

Abstract
Nitric oxide has been shown to be an important component of the human immune response, and as such, it is important to understand how pathogenic organisms respond to its presence. In Neisseria gonorrhoeae, recent work has revealed that NsrR, an Rrf2-type transcriptional repressor, can sense NO and control the expression of genes responsible for NO metabolism. A highly pure extract of epitope tagged NsrR was isolated and mass spectroscopic analysis suggested that the protein contained a [2Fe-2S] cluster. NsrR/DNA interactions were thoroughly analyzed in vitro. Using EMSA analysis, NsrR::FLAG was shown to interact with predicted operators in the norB, aniA, and nsrR upstream regions with a Kd of 7 nM, 19 nM, and 35 nM respectively. DNase I footprint analysis was performed on the upstream regions of norB and nsrR, where NsrR was shown to protect the predicted 29 bp binding sites. The presence of exogenously added NO inhibited DNA binding by NsrR. Alanine substitution of C90, C97, or C103 in NsrR abrogated repression of norB::lacZ and inhibited DNA binding, consistent with their presumed role in coordination of a NO-sensitive Fe-S center required for DNA binding.
Keywords: Neisseria gonorrhoeae, nitric oxide, norB, NsrR, IscR, Rrf2
INTRODUCTION
Neisseria gonorrhoeae, the causative agent of the sexually transmitted infection gonorrhea, is a considerable global threat. It is estimated that worldwide, 62 million new infections occur each year, and antimicrobial resistance remains a major concern (www.cdc.gov, Seib et al., 2006). Prolonged gonococcal infection may lead to serious complications, including pelvic inflammatory disease (PID), a major cause of female sterility, and disseminated gonococcal infection (DGI), which can lead to septicemia, dermatitis, and migratory polyarthritis (Garnett et al., 1999, Edwards & Apicella, 2004).
As an obligate human pathogen, the gonococcus is extremely well adapted for growth on a variety of mucosal surfaces (Edwards & Apicella, 2004, Ghosn & Kibbi, 2004). N. gonorrhoeae is also a facultative anaerobe capable of nitrite (NO2−) respiration under microaerobic and anaerobic conditions, and nitric oxide (NO) respiration in the presence of NO (Knapp & Clark, 1984, Overton et al., 2006). Anaerobic growth is accomplished through utilization of a truncated denitrification pathway encoded within the gonococcal genome as a pair of divergently transcribed genes, aniA, encoding a copper-containing nitrite reductase, and norB, encoding a single subunit nitric oxide reductase (Householder et al., 1999, Householder et al., 2000). Both of these genes are subject to regulation by NsrR, a nitric oxide sensing Rrf2-type transcriptional repressor in the winged helix superfamily (Rodionov et al., 2005, Overton et al., 2006, Seib et al., 2006, Nakano et al., 2006, Filenko et al., 2007, Gilberthorpe et al., 2007, Rock et al., 2007, Rogstam et al., 2007, Isabella et al., 2008).
Recent work has demonstrated that NsrR controls the expression of nitric oxide detoxification systems in several pathogenic and environmental organisms. Also, genes in the NsrR regulon contribute to resistance to nitrosative stress (Rodionov et al., 2005, Bodenmiller & Spiro, 2006, Overton et al., 2006, Filenko et al., 2007, Nakano et al., 2006; Gilberthorpe et al., 2007, Rock et al., 2007, Rogstam et al., 2007). Aside from aniA and norB, gonococcal NsrR has been shown to regulate the expression of dnrN, a gene involved in the repair of RNS or ROS damaged Fe-S centers, mobA, a gene involved in molybdopterin guanine dinucleotide biosynthesis, and the nsrR gene itself (Justino et al., 2007, Heurlier et al., 2008, Isabella et al., 2008, Overton et al., 2008).
The ability of pathogenic organisms to sense and respond to the presence of NO during infection may have profound effects on disease, thus making NsrR an essential target of study (for a review on the role of NO in immunity see: Stefano et al., 2000, Bogdan, 2001, Guzik et al., 2003). In Neisseria meningitidis, reduction of host-produced NO results in modulation of cytokine responses, enhanced intracellular survival, and inhibition of apoptosis in a macrophage model (Stevanin et al., 2005, Tunbridge et al., 2006, Stevanin et al., 2007). The effect of NO reduction during gonococcal infection is still unclear, though recent evidence suggests that the reduction of host-produced NO to anti-inflammatory levels may play a role in the high rates of asymptomatic infection observed in women (Cardinale & Clark, 2005).
A closely related Rrf2 protein, IscR, has been characterized in E. coli and is reported to sense the Fe-S cluster status of the cell by reversible binding of an Fe-S cluster (Schwartz et al., 2001, Giel et al., 2006, Yeo et al., 2006, Rock et al., 2007). NsrR contains three conserved cysteine residues that, in IscR, are presumed to coordinate the Fe-S cluster. It is predicted that NsrR also coordinates an Fe-S cluster, and that NO interaction with this cluster perturbs the ability of the protein to bind DNA (Rock et al., 2007). NsrR homologs in Bacillus subtilis, Salmonella typhimurium, and E. coli have all been shown to regulate the expression of the gene encoding flavohemoglobin (Hmp), one of the most extensively studied NO-detoxifying proteins (Nakano et al., 2006, Overton et al., 2006, Gilberthorpe et al., 2007, Rogstam et al., 2007). Hmp is present in a broad range of bacterial species, and hmp mutations in most bacteria severely compromise bacterial survival in the presence of NO (Gilberthorpe et al., 2007). However, N. gonorrhoeae does not contain an hmp ortholog, and unlike most bacteria, gonococcal norB mutants incubated anaerobically in the presence of nitrite (NO-generating conditions) are still able to survive (Zumft, 1997, Garnett et al., 1999, Householder et al., 2000). This suggests that N. gonorrhoeae has additional NO detoxification pathways, and that these pathways are novel (Seib et al., 2006).
The potential pathogenic outcome of the bacterial response to NO sensing, as well as the relative lack of information available on the Rrf2 class of transcriptional regulators, gave cause to further analyze NsrR. In this study we prepared an extract of epitope-tagged gonococcal NsrR for use in vitro. A thorough analysis of NsrR/DNA interaction with the upstream regions of norB, aniA, and nsrR was completed in order to calculate binding affinities for the NsrR operators controlling expression of those genes. NsrR binding sites are identified in the upstream regions of norB and nsrR by generating DNaseI footprints. Mass spectroscopy is used to analyze the iron-sulfur cluster content of purified NsrR extracts. Finally, the effects of alanine substitution of conserved cysteine residues in NsrR are reported.
RESULTS
NsrR::FLAG purification
Site directed mutagenesis was used to add the codons of a FLAG peptide tag to the 3’ end of the gonococcal nsrR gene. This fusion gene was overexpressed in E. coli, and NsrR::FLAG protein (17 kD) was subsequently isolated with M2 antibody (α-FLAG) to generate an extract for use in vitro. The recovered extract was shown to be extremely pure, as no contamination was observed in a silver stained sample, and no cross-reactive species were observed by Western blot analysis (Fig. 1A). In a gonococcal strain containing a norB::lacZ translational promoter fusion (RUG7500), the wild-type nsrR gene was replaced with a copy of nsrR::FLAG (RUG7800). Comparison of β-galactosidase levels in the wild-type and nsrR::FLAG strain confirm that NsrR::FLAG is still functional as a repressor and is capable of regulating expression of norB::lacZ under NO-generating conditions (cells grown anaerobically with nitrite; Fig. 1B).
Figure 1. Analysis of NsrR::FLAG extract and functionality in vivo.
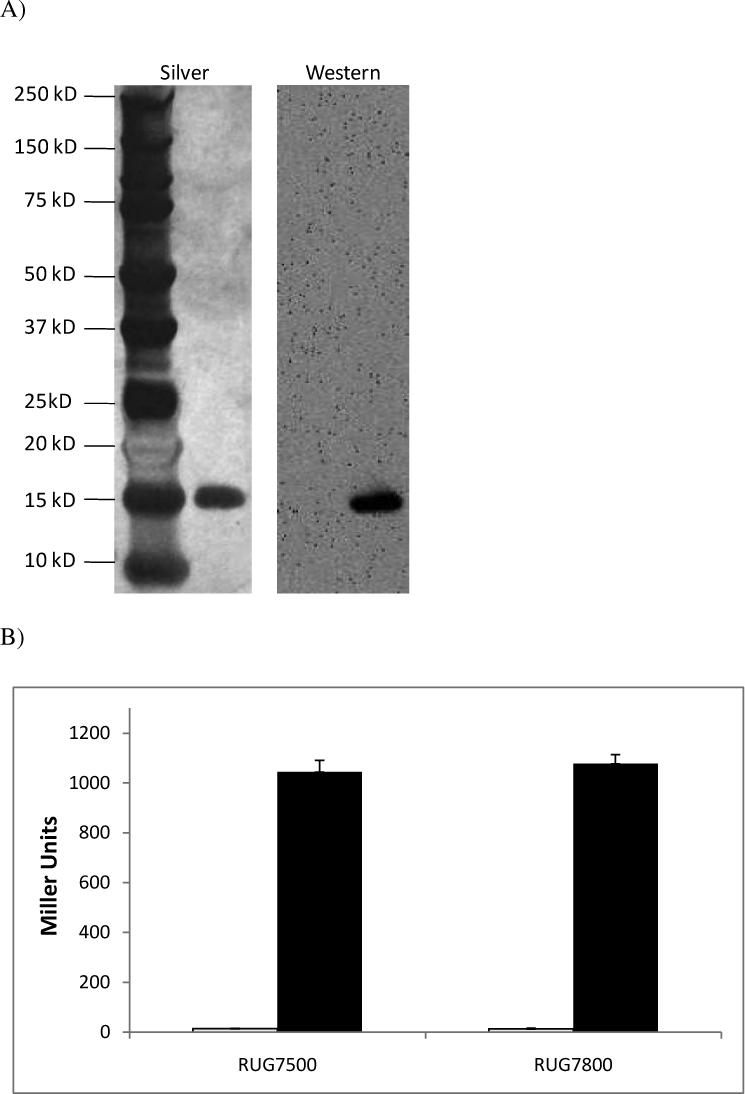

(A) Affinity purified NsrR::FLAG was analyzed by SDS-PAGE and silver stained (left) or probed with M2 antibody in Western blot analysis (right). (B) Wild type (RUG7500) and nsrR::FLAG expressing (RUG7800) gonococcal norB::lacZ reporter fusion strains were grown aerobically (white bars) and anaerobically with nitrite (black bars), and ß–galactosidase activity was measured. These data are the mean of 6 determinations ± one SD. (C) Mass spectrum of undigested NsrR::FLAG in linear positive ion mode. 1800 laser shots were summed to obtain this spectrum. The observed mass peak is equal to the predicted mass of the protein plus the mass of a [2Fe-2S] cluster.
NsrR is predicted to coordinate a [2Fe-2S] cluster involved in NO-sensing (Nakano et al., 2006, Rock et al., 2007). Purification of NsrR::FLAG by immunoprecipitation, though resulting in an extremely pure extract, is not amenable for the production of protein in the concentrations necessary for observation of an iron-sulfur cluster by electron paramagnetic resonance (EPR) or UV-visible spectroscopy. In order to confirm that this NsrR::FLAG extract contained a [2Fe-2S] cluster, mass spectroscopic analysis of undigested protein was performed in linear positive ion mode (Fig. 1C). The predicted mass of a [2Fe-2S] cluster is 175.8 Daltons, while the predicted mass of NsrR with the attached octapeptide FLAG tag is 17,038.8 Daltons. Combining the mass prediction of NsrR::FLAG with that of [2Fe-2S] gives approximately 17,214 Daltons, which was the observed mass peak of the NsrR::FLAG sample. The data also suggests that the extract is fully loaded with [2Fe-2S], as there is no peak visible at 17,038.8 Daltons. The case is different for IscR, where the purified protein was observed to be 26−66% occupied with iron-sulfur cluster (Giel et al., 2006). The observed discrepancy in iron-sulfur cluster content between purified NsrR and IscR may simply be due to the different expression and purification schemes used in each case, or conversely, it may be due to the fact that physiologically, apo-IscR is a functionally relevant state, whereas apo-NsrR has yet to be shown to be involved in gene regulation in vivo (Bodenmiller & Spiro, 2006, Giel et al., 2006, Overton et al., 2006, Rock et al., 2007, Isabella et al., 2008).
NsrR-specific binding of the norB upstream region
An Electrophoretic Mobility Shift Assay (EMSA) was performed using a 120 bp biotin end-labeled fragment of the norB upstream region (−90 to +30 relative to the translation start site). Labeled norB target DNA was shifted to a higher molecular weight upon addition of NsrR::FLAG to the binding reaction (Fig. 2). DNA binding was shown to be NsrR-specific by supershift with M2 antibody. Binding was shown to be sequence-specific by shift inhibition upon the addition of 100 fold higher concentration of unlabeled norB target to the binding reaction.
Figure 2. In vitro analysis of NsrR::FLAG extract.

EMSA analysis was performed on a biotin end-labeled fragment of the norB promoter: Lane (1) No NsrR addition, (2) 50 nM NsrR::FLAG, (3) 50 nM NsrR::FLAG + M2 antibody, (4) 50 nM NsrR::FLAG + 100X concentration (350 fmol) of unlabeled norB fragment.
Binding affinity at the NsrR operator
The gonococcal NsrR operator has been partially characterized in previous work in vivo, by analysis of the ability of NsrR to regulate promoter-lacZ reporter fusions containing variations of the NsrR binding site (Isabella et al., 2008). In order to determine important bases of the 29 bp inverted repeat sequence presumed to make up the NsrR binding site, we determined the in vitro binding affinity of NsrR for its binding sites in the norB, aniA, and nsrR upstream regions. EMSA analysis of the 120 bp biotin end-labeled norB fragment with increasing concentrations of NsrR::FLAG allowed for the estimation of dissociation constant (Kd) between NsrR and its operator in norB, which was equal to the concentration of NsrR::FLAG at which half of the biotinylated DNA was bound. NsrR was found to interact with the norB operator with an estimated Kd of 7 nM (Fig. 3). This measurement was verified by shifting half of the norB fragment with NsrR::FLAG at its estimated Kd in a subsequent EMSA (Fig 3A).
Figure 3. Binding affinity at the NsrR operator.


(A) Increasing concentrations of NsrR::FLAG were used to shift 120 bp biotin end-labeled fragments containing the norB, aniA and norB half-site NsrR operators, as well as a 380 bp fragment of the nsrR upstream region. Panels to the right show fragments shifted to their estimated Kd in subsequent experiments. (*) Positive control containing full length norB NsrR binding site (B) Spot densitometry was used to quantify the ratio of shifted DNA from the EMSA analysis in (A). Kd stimations were calculated by extrapolating the concentration of NsrR::FLAG where 50% of the labeled fragment was shifted. Legend shows the sequence of the NsrR operators from norB, aniA, nsrR, and the norB half-site. Bases shaded in grey are common to the NsrR operator of norB.
The aniA upstream region contains a putative NsrR binding motif that matches the consensus predicted by Rodionov et al. (2005) and that was shown to confer NsrR regulation of aniA (Overton et al., 2006, Rock et al., 2007, Isabella et al., 2008). The putative 29 bp NsrR operator from aniA was cloned into the norB upstream region, replacing the NsrR binding site of norB with that of aniA (Isabella et al., 2008). This fragment, identical to the one used to measure NsrR/norB interaction except for the 29 bp replacement, was used to estimate the Kd between NsrR::FLAG and its operator in aniA. NsrR::FLAG was shown to interact with the aniA operator with an approximate Kd of 19 nM (Fig. 3). These results show that NsrR has a different affinity for the promoters of each of the denitrification genes.
The putative NsrR binding site from the nsrR upstream region has only a low level of similarity to that found in norB (Fig. 3B), however, expression of norB::lacZ in a gonococcal reporter fusion strain in which the norB NsrR operator was replaced with that from nsrR still displayed a weakly repressed phenotype and was induced in the presence of NO, albeit only by four-fold (Isabella et al., 2008). Contrary to this finding, expression of nsrR::lacZ in a gonococcal reporter fusion strain was induced sixty-fold in a ΔnsrR mutant (Isabella et al., 2008). In E. coli, IscR was demonstrated to recognize two distinctly different classes of binding sites (Giel et al., 2006, Yeo et al., 2006). We reasoned that the discrepancy in the aforementioned data could potentially be due to the existence of a second NsrR binding site in the nsrR promoter. To test this hypothesis, we performed an EMSA with a 380 bp biotin end-labeled fragment of the nsrR upstream region to estimate the Kd (−350 to +30 relative to the translation start site; Fig. 3). The 35 nM Kd measurement obtained was higher than that observed for norB or aniA, and no higher order shifted bands were observed, suggesting that there is only one NsrR binding site in the nsrR promoter. A 120 bp fragment of the norB upstream region in which the 29 bp norB NsrR operator was replaced with that from nsrR displayed a Kd comparable to that seen in the 380 bp fragment, suggesting that this low similarity operator was the functional regulatory site (data not shown). IscR was shown to bind one class of operator in its apo-form (Giel et al., 2006, Yeo et al., 2006). The nsrR upstream region could not be shifted by NsrR::FLAG C90A, where we have shown [2Fe-2S] to be absent (data not shown; Fig. 6). Together these results suggest that regulation of nsrR expression is more complex than previously thought and likely involves other regulators or mechanisms of regulation, and that the mechanism of nsrR regulation is different from that observed in iscR.
Figure 6. Role of conserved cysteine residues in NsrR function.
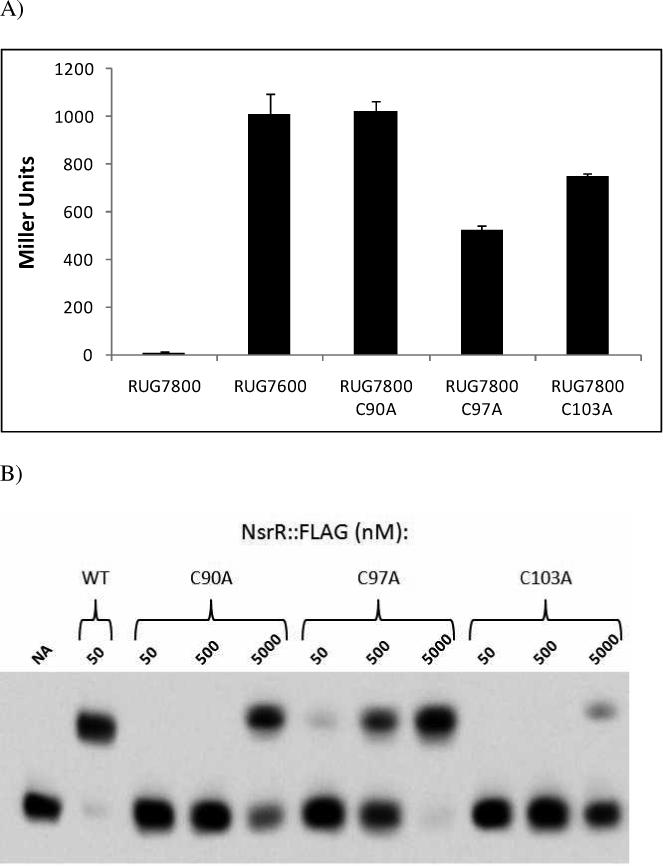

(A) Single C90A, C97A, and C103A substitutions were created in the appropriate codons of the nsrR::FLAG gene. These mutants were monitored for their ability to repress norB::lacZ aerobically in gonoccocal reporter fusion strains. Cells were grown aerobically and ß–galactosidase activity was measured and compared to parental nsrR::FLAG expressing strain (RUG7800) and ΔnsrR strain (RUG7600). These data are the mean of 6 determinations ± one SD. (B) Purified NsrR::FLAG containing C90A, C97A, or C103A substitutions were monitored for their ability to bind to a biotin end-labeled fragment of the norB promoter at 50, 500, and 5000 nM. (C) Mass spectrum of undigested NsrR::FLAG in linear positive ion mode. 1800 laser shots were summed to obtain this spectrum. The observed mass peak is equal to the predicted mass. (D) Purified NsrR::FLAG (wild type or C90A) was monitored for its ability to bind to a biotin end-labeled fragment of the norB promoter with and without the presence of long half life NO-donor DETA-NO. (NA) No addition.
The NsrR binding site of nsrR shows greater similarity in the downstream portion than in the upstream portion compared to the site in norB (Fig. 3B). This suggests that NsrR may recognize only a half-site in the nsrR non-coding region. To test whether NsrR can recognize a half site, we removed the last 14 bp of the 29 bp norB NsrR binding site by substitution with an arbitrary sequence, leaving only the first 15 bp from the original sequence (5’-TTTAACATTCATATT). NsrR::FLAG was unable to cause a significant shift of a labeled target fragment containing this site (Kd > 1 μM; Fig 3), suggesting that NsrR cannot bind to a half-site.
DNaseI footprinting of the norB and nsrR promoters
Rodionov et al. (2005) used computational analysis to predict that NsrR recognized a 19 bp sequence in neisserial genes, while we used genetic analysis that suggested that the NsrR binding site spanned a larger 29 bp sequence (Isabella et al., 2008). To resolve these differences, we performed DNase I footprinting on the norB and nsrR upstream regions, which contained the highest and lowest affinity NsrR binding sites respectively.
As expected, the footprint created by binding of NsrR::FLAG to the norB upstream region spanned the 29 bp inverted repeat sequence, protecting nucleotides −31 to −59 relative to the translation start site (Fig. 4A). These data definitively show that this inverted repeat sequence is the NsrR binding site. The footprint generated upon the interaction of NsrR::FLAG with the nsrR upstream region also protected the previously identified region, spanning nucleotides −42 to −68 relative to the translation start site (Fig. 4B). Though the low affinity binding site in the nsrR upstream region reveals only a half-site in comparison to the site in norB (Fig. 3B), NsrR still retains the capacity to bind DNA across an extended region.
Figure 4. DNaseI footprint analysis of the norB and nsrR upstream regions.

(A) A DNaseI footprint was generated from a radiolabeled fragment of the norB upstream region using increasing concentrations of NsrR::FLAG. Binding by NsrR::FLAG was shown to protect nucleotides −31 through −59 relative to the translational start site. In the sequence data, the large bracket above the sequence represent nucleotides that make up the predicted NsrR binding site (Isabella et al., 2008), and the small bracket makes up the consensus predicted by Rodionov et al. (2005). Underlined bases represent nucleotides protected from DNaseI digestion. (B) DNaseI footprint analysis of the nsrR upstream region using increasing concentrations of NsrR::FLAG. Protein binding was shown to protect nucleotides −42 through −68 relative to the translational start site. Brackets above the sequence show the predicted NsrR binding site. Underlined bases represent nucleotides protected from DNaseI digestion. (NA) No addition.
NsrR responds directly to Nitric Oxide
It has previously been shown that NsrR derepresses the genes in its regulon in response to NO in vivo (Rock et al., 2007, Isabella et al., 2008). To show that NO directly inhibits the ability of NsrR to bind to its operator in vitro, we performed an EMSA with increasing concentrations of the long half-life (∼20 h) NO donor DETA-NO. Increasing concentrations of DETA-NO in the binding reactions inhibited the shift of the biotin-labeled norB target DNA, showing that NsrR senses NO by a direct mechanism (Fig.5).
Figure 5. Nitric Oxide sensing by purified NsrR::FLAG using long half-life NO donor DETA/NO.
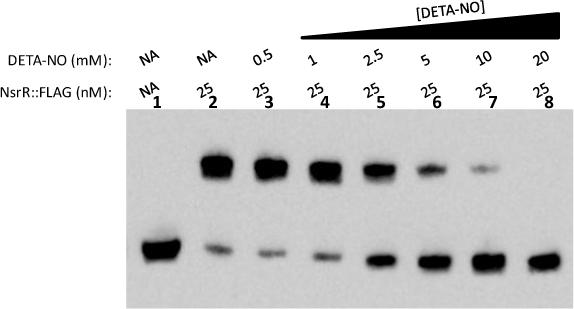
EMSA analysis was performed on a biotin end-labeled fragment of the norB promoter. Lane (1) contains DNA only. All other lanes contain 25 nM NsrR::FLAG with increasing concentrations of the long half-life (∼20 h) NO donor DETA/NO. (NA) No addition.
Role of conserved cysteine residues in NsrR function
The gonococcal NsrR protein contains three conserved cysteines that correspond to residues suggested to coordinate a [2Fe-2S] cluster in IscR (Yeo et al., 2006). To investigate the role of these cysteine residues in NsrR function, we constructed single C90A, C97A, and C103A mutations in the nsrR::FLAG gene in gonococcal strain RUG7800. These strains were monitored for their ability to repress norB::lacZ expression in the absence of NO. A C90A substitution resulted in a level of norB::lacZ expression comparable to that seen in a ΔnsrR mutant (RUG7600), illustrating the importance of this residue in NsrR function and indicative of a potential role in Fe-S coordination (Fig 6A). Expression of norB::lacZ in the C97A and C103A mutants were 52% and 74% of that observed in the ΔnsrR mutant respectively, showing that these residues are also important in the presumed coordination of Fe-S.
In order to further investigate the cysteine mutants of NsrR::FLAG, purified extracts of each modified protein were isolated and used in EMSA analysis in order to determine their capacity to bind to the norB upstream region. A 50 nM concentration of wild type NsrR::FLAG was enough to shift all DNA in the binding reaction (Fig. 6B). The cysteine mutants of NsrR::FLAG were not as effective as the wild type at shifting the norB fragment. The C97A mutant displayed very little binding at 50 nM and only shifted the DNA completely when the NsrR concentration was in the micromolar range. The C90A and C103A mutants only showed a measureable shift of the norB fragment at a concentration of 5 μM NsrR::FLAG, far in excess of what would be encountered in vivo.
To confirm that the loss of norB regulation and decreased binding affinity associated with the C90A mutation was concomitant with lack of [2Fe-2S] coordination, mass spectroscopy of undigested NsrR::FLAG C90A protein was performed in linear positive ion mode (Fig. 6C). Unlike the wild type protein, which displayed a 175.8 Dalton increase above the predicted mass attributable to the presence of [2Fe-2S] (Fig. 1C), the observed mass peak of NsrR::FLAG C90A matched the predicted mass of 17,006 Daltons, suggesting the absence of [2Fe-2S].
High NsrR::FLAG C90A concentrations were able to shift a biotinylated fragment of the norB promoter (Fig. 6B). However, unlike the wild type NsrR::FLAG protein, the mobility shift observed with NsrR::FLAG C90A was not inhibited by the addition of a NO donor (Fig. 6D). The NO insensitivity of NsrR::FLAG C90A shows that the ability of NsrR to sense and respond to NO is likely dependent upon the presence of [2Fe-2S].
DISCUSSION
In this study we have extensively characterized the interactions between the gonococcal NsrR protein and its operator sequence. We demonstrated that NsrR binding could be directly inhibited by the presence of nitric oxide. We also showed that DNA binding by NsrR in vivo is likely dependent on the coordination of a [2Fe-2S] cluster through the concerted action of three conserved cysteine residues. This work has furthered our understanding of the biochemistry of Rrf2 type proteins, a subfamily of winged helix regulators about which relatively little is known (Aravind et al., 2005). We will discuss the relationship between gonococcal NsrR and the related IscR protein in E. coli to illustrate the similarities and differences between these two Rrf2 repressors.
Iron-sulfur clusters are essential cofactors in many different types of proteins, and these clusters can be involved in any number of cellular processes, including electron transfer, nitrogen fixation, and gene regulation to name just a few (Johnson et al., 2005). In E. coli IscR is predicted to utilize three cysteine residues to coordinate a labile Fe-S cluster capable of sensing the Fe-S status of the cell by reversible binding of a [2Fe-2S] cluster (Giel et al., 2006, Yeo et al., 2006). IscR has also been demonstrated to regulate genes involved in biogenesis and repair of Fe-S clusters in times of oxidative stress (Giel et al., 2006, Yeo et al., 2006, Hyduke et al., 2007, Pullan et al., 2007). This observation comes as no surprise, as reactive oxygen species are known to damage or destroy Fe-S clusters, explaining the need for their high turnover rate under stressful conditions (Giel et al., 2006). There is a great deal of similarity in secondary structure between NsrR and IscR (Fig. S1), and there is likely some functional redundancy as well. There are various examples of crosstalk between the regulons of each regulator. NO is capable of causing damage to Fe-S clusters, and IscS, a cysteine desulfurase in the IscR regulon, has been demonstrated to play a role in the repair of Fe-S clusters damaged by nitrosative stress (Yang et al., 2002). IscR, like NsrR, is expected to sense NO by a direct mechanism, and microarray data shows that genes in the IscR regulon are indeed regulated in response to NO (Hyduke, et al., 2007, Pullan et al., 2007). Also, the NsrR protein of several organisms, including Neisseria spp. and E. coli, has been shown to regulate dnrN, a second protein demonstrated to repair the Fe-S clusters of proteins damaged by reactive oxygen or nitrogen (Justino et al., 2007, Heurlier et al., 2008, Isabella et al., 2008, Overton et al., 2008). Further evidence of crosstalk between the two regulons is observed in a gonococcal iscR mutant. In the absence of NO, expression of norB::lacZ was shown to be increased 8 fold in a gonococcal reporter strain harboring a ΔiscR mutation (Fig. S2). The cause of this increase is uncertain, though perturbation of the Fe-S biogenesis pathway by the ΔiscR mutation may inhibit proper loading of NsrR with Fe-S, which we have shown to be important for NsrR/operator interaction.
There are several differences between DNA binding by NsrR and IscR. IscR is capable of interacting with one class of operator when coordinating an Fe-S cluster and to a second class in its apo-form (Giel et al., 2006, Yeo et al., 2006). IscR was also shown to act as a direct activator of the suf operon in its apo-form (Yeo et al., 2006). Conversely, NsrR does not seem to be as broad acting as IscR. Microarray studies comparing nsrR+ and ΔnsrR strains in N. meningitidis have suggested that the Neisserial NsrR regulon is small (Heurlier et al., 2008). Furthermore, only one class of NsrR binding site in both Neisseria spp. and E. coli has been identified to date, and there is no evidence that NsrR is able to act as a direct activator of gene transcription at any promoter (Bodenmiller & Spiro, 2006, Heurlier et al., 2008, Isabella et al., 2008). DNaseI footprint analysis of the E. coli iscR promoter revealed two copies of a class I IscR binding site overlapping the −35 region (Giel et al., 2006), while footprinting of the gonococcal nsrR promoter revealed just a single low affinity NsrR binding site located downstream of the proposed −10 region. The footprint of iscR also revealed a second region of protection that was unique to this promoter, however no additional regions of protection were observed in the nsrR footprint (Giel et al., 2006). The NsrR footprints of norB and nsrR fail to show any bases that are hypersensitive to DNase cleavage, whereas in E. coli, the IscR footprints of several promoter regions contained bands that corresponded to hypersensitivity, suggesting that binding by IscR may have a larger effect on DNA structure and cause more DNA bending than NsrR (Giel et al., 2006).
The dissociation constants estimated from EMSA analysis using the norB, aniA, and nsrR upstream regions may not be the same as that observed in vivo, under differing ionic conditions and in the limiting environment of the cell, rather they should be taken as relative values for comparison between each other. The actual concentration of NsrR in vivo is currently under investigation, however, it is expected to be quite low as the gonococcal NsrR regulon is likely small. The use of a streptavidin purified biotinylated primer for generating a target DNA for EMSA analysis made it possible to perform this assay. Radiolabeling is not as efficient and cannot guarantee that all target DNA used in the binding reaction is labeled, whereas the biotin labeled primer ensures that all target DNA is labeled and can be easily quantitated. As expected from previous work (Isabella et al., 2008), the nsrR promoter displayed the least affinity for NsrR. This comes as no surprise, as basal levels of this protein should ensure that the binding sites of other NsrR-repressed genes are occupied before switching off its own expression. However, the sixty fold increase in expression of nsrR::lacZ seen in a gonococcal ΔnsrR mutant cannot be explained entirely as an NsrR-dependent effect localized at this weak binding site (Isabella et al., 2008). For this reason, it would be appealing to further elucidate the mechanism of nsrR regulation, as these studies may reveal novel, as yet undiscovered targets or mechanisms of NsrR regulation, whether they are direct or indirect.
During the course of this study we attempted to use mass spectroscopy to observe the nature of the interaction between NsrR and NO. Treatment of purified NsrR with NO resulted in conversion of the protein from a charge of +1 (Fig. 1C) to a charge of +3 (data not shown), and the resultant mass peak was a broad triplet of which an accurate mass could not be determined. It is not known if this charge increase is due to the intrinsic characteristics of a NO-modified NsrR or the addition of chemical adducts to NsrR that are hyper-reactive to matrix-assisted laser desorption/ionization. A possible cause of the charge increase is the addition of dinitrosyl adducts to each Fe atom in the [2Fe-2S] cluster, as is the mechanism of NO modification of SoxR, another [2Fe-2S] cluster containing transcription regulator (Ding & Demple, 2000). Dinitrosyl addition to Fe in analogs of the iron sulfur complex cause a +1 increase in charge (Harrop et al., 2007), and dinitrosyl iron is a NO-derived intermediate with the redox equivalence of NO+ (van der Vliet et al., 1999). However, D'Autréaux et al. (2004) have shown that the presence of dinitrosyl iron in purified Fur did not cause a charge increase in the protein, though the active site Fe in Fur is not part of an Fe-S cluster. In any case, we are currently exploring several alternative routes to determine the mechanism by which NO inactivates NsrR.
In conclusion, this study extends recent work dealing with the NO-sensitive repressor protein NsrR. The response to NO sensing in some organisms is beginning to emerge as a instrument of virulence, and it will be important to be aware of exactly what changes occur when a pathogen encounters NO (i.e. as part of the human immune response). Future work will attempt to elucidate the overall response of N. gonorrhoeae upon encountering NO, and to determine what that response means for the host.
EXPERIMENTAL PROCEDURES
Construction of an nsrR:: FLAG fusion gene
The bases encoding a FLAG epitope tag (Sigma) were linked in-frame to the 5'-end of primer that annealed to the 3’ end of the gonococcal nsrR gene and the wild type stop codon was removed (Fig. 8). The complementary FLAG bases and a stop codon, TAA, were added to the 5’ end of a primer that annealed to the 3’ end of a kanamycin resistance cassette (aph). An nsrR fragment (beginning at −342 nucleotides relative to the nsrR start codon) and an aph cassette were separately amplified using these primers with their suitable pairs. These fragments were then spliced to make a single nsrR::FLAG-aph fragment using SOE PCR (Ho et al., 1989). This fragment was cloned into the SstI and HindIII restriction sites in the E. coli expression vector pEXT20 to make pVI28 (Dykxhoorn et al., 1996). Cysteine mutations in the nsrR::FLAG gene were also constructed by SOE PCR using suitable primers and pVI28 as a template.
Construction of gonococcal strains expressing nsrR::FLAG
All gonococcal strains were derived from strain F62 and were grown as previously described (Isabella et al., 2008). An nsrR::FLAG-aph fusion gene was obtained by digestion of pVI28 with SstI and HindIII. PCR was used to amplify another fragment that contained the 915 bp chromosomal region immediately downstream of the nsrR stop codon, and was joined to the free end of the kanamycin resistance cassette by restriction with HindIII and subsequent ligation. This fragment was used to transform the gonococcal strain RUG7500 by methods described previously (Isabella et al., 2008). Cysteine mutants of the nsrR::FLAG gene were used to transform RUG7500 in a similar manner. Presence of fusion gene, as well as insertional mutagenesis of the wild-type nsrR gene, was confirmed by PCR. Gene reporter activity was determined by β-galactosidase assays from cultures grown aerobically and anaerobically with nitrite (Miller, 1972; Isabella et al., 2008).
Isolation and purification of NsrR::FLAG
E. coli strain DH10B was transformed with pVI28, the expression vector containing the nsrR::FLAG fusion gene under control of the IPTG-inducible Ptac promoter (Dykxhoorn et al., 1996). A 20 mL volume of an overnight culture of DH10B harboring pVI28 was used to inoculate 500 mL of LB containing kanamycin at 50 μg ml−1 and cells were grown shaking at 37 °C. When the culture reached an OD600 of 0.5, 1 mM IPTG was added and cells were grown out for 4 hours, at which point cells were harvested and the pellet was frozen overnight at −20 °C. The cell pellet was resuspended in 10 mL of TBS (10 mM Tris, pH 8, 150 mM NaCl), 0.4 mM PEFA-BLOC (Roche), 1 mM DTT, and DNaseI at 200 μg/mL (Sigma, D-5025). Cells were lysed in a French pressure cell press (SLM Instruments), and the resulting lysate was spun at 20,000 × g twice to remove the insoluble fraction. A 90 μL volume of M2 affinity resin (Sigma) was added to the cleared lysate, followed by rocking incubation for 2 h at 4 °C. M2 affinity resin was collected by centrifugation at 500 × g and subsequently washed four times with 10 mL of TBS. Bound resin was added to Handee Spin-columns (Thermo) and incubated for 15 minutes with 400 μL 3X FLAG peptide (Sigma) in TBS at 1 mg/mL before centrifugation to collect released protein. Recovered FLAG fusion protein was stored in TBS, 1 mM DTT, and 50% glycerol at −20 °C.
Electrophoretic Mobility Shift Assays (EMSA)
EMSAs were performed with a LightShift Chemiluminescent EMSA kit (Thermo). Briefly, PCR was used in conjunction with a biotin end-labeled primer (Invitrogen) and unlabeled primer pair to generate biotin end-labeled targets to measure protein binding. Binding reactions contained 3.5 fmol biotin labeled DNA, 2 μl 10× binding buffer (100 mM Tris, 500 mM KCl, 10 mM DTT, pH 7.5), 1 μl 50% glycerol, 1 μl 1% NP-40, 1 μl MgCl2 (100 mM), 0.5 μg poly (dI•dC), varied NsrR::FLAG concentrations, and dH2O up to a total volume of 20 μl. Binding reactions were incubated at room temperature for 15 min and run on 6% 0.5× TBE gels. Gels were transferred to nylon membrane, crosslinked in a UV crosslinker (Stratagene), and biotin was detected following manufacturer's protocol (Thermo). For EMSAs performed with the long half-life NO donor diethylenetriamine/NO adduct (DETA-NO; Sigma), binding reactions were increased to 1 hour. For supershift reaction, 1 μl of M2 antibody (Sigma, F1804) was added to the binding reaction. Quantitation of shifted DNA was calculated by spot densitometry using a FlourChem IS-5500 imager (Alpha Innotech).
DNaseI footprinting
NsrR::FLAG extract was used to generate DNaseI footprints from the upstream regions of norB (nucleotides −92 to +13 relative to the translation start site) and nsrR (−119 to +22 nucleotides relative to the translation start site). Briefly, plasmids containing the relevant upstream regions were cut singly with one restriction enzyme, treated with cow intestine alkaline phosphatase (CIP, New England Biolabs), and radiolabeled on the non-coding strand with [γ-32P]-ATP using T4 polynucleotide kinase (Fermentas). After phenol/chloroform extraction, single end-labeled fragment of these upstream regions were released by digestion at a second restriction site, and fragments were gel purified (Qiagen). Binding reactions were performed as described for EMSA except that 20,000 cpm of radiolabeled DNA was used rather than biotin labeled DNA. After 15 minutes of preincubation, 1 μl of DNaseI at 40 μg/mL (Sigma; D5025), suspended in TBS plus 50 mM CaCl2, was added to each reaction. After 90 sec incubation, reactions were stopped by the addition of 20 μl of 2X stop loading buffer (80% dimethyformamide, 1 mg/mL bromophenol blue, 1 mg/mL xylene cyanol FF, 2% SDS, and 40 mM EDTA, pH 8.0 in 0.5X TBE) and immediately incubated at 95 °C for 20 minutes. A+G sequencing ladders were generated as previously described (Maxam & Gilbert, 1980). A volume of 5 μl of each reaction was loaded onto 8% TBE-Urea sequencing gels. Reaction products were visualized by phosphorimaging (Bio-Rad).
Mass spectroscopy
Undigested samples were analyzed on an AutoflexIII TOF/TOF MALDI mass spectrometer (Bruker Daltonics, Billerica, MA, USA). Spectra were collected in linear positive ion mode (1800 shots accumulated in m/z range 3000−20000). An external calibrant, Protein 1 CalibStandard (Bruker Daltonics, Billerica, MA, USA), was used to ensure mass accuracy. This calibrant ranged from m/z 5734−16952. Spectra were processed by baseline subtraction and analyzed with flexAnalysis (Bruker Daltonics, Billerica, MA, USA). SequenceEditor (Bruker Daltonics, Billerica, MA, USA) was used to predict the m/z of the proteins used in this experiment.
Oligonucleotide and DNA sequencing
All synthesized oligonucleotides were obtained from Invitrogen, and confirmatory DNA sequencing was performed at ACGT Inc. (Wheeling, IL). Primer sequences are available upon request.
Molecular biology techniques
Cloning and PCR techniques were performed in accordance to standard protocols (Ausubel et al., 1987; Ausubel et al., 1992; Sambrook et al., 1989). Plasmid preparations were obtained with Wizard Plus SV Minipreps kits (Promega Corp., Madison, Wis.). DNA fragments were purified with QIAquick PCR Purification or QIAquick Gel Extraction kits (QIAGEN, Valencia, CA).
Figure 7. Construction of an nsrR::FLAG fusion gene.

The 5’ end of primer P2 contains nucleotides coding for a FLAG epitope tag. The 3’end of P2 anneals to the 3’ end of the nsrR coding region and excludes the wild type stop codon. The nsrR gene and upstream region were amplified using primers P1 and P2. The 5’ end of primer P3 contains the codons of a FLAG tag that are complementary to those in P2 followed by the addition of stop codon TAA. The 3’ end of P3 anneals to the 5’ end of aph (Kanr cassette). The aph gene was amplified using primers P3 and P4. After amplification, these fragments were spliced together in a SOE PCR reaction to make nsrR::FLAG-aph, which was subsequently cloned into an E. coli expression vector to make pVI28. Alternatively, the region downstream of nsrR was amplified and ligated downstream of aph for making a fragment for transformation of a gonococcal strain in order to replace wild type nsrR with nsrR::FLAG. Small bent arrows indicate direction of transcription. Fragments are not drawn to scale.
Table 1.
Plasmids and Bacterial strains used in this study
| Constructs: | Relevant genotype or properties | Source or Reference |
|---|---|---|
| Plasmids: | ||
| pEXT20 | E.coli expression vector, Apr | Dykxhoorn, et al. (1996) |
| pVI28 | nsrR::FLAG-aph fragment, pEXT20 backbone | This study |
| pLES94 | promoterless lacZ vector, Apr, Cmr | Silver et al. (1995) |
| pEF1 | pLES94/full norB upstream from −150 to +9 | Householder et al. (1999) |
| pVI51 | nsrR::FLAG-aphfragment, C97A, pEXT20 backbone | This study |
| pVI52 | nsrR::FLAG-aphfragment, C100A, pEXT20 backbone | This study |
| pVI53 | nsrR::FLAG-aphfragment, C103A, pEXT20 backbone | This study |
| Strains: | ||
| F62 | pro− | Laboratory collection |
| RUG7500 | F62, transformed with pEF1 | Householder et al. (1999) |
| RUG7600 | F62, ΔnsrR, transformed with pEF1 | Isabella et al. (2008) |
| RUG7800 | F62, transformed with nsrR::FLAG-aph gene and pEF1 | This Study |
| RUG7801 | RUG7800, C97A, transformed with pEF1 | This Study |
| RUG7802 | RUG7800, C100A, transformed with pEF1 | This Study |
| RUG7803 | RUG7800, C103A, transformed with pEF1 | This Study |
Acknowledgements
This study was supported by Public Health Service grant R01 AI 11709 from the National Institutes of Health. In addition, V.M.I and E.M.K are supported by NIH grant T32 AI07362. Mass spectrometry and J. D. L. were supported by the University of Rochester Clinical Translational Sciences grant UL1 RR024160-1.
REFERENCES
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. John Wiley & Sons; New York, NY: 1987. [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short Protocols in molecular biology. 2nd edn. John Wiley & Sons; New York, NY: 1992. [Google Scholar]
- Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Bodenmiller DM, Spiro S. The yjeB (nsrR) gene of Escherichia coli encodes a nitric oxide-sensitive transcriptional regulator. J Bacteriol. 2006;188:874–881. doi: 10.1128/JB.188.3.874-881.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response-review. Nat Immun. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale JA, Clark VL. Determinants of nitric oxide steady-state levels during anaerobic respiration by Neisseria gonorrhoeae. Mol Microbiol. 2005;58:177–188. doi: 10.1111/j.1365-2958.2005.04807.x. [DOI] [PubMed] [Google Scholar]
- D'Autréaux B, Horner O, Oddou JL, Jeandey C, Gambarelli S, Berthomieu C, Latour JM, Michaud-Soret I. Spectroscopic detection of the two nitrosyl-tron complexes responsible for Fur inhibition by nitric oxide. J Am Chem Soc. 2004;126:6005–6016. doi: 10.1021/ja031671a. [DOI] [PubMed] [Google Scholar]
- Ding H, Demple B. Direct nitric oxide signal transduction via nitrosylation of iron-sulfur centers in the SoxR transcription activator. Proc Natl Acad Sci USA. 2000;97:5146–5150. doi: 10.1073/pnas.97.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykxhoorn DM, St Pierre R, Linn T. A set of compatible tac promoter expression vectors. Gene. 1996;177:133–136. doi: 10.1016/0378-1119(96)00289-2. [DOI] [PubMed] [Google Scholar]
- Edwards JL, Apicella MA. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin Microbiol Rev. 2004;17:965–981. doi: 10.1128/CMR.17.4.965-981.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filenko N, Spiro S, Browning DF, Squire D, Overton TW, Cole J, Constantinidou C. The NsrR regulon of Escherichia coli K-12 includes genes encoding the hybrid cluster protein and the periplasmic, respiratory nitrite reductase. J Bacteriol. 2007;189:4410–4417. doi: 10.1128/JB.00080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett GP, Mertz KJ, Finelli L, Levine WC, St Louis ME. The transmission dynamics of gonorrhoea: modelling the reported behaviour of infected patients from Newark, New Jersey. Philos Trans R Soc Lond B Biol Sci. 1999;354:787–797. doi: 10.1098/rstb.1999.0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosn SH, Kibbi AG. Cutaneous gonococcal infections. Clin Dermatol. 2004;22:476–480. doi: 10.1016/j.clindermatol.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol. 2006;60:1058–1075. doi: 10.1111/j.1365-2958.2006.05160.x. [DOI] [PubMed] [Google Scholar]
- Gilberthorpe NJ, Lee ME, Stevanin TM, Read RC, Poole RK. NsrR: a key regulator circumventing Salmonella enterica serovar Typhimurium oxidative and nitrosative stress in vitro and in IFN-gamma-stimulated J774.2 macrophages. Microbiology. 2007;153:1756–1771. doi: 10.1099/mic.0.2006/003731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54:469–487. [PubMed] [Google Scholar]
- Harrop TC, Song D, Lippard SJ. Reactivity pathways for nitric oxide and nitrosonium with iron complexes in biologically relevant sulfur coordination spheres. J Inorg Biochem. 2007;101:1730–1738. doi: 10.1016/j.jinorgbio.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Heurlier K, Thomson MJ, Aziz N, Moir JW. The nitric oxide (NO)-sensing repressor NsrR of Neisseria meningitidis has a compact regulon of genes involved in NO synthesis and detoxification. J Bacteriol. 2008;190:2488–2495. doi: 10.1128/JB.01869-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Householder TC, Belli WA, Lissenden S, Cole JA, Clark VL. cis- and trans-acting elements involved in regulation of aniA, the gene encoding the major anaerobically induced outer membrane protein in Neisseria gonorrhoeae. J Bacteriol. 1999;181:541–551. doi: 10.1128/jb.181.2.541-551.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Householder TC, Fozo EM, Cardinale JA, Clark VL. Gonococcal nitric oxide reductase is encoded by a single gene, norB, which is required for anaerobic growth and is induced by nitric oxide. Infect Immun. 2000;68:5241–5246. doi: 10.1128/iai.68.9.5241-5246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyduke DR, Jarboe LR, Tran LM, Chou KJY, Liao JC. Integrated network analysis identifies nitric oxide response networks and dihydroxyacid dehydratase as a crucial target in Escherichia coli. Proc Natl Acad Sci U S A. 2007;104:8484–8489. doi: 10.1073/pnas.0610888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabella V, Wright LF, Barth K, Spence JM, Grogan S, Genco CA, Clark VL. cis- and trans-acting elements involved in regulation of norB (norZ), the gene encoding nitric oxide reductase in Neisseria gonorrhoeae. Microbiology. 2008;154:226–239. doi: 10.1099/mic.0.2007/010470-0. [DOI] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Justino MC, Almeida CC, Teixeira M, Saraiva LM. Escherichia coli di-iron YtfE protein is necessary for the repair of stress-damaged iron-sulfur clusters. J Biol Chem. 2007;282:10352–10359. doi: 10.1074/jbc.M610656200. [DOI] [PubMed] [Google Scholar]
- Knapp JS, Clark VL. Anaerobic growth of Neisseria gonorrhoeae coupled to nitrite reduction. Infect Immun. 1984;46:176–181. doi: 10.1128/iai.46.1.176-181.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- Nakano MM, Geng H, Nakano S, Kobyashi K. The nitric oxide-responsive regulator NsrR controls ResDE-dependent gene expression. J Bacteriol. 2006;188:5878–5887. doi: 10.1128/JB.00486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton TW, Justino MC, Li Y, Baptista JM, Melo AM, Cole JA, Saraiva LM. Widespread distribution in pathogenic bacteria of di-iron proteins that repair oxidative and nitrosative damage to iron-sulfur centers. J Bacteriol. 2008;190:2004–2013. doi: 10.1128/JB.01733-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton TW, Whitehead R, Li Y, Snyder LA, Saunders NJ, Smith H, Cole JA. Coordinated regulation of the Neisseria gonorrhoeae-truncated denitrification pathway by the nitric oxide-sensitive repressor, NsrR, and nitrite-insensitive NarQ-NarP. J Biol Chem. 2006;281:33115–33126. doi: 10.1074/jbc.M607056200. [DOI] [PubMed] [Google Scholar]
- Pullan ST, Gidley MD, Jones RA, Barrett J, Stevanin TM, Read RC, Green J, Poole RK. Nitric oxide in chemostat-cultured Escherichia coli is sensed by Fnr and other global regulators: unaltered methionine biosynthesis indicates lack of S nitrosation. J Bacteriol. 2007;189:1845–1855. doi: 10.1128/JB.01354-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JD, Thomson MJ, Read RC, Moir JW. Regulation of denitrification genes in Neisseria meningitidis by nitric oxide and the repressor NsrR. J Bacteriol. 2007;189:1138–1144. doi: 10.1128/JB.01368-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Dubchak IL, Arkin AP, Alm EJ, Gelfand MS. Dissimilatory metabolism of nitrogen oxides in bacteria: comparative reconstruction of transcriptional networks. PLoS Comput Biol. 2005;1:e55. doi: 10.1371/journal.pcbi.0010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogstam A, Larsson JT, Kjelgaard P, von Wachenfeldt C. Mechanisms of adaptation to nitrosative stress in Bacillus subtilis. J Bacteriol. 2007;189:3063–3071. doi: 10.1128/JB.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz CJ, Giel JL, Patschkowski T, Luther C, Ruzicka FJ, Beinert H, Kiley PJ. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc Natl Acad Sci U S A. 2001;98:14895–14900. doi: 10.1073/pnas.251550898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, Wu HJ, Kidd SP, Apicella MA, Jennings MP, McEwan AG. Defenses against oxidative stress in Neisseria gonorrhoeae: a system tailored for a challenging environment. Microbiol Mol Biol Rev. 2006;70:344–361. doi: 10.1128/MMBR.00044-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LE, Clark VL. Construction of a translational lacZ fusion system to study gene regulation in Neisseria gonorrhoeae. Gene. 1995;166:101–104. doi: 10.1016/0378-1119(95)00605-6. [DOI] [PubMed] [Google Scholar]
- Stefano GB, Gouman Y, Bilfinger TV, Welters ID, Cadet P. Basal nitric oxide limits immune, nervous, and cardiovascular excitation: human endothelia express a mu opiate receptor. Prog Neurobiol. 2000;60:513–530. doi: 10.1016/s0301-0082(99)00038-6. [DOI] [PubMed] [Google Scholar]
- Stevanin TM, Laver JR, Poole RK, Moir JW, Read RC. Metabolism of nitric oxide by Neisseria meningitidis modifies release of NO-regulated cytokines and chemokines by human macrophages. Microbes Infect. 2007;9:981–987. doi: 10.1016/j.micinf.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Stevanin TM, Moir JW, Read RC. Nitric oxide detoxification systems enhance survival of Neisseria meningitidis in human macrophages and in nasopharyngeal mucosa. Infect Immun. 2005;73:3322–3329. doi: 10.1128/IAI.73.6.3322-3329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunbridge AJ, Stevanin TM, Lee M, Marriott HM, Moir JW, Read RC, Dockrell DH. Inhibition of macrophage apoptosis by Neisseria meningitidis requires nitric oxide detoxification mechanisms. Infect Immun. 2006;74:729–733. doi: 10.1128/IAI.74.1.729-733.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vliet A, Eiserich JP, Shigenaga MK, Cross CE. Reactive nitrogen species and tyrosine nitration in the respiratory tract: epiphenomena or a pathobiologic mechanism of disease? Am J Respir Crit Care Med. 1999;160:1–9. doi: 10.1164/ajrccm.160.1.9807044. [DOI] [PubMed] [Google Scholar]
- Yang W, Rogers PA, Ding H. Repair of nitric oxide-modified ferredoxin [2Fe-2S] cluster by cysteine desulfurase (IscS). J Biol Chem. 2002;277:12868–12873. doi: 10.1074/jbc.M109485200. [DOI] [PubMed] [Google Scholar]
- Yeo WS, Lee JH, Lee KC, Roe JH. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol Microbiol. 2006;61:206–218. doi: 10.1111/j.1365-2958.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]