for intracellular sodium, apical-to-basolateral glucose transport studies were tested. We observed an increase in glucose transport with 12 h of OGD that was sensitive to the Na,K,2Cl-cotransporter inhibitor bumetanide, suggesting the reliance of SGLT on the Na,K,2Cl-cotransporter (Fig. 5). We also confirmed the presence of SGLT activity at the BBB, with different levels of in vitro ischemia. Hypoxia alone or combined with hypoglycemia or aglycemia models the various stages of nutrient deficit going from the core to the penumbral regions. We determined that SGLT plays a role in the uptake and transport of glucose from blood to brain along with GLUT1.
To further strengthen the presence of SGLT at the BBB, we designed focal ischemia experiments in mice under 12 h. Our studies with [14C]sucrose, an impermeable marker, and studies in the literature have shown that significant paracellular BBB breakdown does not occur under 12 h of permanent MCAO (Hatashita and Hoff, 1990). In addition, increased expression of matrix metalloproteinases does not occur until after 12 h of MCAO (Romanic et al., 1998). Therefore, our experiments were conducted at time points ≤12 h, where the paracellular barrier is believed to remain intact. A significant increase in the brain uptake of [14C]AMG was seen with all the time points in total brain, with 6 and 12 h in the ipsilateral hemisphere and 12 h in contralateral hemisphere compared with the control conditions (Fig. 6A). These results are consistent with another group that observed a bilateral up-regulation of SGLT1 immunoreactivity after MCAO (Elfeber et al., 2004a). We speculate that the observed changes in both ischemic and nonischemic hemispheres at the 12-h time point could be because of the release of mediators into the circulation. Other researchers have also reported bilateral up- and down-regulation of brain-specific proteins (Focking et al., 2006), including brain endothelial cell interleukin-1β (Zhang et al., 1998) with focal ischemia. Future investigations will decipher these interesting pathophysiologic results. In addition, we observed that the influx rate constant, corrected for vascular space for [14C]AMG after 6 h of focal ischemia, was significantly decreased in the presence of phlorizin, further strengthening the function of SGLT at the BBB (Fig. 6B). These in vivo experiments help to verify the function of SGLT-mediated transport at the BBB during ischemia and validate that it takes part in both apical and basolateral transport across the BBB. Because [14C]AMG has specificity for SGLT and not GLUT1 and is a nonmetabolizable substrate (Poppe et al., 1997; Bormans et al., 2003), these data strongly support that SGLT mediates a significant amount of glucose transport across the BBB during ischemia.
OGD-induced SGLT activity could be detrimental during ischemia because it could cause more sodium and calcium influx and contribute to ischemic neuronal damage (Elfeber et al., 2004a). To test this possible hypothesis, we administered phlorizin 1 h after focal ischemia, and we observed a significant decrease in ischemia and edema ratios as judged by TTC staining of viable cells and from measurements of hemispheric enlargement after 6 h of focal ischemia (Fig. 7). We believe that this action of phlorizin on BBB SGLT may have several potential implications. For example, inhibition of SGLT could decrease the continued delivery of glucose, which has been shown to be detrimental during ischemia because of cerebral acidosis and free radical production resulting from anaerobic metabolism of glucose. Another result of SGLT inhibition could be reduction of brain edema associated with ischemia because this transporter is known to transfer approximately 210 water molecules for each Na+ and glucose molecule (Wolkoff et al., 1998); therefore, this could reduce edematous conditions associated with ischemia. Future investigations will decipher the neuroprotective mechanism(s) involved in SGLT inhibition at the neurovascular unit because this transporter is known to be present on both the astrocytes and neurons (Poppe et al., 1997; Véga et al., 2006).
Our results suggest the presence of SGLT at the BBB. Based on the current data, we proposed a working hypothesis for SGLT at the BBB (Fig. 8). The Na,K,2Cl-cotransporter (green) may provide a sodium gradient (blue arrows) to fuel sodium-dependent brain glucose transport during ischemia by SGLT (yellow) from the inside of brain endothelial cells to the brain extracellular fluid. In addition, the abluminal Na,K-ATPase (blue), which aids in the efflux of sodium from the brain endothelial cell into the brain extracellular fluid, has been shown to be deactivated during models of ischemia (Kawai et al., 1996; Abbruscato et al., 2004). The net efflux of sodium from brain endothelial cells is comparatively less, and this would further contribute to the accumulation of sodium in brain endothelial cells during stroke conditions. As a proposed operative mechanism in intestinal and kidney epithelial cells (Eskandari et al., 2005), SGLT in the brain might also have the capability to cotransport sodium and glucose into and out of the cells when provided with a sodium-driving force during conditions of OGD. Future immunohistochemical or electron microscopy experiments are needed to validate the expression polarity of SGLT at the BBB during ischemia.
Fig. 8.

Working hypothesis for enhanced sodium-dependent glucose transport across the blood-brain barrier during ischemia. SGLT is activated at the BBB during ischemia and aids in the transport of glucose along with GLUT1 from blood to brain. The Na,K,2Cl-cotransporter, which is activated on the luminal and the abluminal sides in conditions of low oxygen and glucose, could provide a driving sodium force (blue arrows) that would fuel SGLT for sodium-coupled glucose transport into the brain extracellular fluid. In addition, when the abluminal Na+,K+-ATPase is deactivated at the BBB during H/A, there would be a decrease in the net efflux of sodium from the brain endothelial cell, thus a driving sodium force for the net movement of glucose from the endothelial cell interior to the brain extracellular fluid.
In summary, we have determined the luminal activity of SGLT and confirmed its role in the transport of glucose from blood to brain with in vitro and in vivo conditions. Furthermore, we have confirmed its protective role in permanent focal ischemia. Future studies on the expression, regulation, and inhibition of SGLT in vivo with temporary focal ischemia when BBB damage occurs more acutely would lead to further understanding as to how SGLT can be utilized as a therapeutic target in stroke and other central nervous system disorders. Future work is needed to determine the effect of modulating this transporter in association with tissue plasminogen activator therapy. Modulating this novel neurovascular target possibly could provide a means to extend the therapeutic window of tissue plasminogen activator therapy by reducing the edematous condition and preventing the negative effects of hyperglycemia.
Acknowledgments
We thank Dr. Herman Koepsell for the kind gift of the SGLT1 antibody.
This study was supported by the National Institutes of Health [Grant NS-O46526].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.108.146589.
ABBREVIATIONS: BBB, blood-brain barrier; GLUT, glucose transporter; OGD, oxygen glucose deprivation; SGLT, sodium glucose cotransporter; BBMEC, bovine brain microvessel endothelial cell; H, hypoxia; H/Hg, hypoxia/hypoglycemia; H/A, hypoxia/aglycemia; AMG, α-methyl glucoside; MCAO, middle cerebral artery occlusion; TTC, 2,3,5-triphenyltetrazolium chloride.
References
- Abbruscato TJ and Davis TP (1999) Combination of hypoxia/aglycemia compromises in vitro blood-brain barrier integrity. J Pharmacol Exp Ther 289 668-675. [PubMed] [Google Scholar]
- Abbruscato TJ, Lopez SP, Mark KS, Hawkins BT, and Davis TP (2002) Nicotine and cotinine modulate cerebral microvascular permeability and protein expression of ZO-1 through nicotinic acetylcholine receptors expressed on brain endothelial cells. J Pharm Sci 91 2525-2538. [DOI] [PubMed] [Google Scholar]
- Abbruscato TJ, Lopez SP, Roder K, and Paulson JR (2004) Regulation of blood-brain barrier Na,K,2Cl-cotransporter through phosphorylation during in vitro stroke conditions and nicotine exposure. J Pharmacol Exp Ther 310 459-468. [DOI] [PubMed] [Google Scholar]
- Abbruscato TJ, Williams SA, Misicka A, Lipkowski AW, Hruby VJ, and Davis TP (1996) Blood-to-central nervous system entry and stability of biphalin, a unique double-enkephalin analog, and its halogenated derivatives. J Pharmacol Exp Ther 276 1049-1057. [PubMed] [Google Scholar]
- Betz AL, Drewes LR, and Gilboe DD (1975) Inhibition of glucose transport into brain by phlorizin, phloretin and glucose analogues. Biochim Biophys Acta 406 505-515. [DOI] [PubMed] [Google Scholar]
- Bormans GM, Van Oosterwyck G, De Groot TJ, Veyhl M, Mortelmans L, Verbruggen AM, and Koepsell H (2003) Synthesis and biologic evaluation of (11)c-methyl-d-glucoside, a tracer of the sodium-dependent glucose transporters. J Nucl Med 44 1075-1081. [PubMed] [Google Scholar]
- Brillault J, Berezowski V, Cecchelli R, and Dehouck MP (2002) Intercommunications between brain capillary endothelial cells and glial cells increase the transcellular permeability of the blood-brain barrier during ischaemia. J Neurochem 83 807-817. [DOI] [PubMed] [Google Scholar]
- Dehouck MP, Jolliet-Riant P, Brée F, Fruchart JC, Cecchelli R, and Tillement JP (1992) Drug transfer across the blood-brain barrier: correlation between in vitro and in vivo models. J Neurochem 58 1790-1797. [DOI] [PubMed] [Google Scholar]
- Elfeber K, Köhler A, Lutzenburg M, Osswald C, Galla HJ, Witte OW, and Koepsell H (2004a) Localization of the Na+-d-glucose cotransporter SGLT1 in the blood-brain barrier. Histochem Cell Biol 121 201-207. [DOI] [PubMed] [Google Scholar]
- Elfeber K, Stümpel F, Gorboulev V, Mattig S, Deussen A, Kaissling B, and Koepsell H (2004b) Na(+)-d-glucose cotransporter in muscle capillaries increases glucose permeability. Biochem Biophys Res Commun 314 301-305. [DOI] [PubMed] [Google Scholar]
- Enerson BE and Drewes LR (2006) The rat blood-brain barrier transcriptome. J Cereb Blood Flow Metab 26 959-973. [DOI] [PubMed] [Google Scholar]
- Eskandari S, Wright EM, and Loo DD (2005) Kinetics of the reverse mode of the Na+/glucose cotransporter. J Membr Biol 204 23-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focking M, Besselmann M, and Trapp T (2006) Proteomics of experimental stroke in mice. Acta Neurobiol Exp (Wars) 66 273-278. [DOI] [PubMed] [Google Scholar]
- Gjedde A (1981) High- and low-affinity transport of d-glucose from blood to brain. J Neurochem 36 1463-1471. [DOI] [PubMed] [Google Scholar]
- Harik SI, Behmand RA, and LaManna JC (1994) Hypoxia increases glucose transport at blood-brain barrier in rats. J Appl Physiol 77 896-901. [DOI] [PubMed] [Google Scholar]
- Harik SI, Lust WD, Jones SC, Lauro KL, Pundik S, and LaManna JC (1995) Brain glucose metabolism in hypobaric hypoxia. J Appl Physiol 79 136-140. [DOI] [PubMed] [Google Scholar]
- Hatashita S and Hoff JT (1990) Brain edema and cerebrovascular permeability during cerebral ischemia in rats. Stroke 21 582-588. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals, 7th ed, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC.
- Kalaria RN and Harik SI (1989) Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53 1083-1088. [DOI] [PubMed] [Google Scholar]
- Kawai N, Keep RF, Betz AL, and Nagao S (1998) Hyperglycemia induces progressive changes in the cerebral microvasculature and blood-brain barrier transport during focal cerebral ischemia. Acta Neurochir Suppl 71 219-221. [DOI] [PubMed] [Google Scholar]
- Kawai N, McCarron RM, and Spatz M (1996) Effect of hypoxia on Na(+)-K(+)-Cl-cotransport in cultured brain capillary endothelial cells of the rat. J Neurochem 66 2572-2579. [DOI] [PubMed] [Google Scholar]
- Kumagai AK, Kang YS, Boado RJ, and Pardridge WM (1995) Upregulation of blood-brain barrier GLUT1 glucose transporter protein and mRNA in experimental chronic hypoglycemia. Diabetes 44 1399-1404. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Peterson DR, Sukowski EJ, and Hawkins RA (1997) Glucose transport by isolated plasma membranes of the bovine blood-brain barrier. Am J Physiol 272 C1552-C1557. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Loo DD, Panayotova-Heiermann M, and Wright EM (1996) Biophysical characteristics of the pig kidney Na+/glucose cotransporter SGLT2 reveal a common mechanism for SGLT1 and SGLT2. J Biol Chem 271 32678-32683. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Loo DD, and Wright EM (1998) Relationships between Na+/glucose cotransporter (SGLT1) currents and fluxes. J Membr Biol 162 101-106. [DOI] [PubMed] [Google Scholar]
- Mdzinarishvili A, Geldenhuys WJ, Abbruscato TJ, Bickel U, Klein J, and Van der Schyf CJ (2005) NGP1–01, a lipophilic polycyclic cage amine, is neuroprotective in focal ischemia. Neurosci Lett 383 49-53. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Kammesheidt A, Sumikawa K, Asada T, and Okada Y (1995) A sodium- and energy-dependent glucose transporter with similarities to SGLT1–2 is expressed in bovine cortical vessels. Neurosci Res 22 13-22. [DOI] [PubMed] [Google Scholar]
- Nishizaki T and Matsuoka T (1998) Low glucose enhances Na+/glucose transport in bovine brain artery endothelial cells. Stroke 29 844-849. [DOI] [PubMed] [Google Scholar]
- O'Donnell ME, Tran L, Lam TI, Liu XB, and Anderson SE (2004) Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24 1046-1056. [DOI] [PubMed] [Google Scholar]
- Poppe R, Karbach U, Gambaryan S, Wiesinger H, Lutzenburg M, Kraemer M, Witte OW, and Koepsell H (1997) Expression of the Na+-d-glucose cotransporter SGLT1 in neurons. J Neurochem 69 84-94. [DOI] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, and Barone FC (1998) Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29 1020-1030. [DOI] [PubMed] [Google Scholar]
- Sabolić I, Skarica M, Gorboulev V, Ljubojević M, Balen D, Herak-Kramberger CM, and Koepsell H (2006) Rat renal glucose transporter SGLT1 exhibits zonal distribution and androgen-dependent gender differences. Am J Physiol Renal Physiol 290 F913-F926. [DOI] [PubMed] [Google Scholar]
- Shi H and Liu KL (2006) Effects of glucose concentration on redox status in rat primary cultured neuronal cells under hypoxia. Neurosci Lett 410 57-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, Koehler-Stec EM, Vannucci SJ, and Smith QR (1999) Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem 72 238-247. [DOI] [PubMed] [Google Scholar]
- Véga C, R Sachleben L Jr, Gozal D, and Gozal E (2006) Differential metabolic adaptation to acute and long-term hypoxia in rat primary cortical astrocytes. J Neurochem 97 872-883. [DOI] [PubMed] [Google Scholar]
- Weber SJ, Abbruscato TJ, Brownson EA, Lipkowski AW, Polt R, Misicka A, Haaseth RC, Bartosz H, Hruby VJ, and Davis TP (1993) Assessment of an in vitro blood-brain barrier model using several [Met5]enkephalin opioid analogs. J Pharmacol Exp Ther 266 1649-1655. [PubMed] [Google Scholar]
- Weber SJ, Greene DL, Sharma SD, Yamamura HI, Kramer TH, Burks TF, Hruby VJ, Hersh LB, and Davis TP (1991) Distribution and analgesia of [3H][d-Pen2,d-Pen5]enkephalin and two halogenated analogs after intravenous administration. J Pharmacol Exp Ther 259 1109-1117. [PubMed] [Google Scholar]
- Wolkoff AW, Suchy FJ, Moseley RH, Meier PJ, Gollan JL, Freimer N, Fitz JG, Boyer JL, Berk PD, and Scharschmidt BF (1998) Advances in hepatic transport: molecular mechanisms, genetic disorders, and treatment: a summary of the 1998 AASLD single topic conference. Hepatology 28 1713-1719. [DOI] [PubMed] [Google Scholar]
- Wright EM (1993) The intestinal Na+/glucose cotransporter. Annu Rev Physiol 55 575-589. [DOI] [PubMed] [Google Scholar]
- Wright EM (2001) Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol 280 F10-F18. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chopp M, Goussev A, and Powers C (1998) Cerebral vessels express interleukin 1beta after focal cerebral ischemia. Brain Res 784 210-217. [DOI] [PubMed] [Google Scholar]