

Abstract
The influence of a hydrogen bond to the coordinated imidazole on the geometric and electronic structure of iron has been further studied in new complexes of five-coordinate high-spin imidazole-ligated iron(II) porphyrinates. With 1,10-phenanthroline as the hydrogen-bond acceptor, several new species of OEP and TPP derivatives have been synthesized and characterized by X-ray crystallography and Möossbauer spectroscopy. In all three new structures, the porphyrin molecules and 1,10-phenanthroline molecules have been found with a ratio of 1:1. All the porphyrin derivatives are five-coordinate 2-methylimidazole-ligated iron(II) species. 1,10-phenanthroline is hydrogen bonded to the coordinated imidazole to form two unequal hydrogen bonds. The Fe–Np and Fe–NIm bond lengths, and displacement of the iron atom out of the porphyrin plane are similar to those in imidazole-ligated species. Möossbauer measurements showed remarkable temperature dependence; the analysis of the data obtained in applied magnetic field for [Fe(OEP)(2-MeHIm)]·(1,10-phen) gave a negative quadrupole splitting value and large asymmetry parameters. All the structural and Mössbauer properties suggest that these new hydrogen-bonded species have the same electronic configuration as imidazole-ligated species.
Introduction
Many biologically important proteins have heme (iron porphyrin) as the active site. These heme proteins have a variety of functions, including oxygen carriers in mammals, electron carriers in photosynthesis and respiration, and catalysts for a variety of biochemical reactions involving O2, H2O2 etc. Their functionalities have been influenced by many factors. Hydrogen bonding is one such factor believed to play an important role. One important hydrogen bond in heme proteins is between the coordinated imidazole of a histidine residue and an adjacent amino acid residue. Crystal structures of heme proteins show that such hydrogen bonds are very widespread.1-3 The strength of these hydrogen bonds is thought to vary from very weak proton donation to complete proton donation to form the imidazolate ligand.4-8 There has been substantial speculation that hydrogen bonding in heme proteins could control the relative stability of the oxidation state of iron and thus control reactivity of the heme protein.9
For example, a particularly striking pair of heme protein families are involved in such hydrogen bonds, the globins and the peroxidases. The heme in globins is the same b-type heme in the peroxidase heme, and they both have a coordinated imidazole from a histidyl residue of the protein chain as the proximal ligand. The proximal imidazole in the globins forms a hydrogen bond to a backbone carbonyl oxygen atom. In peroxidases, such as horseradish peroxidase,10,11 the proximal histidine (His170) is ligated to the heme center and also forms a hydrogen bond with a highly conserved aspartate group Asp247, a stronger hydrogen bond acceptor than that of backbone carbonyl oxygen. But they have much different chemistry. The biological function of globins is the reversible transport and storage of dioxygen, whereas peroxidases can catalyze the conversion of hydrogen peroxide to water and/or the oxidation of substrates. It has been postulated that the hydrogen bond stabilizes higher oxidation states of iron and distinctly alters the chemical behavior of the peroxidases relative to the globins. The importance of such hydrogen bond has been commented on and calculated on, but to our knowledge, has not been experimentally investigated in a systematic manner.
We have been studying the nature of various five-coordinate iron(II) porphyrinates.12-17 Much of this work has focused on the nature of high-spin imidazole-ligated species.13, 14, 16 As part of that work, we reported15 the effects of deprotonating the imidazole ligand in five-coordinate iron(II) porphyrinate derivatives of the type [Fe(Por)(2-MeHIm)].18 Both imidazole-and imidazolate-ligated iron(II) porphyrinates exhibit an S = 2 (quintet) state, but the structural parameters of the coordination groups are distinct with both axial and equatorial bond distances differences and large differences in the displacement of iron from the porphyrin plane. Distinctive features in the Mössbauer spectra obtained in applied magnetic fields show that the doubly occupied d orbital is different in imidazole- vs. imidazolate-ligated iron(II) porphyrinates. The positive sign of the quadrupole splitting in the imidazolate derivative shows that the doubly occupied orbital must be the dxy orbital whereas the negative sign in the imidazole derivative is consistent only with a low-symmetry orbital comprised of a hybrid of dxz, dyz, and dxy. This change in the d-electron configuration is clearly consonant with all observed differing features of the two classes.
These differences are similar to those of the globins and the peroxidases. Reduced HRP has been studied by Mössbauer spectroscopy and compared with deoxymyoglobin.19,20 These Mössbauer studies, in a strong magnetic field, showed remarkable differences between reduced HRP and deoxymyoglobin even though both are five-coordinate hemes with histidine as the axial ligand. Reduced HRP has a positive quadrupole splitting constant (Vzz > 0) and a rather small asymmetry constant,20 whereas deoxyMb has a large asymmetry constant and a negative value of the quadrupole splitting constant (Vzz < 0). This strongly argues for different properties of the axial ligand in the five-coordinate iron(II) states of reduced HRP and deoxyMb.
Based on these studies, we have begun further investigations with iron(II) porphyrinates of hydrogen-bonded species to fill the gap between imidazole- and imidazolate-ligated porphyrinates. They will also help us understand how hydrogen bonds influence the molecular and electronic structure of the high-spin species. Recently, we presented the first of our investigations of hydrogen bond formation with a coordinated imidazole in a high-spin iron(II) porphyrinate system, [Fe(TPP)(2-MeHIm)]2·2-MeHIm.21 This species has two independent five-coordinate, high-spin iron(II) porphyrinate sites, one with a strong hydrogen bond to the imidazole and the second with a “neutral” imidazole ligand. This complex has been studied by X-ray and neutron diffraction studies as well as Mössbauer spectroscopy to assess the effects of hydrogen-bonded imidazole as a ligand. These studies show that there is a clear difference in the iron(II) electronic structure between the two iron site, distinctions that can be attributed to the presence of a hydrogen bond to the coordinated imidazole in the one site. But in this case, the two different porphyrin molecules in the asymmetric unit make it difficult to unambiguously assign their electronic configuration. We have attempted to obtain additional hydrogen-bonded imidazole-ligated iron(II) porphyrinates for characterization. Success requires finding suitable hydrogen-bond acceptors that are either weak ligands or nonligands to avoid coordination competition with the imidazole (2-methylimidazole).
Balch and co-workers22,23 have reported that 1,10-phenanthroline forms hydrogen bonds with coordinated imidazole in six-coordinate iron(III) porphyrinates.
They showed that the equilibrium in the reaction above was significantly shifted to the right by the addition of 1,10-phenanthroline. Balch et al. investigated this system by visible spectroscopy and NMR.22 In the NMR experiments, diamagnetic cobalt(III) derivatives were substituted for the paramagnetic iron(III) center. Observed proton shifts are consistent with the formation of a hydrogen bond between imidazole and 1,10-phenanthroline. No complexes were isolated.
In this paper, we report new, isolated, imidazole-ligated, hydrogen-bonded iron(II) species with 1,10-phenanthroline as the hydrogen bond acceptor. The species are [Fe(TPP)(2-MeHIm)]·-(1,10-phen) (two crystalline forms) and [Fe(OEP)(2-MeHIm)]·(1,10-phen). The effects of the hydrogen bond have been investigated by crystallographic characterization and Mössbauer studies. The consequences of the relatively weak hydrogen bonds formed between coordinated imidazole and 1,10-phenanthroline are found to have a relatively small or no effect on the electronic structure of the high-spin iron(II) species.
Experimental Section
General Information
All reactions and manipulations for the preparation of the iron(II) porphyrin derivatives (see below), were carried out under argon using a double-manifold vacuum line, Schlenkware and cannula techniques. Chlorobenzene was washed with concentrated sulfuric acid, then with water until the aqueous layer was neutral, dried with MgSO4, and distilled twice over P2O5 under argon. Hexanes were distilled over sodium benzophenone. 2-Methylimidazole (Aldrich) was recrystallized from toluene/methanol and dried under vacuum. All other chemicals were used as received from Aldrich or Fisher. The free-base porphyrin meso-tetraphenylporphyrin (H2TPP) was prepared according to Adler et al.24 Octaethylporphyrin (H2OEP) was prepared from formaldehyde and 3,4-diethylpyrrole.25 The metalation of the free-base porphyrin to give [Fe(Por)Cl] (Por = TPP, OEP) was done as previously described.26 [Fe(Por)]2O was prepared according to a modified Fleischer preparation.27
Mössbauer measurements were performed on a constant acceleration spectrometer from 4.2 K to 300 K with optional small field and in a 9 T superconducting magnet system (Knox College). Samples for Mössbauer spectroscopy were prepared by immobilization of the crystalline material in Apiezon M grease.
IR spectra were recorded on a Nicolet Nexus 670 FT-IR spectrometer as KBr pellets. Both [Fe(Por)(2-MeHIm)] and [Fe(Por)(2-MeHIm)]·(1,10-phen) crystalline samples were measured.
Synthesis of [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form A)
[Fe(TPP)]2O (32 mg, 0.024 mmol) was mixed with ethanethiol (1 mL) in chlorobenzene (9 mL), and stirred for three days at room temperature. The resulting solution of the four-coordinate [Fe(II)(TPP)] was transferred into a Schlenk flask containing 2-methylimidazole (17 mg, 0.21 mmol) and 1,10-phenanthroline (50 mg, 0.28 mmol). The mixture was stirred for 15 min. X-ray quality crystals were obtained in 8 mm × 250 mm sealed glass tubes by liquid diffusion using hexanes as nonsolvent.
Synthesis of [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form B)
A similar procedure as the above was used. [Fe(TPP)]2O (105 mg, 0.078 mmol), ethanethiol (2.5 mL), chlorobenzene (25 mL), 2-methylimidazole (44 mg, 0.54 mmol) and 1,10-phenanthroline (154 mg, 0.86 mmol) were used.
Synthesis of [Fe(OEP)(2-MeHIm)]·(1,10-phen)
A similar procedure as the above was used. [Fe(OEP)]2O (28 mg, 0.023 mmol), ethanethiol (1 mL), chlorobenzene (7 mL), 2-methylimidazole (18 mg, 0.22 mmol) and 1,10-phenanthroline (46 mg, 0.26 mmol) were used.
UV-vis Spectroscopy
UV-vis spectra were recorded on a Perkin-Elmer Lambda 19 UV/vis/near-IR spectrometer, and were obtained in a specially designed combined 1- and 10-mm inert atmosphere cell. A solution of four-coordinate [Fe(II)(TPP)] (0.5 × 10−3 mol/L) was prepared as the above. UV-vis spectra of iron porphyrinates at different concentration of 2-methylimidazole and 1,10-phenanthroline were measured. The concentrations of 2-methylimidazole for UV-vis measurements range from 0.11 to 1.07 mol/L, the concentrations of 1,10-phenanthroline range from 4.8 × 10−3 to 8.6 × 10−1 mol/L.
X-ray Structure Determinations
Single-crystal experiments were carried out on a Bruker Apex system with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined against F2 using SHELXTL;28,29 subsequent difference Fourier syntheses led to the location of most of the remaining non-hydrogen atoms. All nonhydrogen atoms were refined anisotropically if not remarked otherwise below. Hydrogen atoms were added with the standard SHELXL-97 idealization methods. The program SADABS 30 was applied for the absorption correction. Brief crystal data and intensity collection parameters for the crystalline complexes are shown in Table 1. Complete crystallographic details, atomic coordinates, anisotropic thermal parameters, and fixed hydrogen atom coordinates are given in the Supporting Information.
Table 1.
Brief Crystallographic Data and Data Collection Parameters.
| [Fe(TPP)(2-MeHIm)]·(1,10-phen) (Form A) | [Fe(TPP)(2-MeHIm)]·(1,10-phen) (Form B) | [Fe(OEP)(2-MeHIm)]·-(1,10-phen) | |
|---|---|---|---|
| formula | C48H34FeN6·C12H8N2·C6H5Cl | 2C48H34FeN6·2C12H8N2·2C6H5Cl | 2C40H50FeN6·2C12H8N2 |
| FW | 1043.42 | 2086.83 | 1701.83 |
| a, Å | 11.8059(10) | 24.6264(9) | 25.319(2) |
| b, Å | 21.8824(16) | 18.7689(7) | 19.1907(14) |
| c, Å | 21.3200(17) | 23.2011(8) | 18.3944(13) |
| β, deg | 100.143(4) | 106.726(2) | |
| V, Å3 | 5421.8(7) | 10270.1(6) | 8937.6(12) |
| Z | 4 | 4 | 4 |
| space group | P21/c | P21/c | Pca21 |
| Dc, g/cm3 | 1.278 | 1.350 | 1.265 |
| F(000) | 2168 | 4336 | 3616 |
| μ, mm−1 | 0.377 | 0.398 | 0.383 |
| crystal dimens, mm | 0.49 × 0.43 × 0.14 | 0.34 × 0.21 × 0.19 SADABS 0.71073 Å | 0.44 × 0.28 × 0.20 |
| absorption correction | SADABS | ||
| radiation, MoKα, λ̄ | 0.71073 Å | ||
| T, K | 293(2) | 100(2) | 100(2) |
| total data collected | 59973 | 130581 | 181135 |
| unique data | 14107 (Rint = 0.030) | 23997 (Rint = 0.032) | 24415 (Rint = 0.046) |
| unique obsd data [I > 2 σ(I)] | 8398 | 20261 | 21117 |
| refinement method | on F2 (SHELXL) | ||
| final R indices [I > 2σ(I)] | R1 = 0.0428, wR2 = 0.1101 | R1 = 0.0678, wR2 = 0.1521 | R1 = 0.0499, wR2 = 0.1228 |
| final R indices [for all data] | R1 = 0.0765, wR2 = 0.1195 | R1 = 0.0806, wR2 = 0.1587 | R1 = 0.0605, wR2 = 0.1292 |
[Fe(TPP)(2-MeHIm)]·(1,10-phen) (form A)
A red crystal with the dimensions 0.49 × 0.43 × 0.14 mm3 was used for the structure determination. Crystal data were collected at room temperature as the crystals cracked on cooling. The structure was refined in space group P21/c. The asymmetric unit contains one porphyrinate molecule, one 1,10-phenanthroline, and one chlorobenzene solvate. The C6H5Cl was badly disordered, showing rotation around an axis perpendicular to the molecular plane and passing through the ring center. No models could satisfy this disorder. SQUEEZE 31 was used to model this disordered chlorobenzene. A 1030 Å3 void volume and electron density equivalent to 233.1 e− are consistent with four C6H5Cl in the unit cell, resulting in a 1:1 ratio of solvent to porphyrin.
[Fe(TPP)(2-MeHIm)]·(1,10-phen) (form B)
A red crystal with the dimensions 0.34 × 0.21 × 0.19 mm3 was used for the structure determination. Crystal data were collected at 100 K. The structure was refined in space group P21/c. The asymmetric unit contains two porphyrinate molecules, two 1,10-phenanthrolines, and two chlorobenzene solvates. For both porphyrin molecules, two phenyl rings are disordered, each over two positions with 50% occupancy.
An attempted refinement in a small unit cell containing just one porphyrin required addition disorder in the porphyrin, and resulted in a less satisfactory refinement. To verify the correct choice of cell, the two porphyrins were overlaid, one on the top of the other. This indicated differences in the conformations of the phenyl ring and β carbons, confirming the cell choice.
[Fe(OEP)(2-MeHIm)]·(1,10-phen)
A red crystal with the dimensions 0.44 × 0.28 × 0.20 mm3 was used for the structure determination. Crystal data were collected at 100 K. The structure was refined in space group Pca21. The asymmetric unit contains two porphyrinate molecules and two 1,10-phenanthroline molecules. The methyl carbon atom of one ethyl group in the second porphyrin molecule was found to be disordered over two positions, a major and a minor position (C(22a) and C(22b)). Their distances to C(221) were constrained to be 1.52 Å, and their thermal displacement parameters were constrained to be equal to each other. After the final refinement the occupancy of the major orientation was found to be 75%.
PLATON 31 suggested a possible higher symmetry space group, Pbcn, but several peripheral ethyl groups don’t fit in this symmetry. The refinement as disordered parts under Pbcn gave unsatisfactorily high R1/wR2 values. Thus the space group Pca21 was used in the final refinement, which gave R1 = 0.0499, wR2 = 0.1228.
Results
New five-coordinate iron(II) porphyrin complexes with a hydrogen-bonded 2-methylimidazole as the fifth ligand have been synthesized by reaction with 2-methylimidazole in the presence of excess 1,10-phenanthroline. Three crystalline derivatives of two porphyrins have been obtained. Two of them are polymorphs of [Fe(TPP)(2-MeHIm)]·(l,10-phen) and are called forms A and B; the third is an OEP derivative, [Fe(OEP)(2-MeHIm)]·(l,10-phen). Two of these three crystalline species have two independent porphyrinate molecules in the asymmetric unit, called molecules 1 and 2. In the figures and tables the following atom naming convention has been used: Q(n), Q(ny) and Q(nyy), where Q is the atom type, n refers to molecule 1 or 2 and y or yy are further numbers and letters needed to completely specify the atom. Thus similar atoms in the two molecules except phenyl rings in [Fe(TPP)(2-MeHIm)]·(l,10-phen) (form B) have the same name except for the digit n.
In all three crystalline derivatives, there are both porphyrin molecules and 1,10-phenanthroline molecules. The porphyrin molecules are all five-coordinate imidazole-ligated iron(II) species and a 1,10-phenanthroline molecule is hydrogen bonded to the coordinated 2-methylimidazole. As an example, the ORTEP diagram of [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form B, molecule 1) is illustrated in Figure 1. ORTEP diagrams of the four additional structures and a complete listing of bond distances and angles are given in the Supporting Information.
Figure 1.
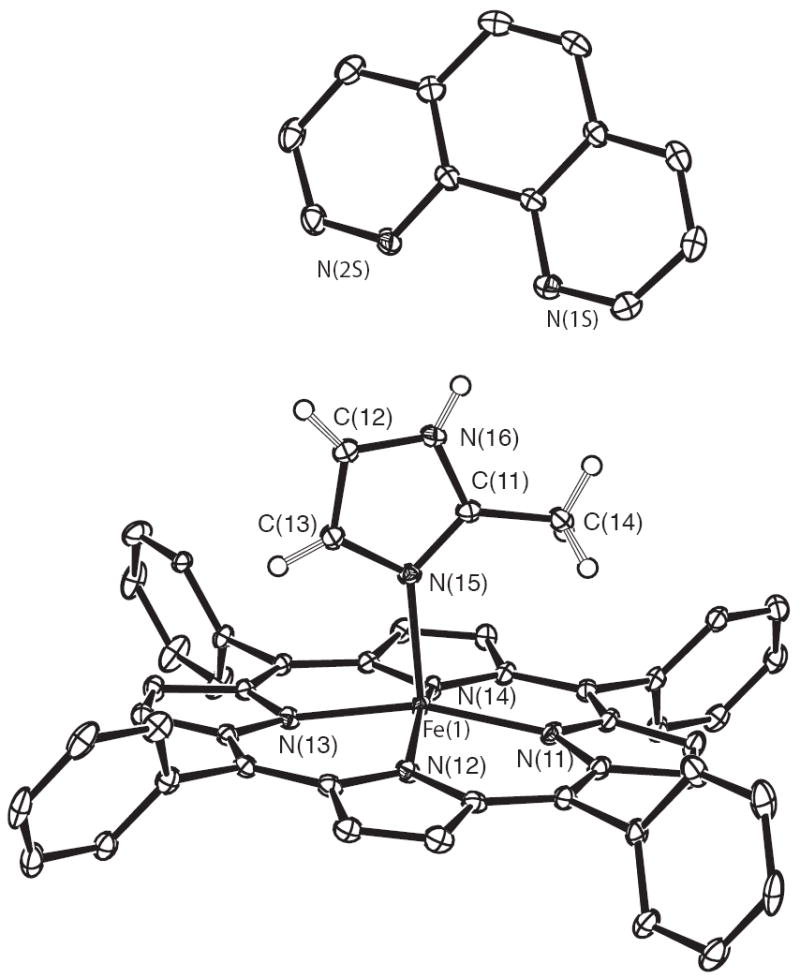
ORTEP diagram of [Fe(TPP)(2-MeHIm)]·(l,10-phen) (form B molecule 1). For clarity, one orientation of the disordered phenyl rings and the hydrogen atoms of the porphyrin ligand have been omitted. 50% probability ellipsoids are depicted.
The average value of the Fe–Np bond distances are 2.084(5) Å for [Fe(TPP)(2-MeHIm)]·(1,10-phen) (A), 2.080(9), 2.081(6) Å for [Fe(TPP)(2-MeHIm)]·(1,10-phen) (B) and 2.088(14), 2.087(14) Å for [Fe(OEP)(2-MeHIm)]·(1,10-phen). The Fe–NIm bond length is 2.1289(13) Å for [Fe(TPP)(2-MeHIm)]·(1,10-phen) (A), 2.125(3), 2.120(3) for [Fe(TPP)(2-MeHIm)]·(1,10-phen) (B) and 2.135(2), 2.131(2) Å for [Fe(OEP)(2-MeHIm)]·(1,10-phen), respectively.
Two crystalline species of TPP and OEP derivatives were studied with variable-temperature Mössbauer spectroscopy. For TPP derivative, the Mössbauer sample was made by the method for [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form A). These Mössbauer parameters at various temperatures are given in Table 2.
Table 2.
Variable-Temperature Mössbauer Parameters (mm/s) for [Fe(TPP)(2-MeHIm)]·(1,10-phen) and [Fe(OEP)(2-MeHIm)]·(1,10-phen)
| [Fe(TPP)(2-MeHIm)]·(1,10-phen) | |||||
| T (K) | 298 | 200 | 100 | 20 | |
| ΔEQ | 1.46 | 1.65 | 2.06 | 2.12 | |
| δFe | 0.80 | 0.86 | 0.88 | 0.90 | |
| [Fe(OEP)(2-MeHIm)]·(1,10-phen) | |||||
| T (K) | 295 | 200 | 100 | 25 | 4.2 |
| ΔEQ | 1.73 | 1.80 | 1.92 | 2.01 | 1.96 |
| δFe | 0.82 | 0.89 | 0.89 | 0.90 | 0.94 |
Discussion
Our previous studies have shown that strong hydrogen bonding 21 to or deprotonation 15 of the coordinated imidazole in five-coordinate iron(II) porphyrinate species significantly influences both the structural and electronic properties of the resulting species. The structural consequences of these effects are schematically illustrated in Figure 2. Limiting behavior is shown by the averaged values displayed by a number of imidazole complexes depicted at the left side of the figure. The other limiting case, deprotonation of the N–H of the coordinated imidazole to yield coordinated imidazolate, is shown at the right. Are these differences meaningful? We believe that the differences in geometry around the iron atom in the two cases reflect a difference in the electronic structure of iron. Experimental study of the electronic structure of iron in these systems is best investigated with Mössbauer spectroscopy. In addition to significant differences in the magnitude of the quadrupole splitting constants, the sign of the quadrupole splitting is an important distinction between imidazole- and imidazolate-ligated iron(II) species. The imidazolate derivatives have both larger values of the quadrupole splitting and a positive value for the sign. This distinction in sign of the quadruple splitting arises from differing symmetry of the doubly occupied orbital, which in the imidazolate species has essentially pure dxy character, i.e., the doubly occupied orbital is in the plane of the heme. This appears to be the usual electronic state for high-spin iron(II), whereas the imidazole-ligated species, including deoxyhemoglobin and deoxymyoglobin, have a very low symmetry orbital comprised of a hybrid dxy, dxz, and dyz set in which the major direction of the electron density is oblique to the heme plane and all Fe–N bond directions.
Figure 2.

Schematic diagrams of the coordination group geometries found for a series of imidazole-ligated high-spin iron(II) porphyrinates (left), an analogous species with a strong hydrogen bond to the coordinated imidazole (center), and imidazolate-ligated high-spin iron(II) porphyrinates (right).
The coordination geometry of the hydrogen-bonded species shown in the center of Figure 2 suggests that the hydrogen bond has a real effect on the structure. The structural parameters suggest that the hydrogen bond leads to an “imidazolate”-like coordinated ligand. The Mössbauer analysis of this system is unfortunately complicated by the presence of a second iron center in the solid-state sample. The assignment of a positive sign for the quadrupole doublet, similar to that of the imidazolate derivatives was judged most likely, but an absolute, unambiguous assessment of the sign was not possible.
In order to further investigate this question, we sought to prepare additional imidazole-ligated iron(II) porphyrinates in which the imidazole was hydrogen bonded to an external acceptor. Suitable hydrogen-bond acceptors in such a system must either be weak ligands or a nonligand so as to avoid coordination competition with 2-methylimidazole. Attempts using species including 2-methylpyridine, 2-chloropyridine, proton sponge, and DBU failed because they failed to give crystalline materials, 32 failed to yield an hydrogen-bonded species with the coordinated imidazole or, most interestingly, yielded the already known species with a 2-methylimidazole solvate hydrogen bonded to the coordinated imidazole.21 One hydrogen bonding acceptor that did yield interesting new materials was 1,10-phenanthroline.
1,10-phenanthroline has been shown as a hydrogen-bond acceptor to coordinated imidazole.22 By addition of excess 1,10-phenanthroline, our experiments gave new species, [Fe(TPP)(2-MeHIm)]·(1,10-phen) and [Fe(OEP)(2-MeHIm)]·(1,10-phen), in which 1,10-phenanthroline is H-bonded to the coordinated imidazole. These species have been characterized by X-ray crystallography, Mössbauer spectroscopy, IR and UV-vis spectroscopic measurements.
IR Spectroscopy
IR has been used to demonstrate the formation of a hydrogen bond to coordinated imidazole in porphyrins 33 or other species.34 In our case, in order to verify the formation of a hydrogen-bonded species, IR spectra of [Fe(Por)(2-MeHIm)] and [Fe(Por)(2-MeHIm)]·(l,10-phen) have been measured.
The spectra of [Fe(OEP)(2-MeHIm)] and [Fe(OEP)(2-MeHIm)]·(l,10-phen) are shown in Figure 3. The sharp band at 3360 cm−1 for [Fe(OEP)(2-MeHIm)] was assigned as the N–H stretch. But in [Fe(OEP)(2-MeHIm)]·(l,10-phen), it has shifted, and two new broad bands arise at lower frequencies. One is at 3050 cm−1 which is the same position as the C–H stretch in 1,10-phenanthroline. The other band is at 3100 cm−1 which can reasonably be assigned the hydrogen-bonded N–H stretch. The spectra of [Fe(TPP)(2-MeHIm)] and [Fe(TPP)(2-MeHIm)]·(l,10-phen) showed similar behavior. Such a change indicates the formation of a hydrogen bond in new species, which is similar to that for an imidazole-coordinated manganese complex.34
Figure 3.
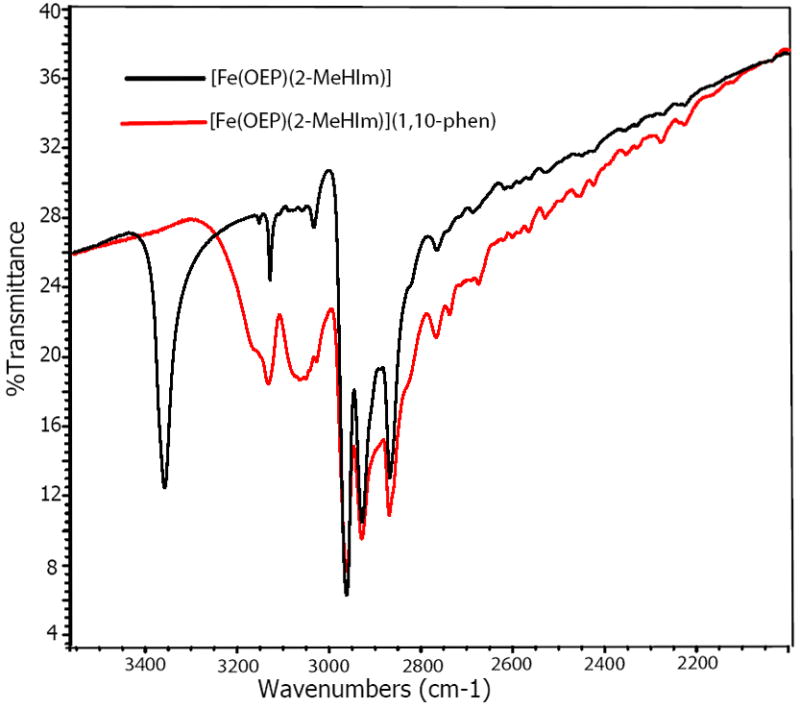
IR spectra of [Fe(OEP)(2-MeHIm)]·(l,10-phen) and [Fe(OEP)(2-MeHIm)] between 3500−2000 cm−1.
Structural Studies
All new porphyrin molecules are five-coordinate 2-methylimidazole-ligated species. 1,10-phenanthroline does not coordinate to the iron center in the solid state, presumably as the result of the steric configuration between 1,10-phenanthroline and the porphyrin ring. But, as expected, 1,10-phenanthroline has formed hydrogen bonds with the coordinated imidazole in a 1:1 ratio of 2-methylimidazole and 1,10-phenanthroline.
Two views of the hydrogen-bonding pattern are shown in Figure 4. The hydrogen bonds between 2-methylimidazole and 1,10-phenanthroline are three-center hydrogen bonds: the hydrogen of the imidazole nitrogen is hydrogen bonded to both nitrogen atoms of 1,10–phenanthroline; two hydrogen bonds are formed. Individual distances and angles for the five independent molecules are listed in Table 3. In all structures, the two hydrogen bonds are not equal, one is stronger with a shorter N⋯N distance (average 2.97(2) versus 3.08(3) Å), shorter intermolecular H⋯N distance (average 2.13(4) versus 2.52(5) Å), and larger N–H⋯N angle (average 162(6) versus 122(2) °). It should be noted that all values involving the hydrogen atom of the imidazole are based on an idealized position of this hydrogen atom. The dihedral angle between 2-methylimidazole plane and the corresponding 1,10-phenanthroline plane ranges from 37.6 to 46.5°.
Figure 4.
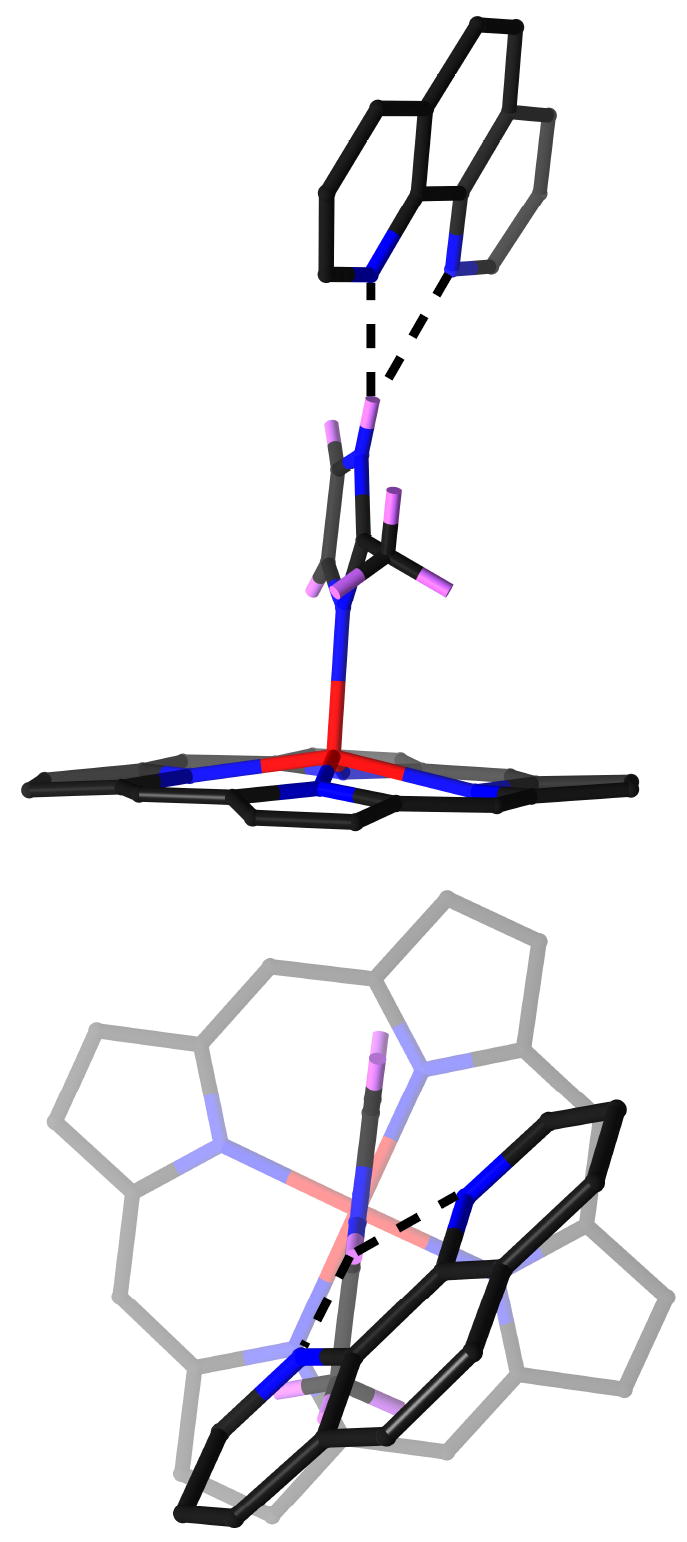
Two views of the hydrogen-bonding interaction between the coordinated imidazole and 1,10-phenanthroline. For clarity, only the 24 atoms of porphyrin core are shown in the figures.
Table 3.
Hydrogen Bond Parameters Between Imidazole and 1,10-Phenanthroline.
| Complex | NIm–H distancea | N⋯H distancea | NIm⋯Nphen distance a | NIm–H⋯Nphen angleb | dihedral angleb,c |
|---|---|---|---|---|---|
| [Fe(TPP)(2-MeHIm)]·- | 0.86 | 2.14 | 2.980(2) | 165.0 | 43.7 |
| (1,10-phen) (form A) | 0.86 | 2.48 | 3.032(2) | 123.0 | |
| [Fe(TPP)(2-MeHIm)]·- | 0.88 | 2.09 | 2.949(4) | 164.1 | 40.8 |
| (1,10-phen) (form B, Mol 1) | 0.88 | 2.52 | 3.089(4) | 122.7 | |
| [Fe(TPP)(2-MeHIm)]·- | 0.88 | 2.20 | 3.006(4) | 151.8 | 37.6 |
| (1,10-phen) (form B, Mol 2) | 0.88 | 2.48 | 3.059(4) | 123.9 | |
| [Fe(OEP)(2-MeHIm)]·- | 0.88 | 2.10 | 2.954(3) | 163.7 | 46.5 |
| (1,10-phen) (Mol 1) | 0.88 | 2.57 | 3.104(3) | 119.6 | |
| [Fe(OEP)(2-MeHIm)]·- | 0.88 | 2.12 | 2.979(3) | 165.2 | 42.4 |
| (1,10-phen)(Mol 2) | 0.88 | 2.57 | 3.113(3) | 121.1 |
in Å.
Value in degrees.
Dihedral angle between the imidazole plane and the corresponding phenanthroline plane.
Is there anything unusual about the hydrogen bonds between the imidazole and the 1,10–phenanthroline? How to evaluate the strength of these hydrogen bonds? One useful comparison is to utilize the Cambridge Structural Database35 to assess the hydrogen bonds formed by 1,10–phenanthroline in a range of crystal structures. We found a total of 60 instances of three-center hydrogen bonds formed by 1,10-phenanthroline to a nitrogen atom hydrogen-bond donor in a solid state structure. Since all of these are X-ray diffraction studies, the hydrogen atom positions are relatively uncertain. Thus, the best distances to compare are the intermolecular N⋯N distances. The correlations between these two N⋯N distances associated with the hydrogen bonds are shown in Figure 5, the shorter N⋯N distance is shown on the x-axis, the longer one is the y-axis. The big black circles are the results for our current samples. The figure shows that the N⋯N distance in these new species are well within the range of hydrogen bonds associated with 1,10-phenanthroline in other species and the observed asymmetry is typical.
Figure 5.
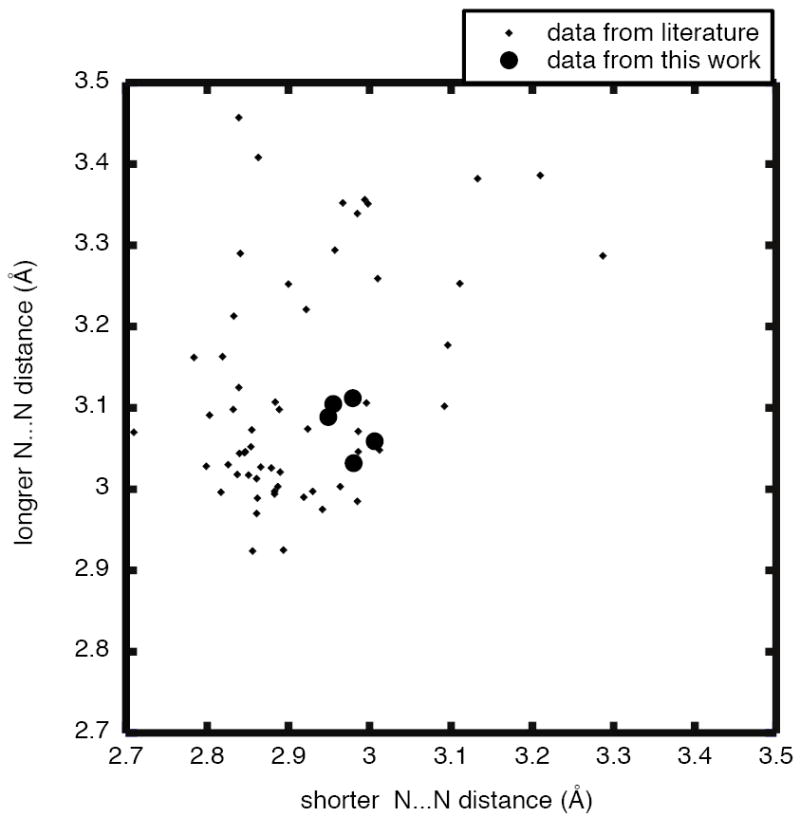
Results from a Cambridge Structural Database survey on the hydrogen bonds formed by 1,10–phenanthroline to a nitrogen atom hydrogen-bond donor in a range of differing crystal structures, showing the correlations between the two intermolecular N⋯N distances associated with the hydrogen bonds. The shorter distance is given on the x-axis, the longer one on the y-axis. The large black circles are the results for our current samples.
Are there structural changes caused by these hydrogen bonds? The overall structural features are those expected for a high-spin iron(II) complex.14, 36 These include large equatorial Fe–Np bond distances, a significant out-of-plane displacement of the iron(II) atom and a radial expansion of the core (an increase in the size of the central hole).37 The average Fe–Np bond distances range from 2.080(9) to 2.088(14) Å and the Fe–NIm bond lengths range from 2.120(3) to 2.135(2) Å; both are similar to the values for other imidazole-ligated porphyrinates as shown in Table 4. As shown in Table 4, the out-of-plane displacement of the iron atom out of the mean 24-atom porphyrin core (Δ) and the plane defined by the four pyrrole nitrogen atoms (ΔN4) also fit the range for those imidazole-ligated porphyrinates. The displacements range from 0.38 to 0.49 Å out of the 24-atom plane and from 0.36 to 0.39 Å out of the four pyrrole nitrogen atom plane. The radii of the porphyrin cores, given by Ct⋯N in Table 4, are nearly identical at 2.049 and 2.051 Å.
Table 4.
Selected Bond Distances (Å) and Angles (deg) for New Structures and Related Species a
| Complexb | Fe–Npc,d | Fe–Nimd | ΔN4d,e | Δd,f | Ct⋯Nd | Fe–N–Cg,h | Fe–N–Cg,i | θg,j | ϕg,k | ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| [Fe(TPP)(2-MeHIm)]·(1,10-phen) (A) | 2:084(5) | 2:1289(13) | 0:38 | 0:47 | 2:049 | 132.03(12) | 123.09(11) | 8:8 | 18:5 | tw |
| [Fe(TPP)(2-MeHIm)]·(1,10-phen) (B) | 2:080(9) | 2:125(3) | 0:36 | 0:39 | 2:049 | 130.8(2) | 123.8(2) | 6:6 | 17:7 | tw |
| 2:081(6) | 2:120(3) | 0:36 | 0:38 | 2:051 | 133.7(2) | 121.5(2) | 5:4 | 7:8 | tw | |
| [Fe(OEP)(2-MeHIm)]·(1,10-phen) | 2:088(14) | 2:135(2) | 0:39 | 0:49 | 2:051 | 132.75(17) | 121.20(16) | 2:7 | 8:1 | tw |
| 2:087(14) | 2:131(2) | 0:39 | 0:47 | 2:051 | 133.43(17) | 121.66(17) | 2:1 | 13:8 | tw | |
| [Fe(OEP)(1,2-Me2Im)] | 2:080(6) | 2:171(3) | 0:37 | 0:45 | 2:047 | 132.7(3) | 121.4(2) | 3:8 | 10:5 | 16 |
| [Fe(OEP)(2-MeHIm)] | 2:077(7) | 2:135(3) | 0:34 | 0:46 | 2:049 | 131.3(3) | 122.4(3) | 6:9 | 19:5 | 16 |
| [Fe(TPP)(1,2-Me2Im)] | 2:079(8) | 2:158(2)l | 0:36 | 0:42 | 2:048 | 129.3(2) | 124.9(2) | 11:4 | 20:9 | 14 |
| [Fe(TTP)(2-MeHIm)] | 2:076(3) | 2:144(1) | 0:32 | 0:39 | 2:050 | 132.8(1) | 121.4(1) | 6:6 | 35:8 | 14 |
| [Fe(Tp-OCH3PP)(2-MeHIm)] | 2:087(7) | 2:155(2)l | 0:39 | 0:51 | 2:049 | 130.4(2) | 123.4(2) | 8:6 | 44:5 | 14 |
| [Fe(Tp-OCH3PP)(1,2-Me2Im)] | 2:077(6) | 2:137(4) | 0:35 | 0:38 | 2:046 | 131.9(3) | 122.7(3) | 6:1 | 20:7 | 14 |
| [Fe(TPP)(2-MeHIm)](2-fold) | 2:086(8) | 2:161(5) | 0:42 | 0:55 | 2:044 | 131.4(4) | 122.6(4) | 10:3 | 6:5 | 38 |
| [Fe(TPP)(2-MeHIm)]·1.5C6H5Cl | 2:073(9) | 2:127(3)l | 0:32 | 0:38 | 2:049 | 131.1(2) | 122.9(2) | 8:3 | 24:0 | 13 |
| average of the eight | 2:080(5) | 2:147(16) | 0:36(3) | 0:44(6) | 2:048(2) | 131.4(12) | 122.7(11) | 7:8(24) | ||
| [Fe(TPP)(2-MeHIm)] (Mol. 1) | 2:080(8) | 2:120(2) | 0:36 | 0:41 | 2:050 | 131.6(1) | 122.4(1) | 9:2 | 16:0 | 21 |
| [Fe(TPP)(2-MeHIm)] (Mol. 2) | 2:099(7) | 2:099(2) | 0:49 | 0:55 | 2:040 | 129.0(1) | 125.7(1) | 7:6 | 22:9 | 21 |
| [K(222)][Fe(OEP)(2-MeIm −)] | 2:113(4) | 2:060(2) | 0:56 | 0:65 | 2:036 | 136.6(2) | 120.0(2) | 3:6 | 37:4 | 15 |
| [K(222)][Fe(TPP)(2-MeIm −)] | 2:118(13) | 1:999(5) | 0:56 | 0:66 | 2:044 | 129.6(3) | 126.7(3) | 9:8 | 23:4 | 15 |
| 2:114(5) | 133.6(4) | 121.9(4) | 6:5 | 21:6 | ||||||
| [Fe(TpivPP)(2-MeIm−)] − | 2:11(2) | 2:002(15) | 0:52 | 0:65 | 2:045 | NRm | NR | 5:1 | 14:7 | 39 |
| average of the three | 2:114(4) | 2:044(54) | 0:55(2) | 0:65(1) | 2:042(5) |
Estimated standard deviations are given in parentheses.
All complexes are high spin.
Averaged value.
in Å.
Displacement of iron from the mean plane of the four pyrrole nitrogen atoms.
Displacement of iron from the 24-atom mean plane of the porphyrin core.
Value in degrees.
2-carbon, methyl substituted.
Imidazole 4-carbon.
off-axis tilt (deg) of the Fe–NImbond from the normal to the porphyrin plane.
Dihedral angle between the plane defined by the closest Np–Fe–NIm and the imidazole plane in deg.
Major imidazole orientation.
Not reported.
The steric bulk of the imidazole 2-methyl group leads to, in all derivatives examined to date, an off-axis tilt of the axial Fe–NIm bond and a rotation of the imidazole ligand that leads to unequal Fe–NIm–CIm angles. The tilt angles range from 2.1° for [Fe(OEP)(2-MeHIm)]·(l,10-phen) (Mol 2) to 8.8 ° for [Fe(TPP)(2-MeHIm)]·(1,10-phen) (A). This tilting is the partial result of minimizing the interaction between the bulky imidazole methyl group and the porphyrin core. Two important angles associated with the imidazole ligands (Fe∓NIm∓CIm) are given in Table 4. They range from 130.8(2) to 133.7(2)° on the methyl side and 121.2(16) to 123.8(2)° on the other side. The off-axis tilt and imidazole rotation are correlated so as to maximize the distance between the 2-methyl group and porphyrin core atoms, the values are similar to those for other imidazole-ligated species.
The above structural data suggests these new species are essentially identical to those of other imidazole-ligated species and indicates that the imidazole⋯1,10-phen hydrogen bonds do not have an obvious influence on the structure at iron. This is quite distinct from the strong hydrogen bond in [Fe(TPP)(2-MeHIm)]2·2-MeHIm,21 which has remarkable effects on the structure at iron, including much larger Fe–Np distances (difference of ~0.02 Å) and iron displacement (difference of ~0.10 Å). These data allow us to tentatively conclude that there is a gradation of effects from hydrogen bonding on the structure of the high-spin, imidazole-ligated iron porphyrinates.
Figure 6 gives formal diagrams of the porphyrin cores of the new iron(II) structures. Given are the displacements of each atom from the mean plane of the four pyrrole nitrogen in units of 0.01 Å. An analogous diagram showing atomic displacements from the mean plane of the 24-atom core is given in the supporting information (Figure S5). The orientation of the imidazole ligand with respect to the core atoms are shown by the line with the circle representing the methyl group bound at the 2-carbon atom position. The dihedral angles (ϕ) between the imidazole ligand and the plane defined by N(n1), Fe(n), N(n5) are all less than 20°. Also included on the diagrams of Figure 6 are the individual Fe–Np bond lengths. The pair of Fe–Np distances closer to imidazole plane are longer than the other pair (average 2.090(8) versus 2.078(7) Å), which could be caused by a repulsive interaction between imidazole group and the close N(1) and N(3) atoms when the dihedral angle is small.
Figure 6.

Formal diagrams of the porphyrinato cores of a) [Fe(TPP)(2-MeHIm)]·(l,10-phen) (A); b) [Fe(TPP)(2-MeHIm)]·(l,10-phen) (B, Molecule 1); c) [Fe(TPP)(2-MeHIm)]·(l,10-phen) (B, Molecule 2); d) [Fe(OEP)(2-MeHIm)]·(l,10-phen) Molecule 1; e) [Fe(OEP)(2-MeHIm)]·(l,10-phen) Molecule 2. Illustrated are the displacements of each atom from the mean plane of the four pyrrole nitrogen in units of 0.01 Å. Positive values of displacement are toward the imidazole ligand. The diagrams also show the orientation of the imidazole ligand with respect to the atoms of the porphyrin core. The location of the methyl group at the 2-carbon position is represented by the circle.
As can be seen in Figure 6, the porphyrin cores have somewhat different conformations even within the same crystal. This is consistent with our previous observation that there is not a single preferred core conformation for imidazole-ligated high-spin iron(II) complexes.14,16 Core conformations have also been analyzed by the normal structural decomposition (NSD) method provided by Shelnutt et al.40 Table S1 lists the NSD out-of-plane displacements of known high-spin iron(II) porphyrinates ligated with a 2-methylimidazole or 1,2-dimethylimidazole axial ligand. This further confirms that there was no common pattern for the core conformations.
In addition to the hydrogen bonds, there are remarkable π - π stacking interactions associated with 1,10-phenanthroline. For [Fe(TPP)(2-MeHIm)]·(1,10-phen) (A), as shown in Figure 7a, one aromatic ring of a symmetry related phenanthroline (symmetry operator #1:x,1.5 − y,−0.5+z) is overlapped with a six-membered chelating ring in porphyrin (Fe(1), N(1), N(2), C(a2), C(m1), C(a3)) and the dihedral angle between them is 15.9°. The shortest intermolecular distance is Fe(1)⋯C(9S#1) = 3.261 Å. For [Fe(OEP)(2-MeHIm)]·(1,10-phen), phenanthroline hydrogen bonded to molecule 1 stacks over another phenanthroline hydrogen bonded to molecule 2 from a symmetry related unit (symmetry operator #2: 1.5−x,y,−0.5+z). They are almost parallel to each other with the small dihedral angle of 0.7°. The closest intermolecular distance is C(7S)⋯C(23S#2) = 3.479 Å.
Figure 7.
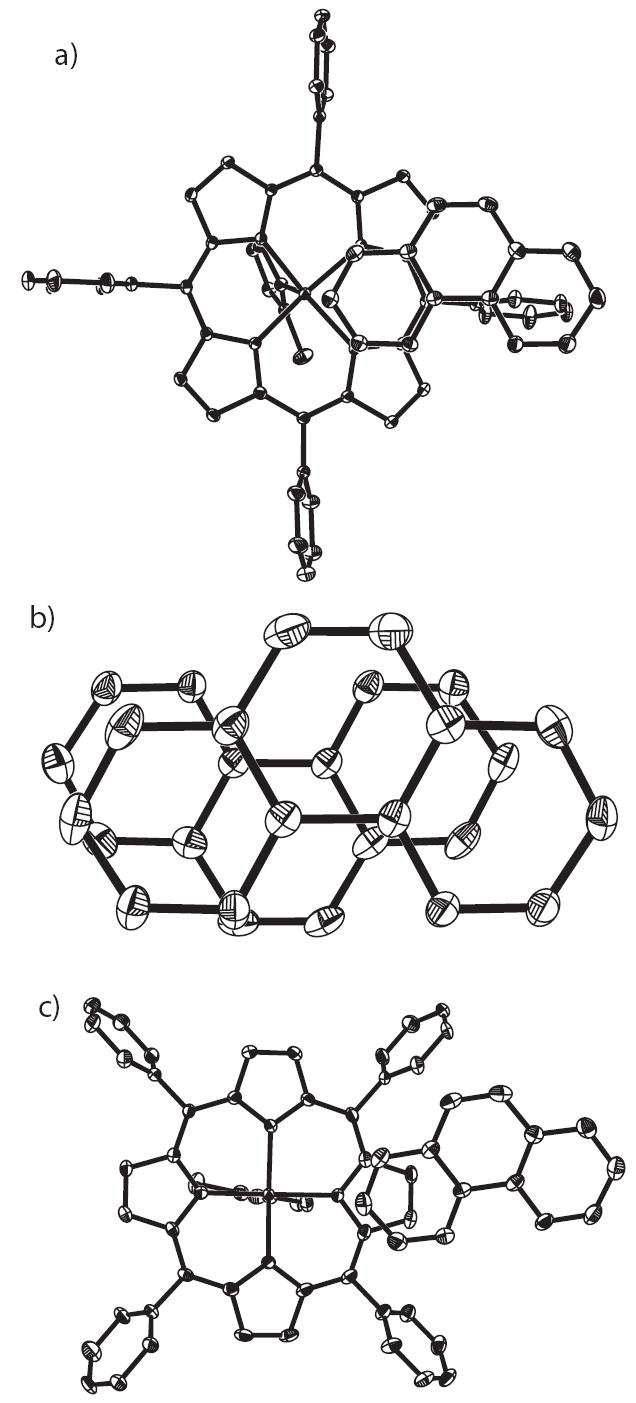
Stacking diagrams showing the π - π interactions: a) in [Fe(TPP)(2-MeHIm)]·(l,10-phen) (Form A), all atoms are contoured at the 10% probability level for clarity; b) in [Fe(OEP)(2-MeHIm)]·(l,10-phen), all atoms are contoured at the 50% probability level; c) in [Fe(TPP)(2-MeHIm)]·(l,10-phen) (Form B), all atoms are contoured at the 50% probability level.
For [Fe(TPP)(2-MeHIm)]·(1,10-phen) (B), there are two kinds of π - π interactions. One is like that for [Fe(OEP)(2-MeHIm)]·(1,10-phen), a phenanthroline hydrogen bonded to molecule 1 stacks over another phenanthroline hydrogen bonded to molecule 2 from a symmetry related unit (symmetry operator #3: x, 1.5 −y, 0.5+z). They are almost parallel to each other with a small dihedral angle of 2.6°. The closest intermolecular distance is C(1S)⋯C(17S#3) = 3.516 Å. Another π - π interaction is between phenanthroline and one pyrrole ring of porphyrin as shown in Figure 7c. One pyrrole ring in molecule 1 (consisting of N(11), C(1a1), C(1a2), C(1b1), C(1b2)) is overlapped with a phenyl ring (consisting of N(3S), C(19S), C(20S), C(21S), C(22S), C(23S)) in a symmetry related phenanthroline (symmetry operator #4: x,y−1,z). The dihedral angle between them is 18.1°. The closest intermolecular distance is C(lal)⋯C(22S#4) = 3.311 Å. Another pyrrole ring in molecule 2 (consisting of N(23), C(2a5), C(2a6), C(2b5), C(2b6)) is overlapped with a phenyl ring of phenanthroline (consisting of N(1S), C(1S), C(2S), C(3S), C(4S), C(12S)). The corresponding dihedral angle is 10.3°, and the closest intermolecular distance is C(2b5)⋯C(1S) = 3.322 3 Å. Obviously, these π - π interactions are important factors to stabilize the solid-state structures.
Electronic Structure
The X-ray structures show that the new phenanthroline species are strongly similar to the imidazole-ligated species. Our previous studies suggest that imidazole-and imidazolate-ligated five-coordinate iron(II) porphyrinates form two distinct groups with different electronic configurations. 14, 15 Mössbauer spectra of the imidazolate-ligated species show a large positive value of the quadrupole splitting with a small asymmetry parameter (η), consistent with a ground-state doubly-occupied dxy orbital, (dxy)2(dxz)1(dyz)1(dz2)1(dx2−y2)1, i.e., an orbital in the heme plane. On the other hand, Mössbauer spectra of the imidazole-ligated species, including deoxymyoglobin and deoxyhemoglobin, show a negative value of the quadrupole splitting with large asymmetry parameters. The best interpretation is that these species have an unusual ground state in which the doubly-occupied d orbital can be best described as a hybrid orbital comprised of the dxz, dyz, and dxy orbitals, i.e., a very low symmetry orbital. Our recent study shows that a hydrogen bond to imidazole in [Fe(TPP)(2-MeHIm)]2·2-MeHIm 21 can have significant effects on Mössbauer properties as well as on structure, yielding a species with properties very much like that of an imidazolate.
Accordingly, the new hydrogen bonded species, [Fe(TPP)(2-MeHIm)]·(1,10-phen) and [Fe-(OEP)(2-MeHIm)]·(1,10-phen), were characterized by Mössbauer spectroscopy. For the TPP derivative, the value of quadrupole splitting (ΔEq) observed at 20 K is 2.12 mm/s, which is similar to the previously reported high-spin iron(II) species as shown in Table 5;14,41 the large value of the isomer shift (δ ~0.90 mm/s) is also consistent with high-spin iron(II).42 There is a substantial temperature variation of the quadrupole splitting values as shown in Table 2. The value of ΔEq decreases significantly as the temperature is increased. The observed temperature dependence of ΔEq is similar to the variation seen for imidazole-ligated samples. 14,15,41,43-49 As discussed previously,14 the explanation for this temperature variation is that there are closelying excited states. The excited states could have the same or differing spin multiplicity relative to the ground state. These data clearly suggest that the new hydrogen bonded species has the same electronic structure as the imidazole-ligated species. The quadrupole splitting values of the OEP derivative, as shown in Table 2, had similar values, but a smaller temperature variation than many other imidazole-ligated species. In order to further study its electronic structure, the Mössbauer spectra were measured in applied magnetic fields.
Table 5.
Solid-State Mössbauer Parameters for Five-Coordinate, High-Spin Imidazole-Ligated Iron(II) Porphyrinates and Related Species.
| Complex | ΔEQa | δFea | ηb | Γc | T, K | ref. |
|---|---|---|---|---|---|---|
| [Fe(OEP)(2-MeHIm)]·(1,10-phen) | −1.93 | 0.94 | 0.76 | 0.43 | 4.2 | tw |
| [Fe(TPP)(2-MeHIm)]·(1,10-phen) | −2.12c | 0.90 | 0.30 | 20 | tw | |
| [Fe(OEP)(1,2-Me2Im)] | −2.19 | 0.92 | 0.50 | 0.37 | 4.2 | 16 |
| [Fe(OEP)(2-MeHIm)] | −1.94 | 0.90 | 0.48 | 0.41 | 4.2 | 16 |
| [Fe(Tp-OCH3PP)(1,2-Me2Im)] | −2.44 | 0.95 | 0.68 | 0.46 | 4.2 | 14 |
| [Fe(Tp-OCH3PP)(2-MeHIm)] | −2.18 | 0.94 | 0.58 | 0.58 | 4.2 | 14 |
| [Fe(TPP)(1,2-Me2Im)] | −1.93 | 0.92 | 0.53 | 0.44 | 4.2 | 14 |
| [Fe(TTP)(2-MeHIm)] | −1.95 | 0.85 | 0.63 | 0.42 | 4.2 | 14 |
| [Fe(TTP)(1,2-Me2Im)] | −2.06 | 0.86 | 0.58 | 0.43 | 4.2 | 14 |
| [Fe(TPP)(2-MeHIm)] | −2.40 | 0.92 | 0.8 | 0.50 | 4.2 | 13 |
| [Fe(TPP)(2-MeHIm)(2-fold) | −2.28 | 0.93 | 0.8 | 0.31 | 4.2 | 41 |
| [Fe(TPP)(1,2-Me2Im)] | −2.16 | 0.92 | 0.7 | 0.25 | 4.2 | 41 |
| [Fe((Piv2C8P)(1-MeIm)] | −2.3d | 0.88 | 0.40 | 0.40 | 4.2 | 50 |
| deoxyHb | −2.40 | 0.92 | 0.7 | 0.30 | 4.2 | 41 |
| deoxyMb | −2.22 | 0.92 | 0.7 | 0.34 | 4.2 | 41 |
| [Fe(TPP)(2-MeHIm)] (Mol. 1) | −2.40e | 0.92 | 0.90 | 0.37 | 4.2 | 21 |
| [Fe(TPP)(2-MeHIm)] (Mol. 2) | +2.94e | 0.97 | 0.71 | 0.58 | 4.2 | 21 |
| [K(222)][Fe(OEP)(2-MeIm −)] | +3.71 | 1.00 | 0.22 | 0.31 | 4.2 | 15 |
| [K(222)][Fe(TPP)(2-MeIm−)] | +3.60 | 1.00 | 0.02 | 0.32 | 4.2 | 15 |
| [Fe(TPpivP)(2-MeIm)]− | +3.51f | 0.97 | 77 | 39 | ||
| [Fe(OC6H5)(TPP)]− | +4.01 | 1.03 | 0.25 | 0.38 | 4.2 | 51 |
| [Fe(O2CCH3)(TPpivP)]− | +4.25 | 1.05 | 0.30 | 0.30 | 4.2 | 52 |
| [Fe(OCH3)(TPpivP)]− | +3.67f | 1.03 | 0.40 | 4.2 | 53 | |
| [Fe(OC6H5)(TPpivP)]− | +3.90f | 1.06 | 0.38 | 4.2 | 53 | |
| [NaC12H24O6][Fe(TPpivP)(SC6HF4)] | +2.38f | 0.84 | 0.28 | 4.2 | 54 | |
| [Na(222)][Fe(TpivPP)(SC6HF4)] | +2.38f | 0.83 | 0.32 | 4.2 | 54 | |
| [Fe(TpivPP)(SC2H5)]− | +2.18 | 0.83 | 0.80 | 0.30 | 4.2 | 54 |
| [Fe(TpivPP)Cl]− | +4.36f | 1.01 | 0.31 | 77 | 55 |
mm/s.
Asymmetry parameter.
Line width, FWHM.
Sign not determined experimentally, presumed negative.
Sign is based on a best fit. Because of the complexity of this two-site model, the set of parameters that result in this fit are not necessarily unique.
Sign not determined experimentally, presumed positive.
The application of applied magnetic field Mössbauer spectroscopy provides more detailed information concerning the electronic ground state. The Mössbauer data in strong magnetic fields shown in Figure 8 were fit with the spin Hamiltonian model used by Kent et al.41
Figure 8.
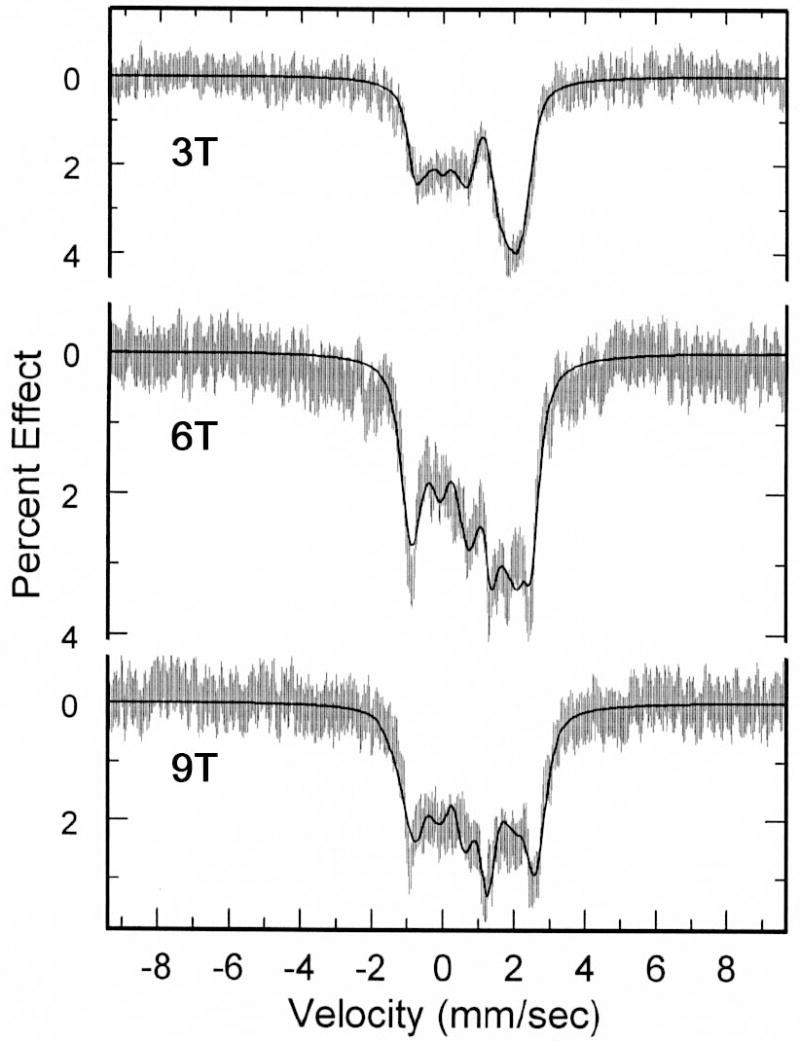
Data and fits obtained for [Fe(OEP)(2-MeHIm)]·(l,10-phen) at a) 3 T, b) 6 T, and c) 9 T applied magnetic field.
where D and E are the axial and rhombic zero-field splitting parameters that describe the fine structure of the S = 2 multiplet, Ã* is the magnetic hyperfine tensor and HQ gives the nuclear quadrupole interaction:
Q is the quadrupole moment of the 57Fe nucleus and η = (Vxx − Vyy)/Vzz, where Vii are components of the electric field gradient. The quadrupole splitting and isomer shift were constrained to the values determined from the zero-field data. Complete fitting data are given in Table S2 of the Supporting Information.
An analysis of the spectra shows the largest component of the electric field gradient, Vzz, has a negative value and hence the sign of the quadrupole splitting value is also negative. The asymmetry parameters η for [Fe(OEP)(2-MeHIm)]·(l,10-phen) (0.76) is relatively large, which is consistent with those for imidazole-ligated species.14,41,44-49 The large asymmetry parameter indicates the low symmetry of the electric field gradient (EFG), which is also reffected in the solid-state structure. The results are consistent with our earlier studies of imidazole-ligated species and we can conclude that the moderate hydrogen bonding in these phenanthroline derivatives is inadequate to lead to the imidazolate-like character observed in [Fe(TPP)(2-MeHIm)]2·2-MeHIm.21
An attempted preparation of a bulk Mössbauer sample of [Fe(TPP)(2-MeHIm)]·(1,10-phen) led to an interesting observation. The sample was prepared with same reaction conditions that were used for the form B preparation except for the use of 20 mL of chlorobenzene rather than 25 mL. The product was then crashed out of solution by the addition of 120 mL of hexanes. This yielded a microcrystalline solid, the Mössbauer spectrum of which displayed two quadrupole doublets overlapped at most temperatures. These additional Mössbauer data are given in Table S3 of the Supporting Information. The quadrupole splitting values for the major component showed very little temperature dependence, whereas the minor component showed strong temperature dependence. The minor component has quadrupole splitting and isomer shift values very similar to those for five-coordinate species, [Fe(TPP)(2-MeHIm)]·(1,10-phen), as shown in Table 2. For the major component, the values are very similar to those for low-spin [Fe(TMP)(2-MeHIm)2],56 and thus most likely to be six-coordinate [Fe(TPP)(2-MeHIm)2].However, [Fe(TPP)(2-MeHIm) 2] has been only reported at low temperature,57 suggesting small values for the binding constants. The observation led us to ask how much the hydrogen bonding by 1,10-phenanthroline could increase the binding constants and lead the formation of the six-coordinate species. We have therefore carried out further spectral studies to study this question.
Spectral Studies
Hydrogen bonding of 1,10-phenanthroline with coordinated imidazole in iron(III) and cobalt(III) porphyrinates has been reported to enhance the binding constants of imidazole and thus enhance the formation of six-coordinate species.22,23
In solutions of our system, there are two reactions that must be considered:
| (1) |
| (2) |
Because the three possible iron(II) porphyrinates have substantially different absorption spectra,58 UV-vis spectroscopy can be used to monitor the above reactions. The results of the spectroscopic titrations are shown in Figure 9. As seen, both the Soret band and visible bands show little change from spectrum a to spectrum d. Only in spectrum e, when the concentrations of 2-methylimidazole and 1 ,10-phenanthroline are both extremely large, do the spectra show moderate changes. The relative intensities of the two bands at 535 nm and 565 nm have become more equivalent suggesting an increased intensity of the 535 nm band. The intensity change is consistent with the observations of six-coordinate imidazole-ligated iron(II) porphyrinates.58 Thus the change suggests that a small amount of six-coordinate species is formed. But in the Soret band region, there is no extra band or shoulder shown at 425 nm a typical position for a six-coordinate imidazole-ligated iron(II) species.58 The spectra indicate that hydrogen-bonding by 1,10-phenanthroline to the coordinated imidazole probably causes an equilibrium shift to the right in reaction (2) and the formation of a small amount of six-coordinate species. However, the large amount of a six-coordinate species seen in the bulk Mössbauer sample seems understood only in terms of solubility issues at high iron(II) concentrations.
Figure 9.

UV-vis spectra taken under argon in CH2Cl2 solution. Concentration of [Fe(TPP)] is 0.5×10−3 mol/L; concentrations of 2-methylimidazole are 0.11 mol/L for a), b), c) and d), 1.07 mol/L for e); concentration of 1,10–phenanthroline in mol/L a) 0. b) 4.8×l0−3. c) 0.08. d) 0.16. e) 0.86. The enlarged spectra from 480 to 700 nm are measured in the 1-mm UV cell.
Summary
We have prepared new hydrogen-bonded species, [Fe(TPP)(2-MeHIm)]·(1,10-phen) and [Fe(OEP)(2-MeHIm)]·(1,10-phen), with 1,10-phenanthroline as the hydrogen-bond acceptor. Three new structures have been characterized and showed the formation of hydrogen bonds between 1,10-phenanthroline and the coordinated imidazole. The structures show that the geometric parameters of iron, such as the Fe–Np and Fe–NIm bond lengths, and displacement of the iron atom out of the porphyrin plane, are similar to those in imidazole-ligated species. Further studies of their Mössbauer spectra suggest they have the same electronic configuration as those of the imidazole-ligated species. Unlike the case in [Fe(TPP)(2-MeHIm)]2·2-MeHIm,21 the hydrogen bonds in those new species have little or no effect on the geometric and electronic structures. These studies suggest there is a relatively large gradation of effects from hydrogen bonding on the five-coordinate high-spin, imidazole-ligated iron(II) porphyrinates.
Supplementary Material
Figures S1–S4 show ORTEP diagrams of [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form B molecule 2), [Fe(OEP)(2-MeHIm)]·(1,10-phen) (molecule 1 and 2), and [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form A). Figure S5 shows atomic displacements from the mean plane of the 24-atom core. Table S1 shows analysis results of the core conformations by the normal structural decomposition method. Table S2 gives full details on the fits for applied field Mössbauer measurements of [Fe(OEP)(2-MeHIm)]·(1,10-phen). Table S3 shows variable temperature Mössbauer Parameters for crashed out solid sample of TPP derivative. Tables S4–S21 give complete crystallographic details, atomic coordinates, bond distances and angles, anisotropic temperature factors, and fixed hydrogen atom positions. This information is available as a PDF file. The crystallographic information files (CIF) are also available. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health for support of this research under Grant GM-38401. We thank the NSF for X-ray instrumentation support through Grant CHE-0443233. We also thank a reviewer for suggesting that we discuss the IR data.
References and Notes
- 1.Zerić SD, Popović DM, Knapp E-W. Biochemistry. 2001;40:7914. doi: 10.1021/bi010428q. [DOI] [PubMed] [Google Scholar]
- 2.Valentine JS, Sheridan RP, Allen LC, Kahn P. Proc Natl Acad Sci U S A. 1979;76:1009. doi: 10.1073/pnas.76.3.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salemme FR, Freer ST, Huu Xuong Ng, Alden RA, Kraut J. J Biol Chem. 1973;248:3910. doi: 10.2210/pdb1c2c/pdb. [DOI] [PubMed] [Google Scholar]
- 4.Peisach J, Blumberg WE, Adler A. Ann N Y Acad Sci. 1973;206:310. doi: 10.1111/j.1749-6632.1973.tb43219.x. [DOI] [PubMed] [Google Scholar]
- 5.Nicholls P. Biochim Biophys Acta. 1962;60:217. doi: 10.1016/0006-3002(62)90397-9. [DOI] [PubMed] [Google Scholar]
- 6.Mincey T, Traylor TG. J Am Chem Soc. 1979;101:765. [Google Scholar]
- 7.Teroaka J, Kitagawa T. Biochem Biophys Res Commun. 1980;93:674. doi: 10.1016/0006-291x(80)91133-x. [DOI] [PubMed] [Google Scholar]
- 8.Morrison M, Schonbaum GR. Annu Rev Biochem. 1976;45:861. doi: 10.1146/annurev.bi.45.070176.004241. [DOI] [PubMed] [Google Scholar]
- 9.Peisach J. Ann N Y Acad Sci. 1975;244:187. doi: 10.1111/j.1749-6632.1975.tb41531.x. [DOI] [PubMed] [Google Scholar]
- 10.Berglund GI, Carlsson GH, Smith AT, Szöoke H, Henriksen A, Hajdu J. Nature. 2002;417:463. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 11.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Nat Struct Biol. 1997;4:1032. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 12.Nasri H, Ellison MK, Krebs C, Huynh BH, Scheidt WR. J Am Chem Soc. 2000;122:10795. [Google Scholar]
- 13.Ellison MK, Schulz CE, Scheidt WR. Inorg Chem. 2002;41:2173. doi: 10.1021/ic020012g. [DOI] [PubMed] [Google Scholar]
- 14.Hu C, Roth A, Ellison MK, An J, Ellis CM, Schulz CE, Scheidt WR. J Am Chem Soc. 2005;127:5675. doi: 10.1021/ja044077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu C, Noll BC, Schulz CE, Scheidt WR. J Am Chem Soc. 2005;127:15018. doi: 10.1021/ja055129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu C, An J, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2006;45:4177. doi: 10.1021/ic052194v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nasri H, Ellison MK, Shaevitz B, Gupta GP, Scheidt WR. Inorg Chem. 2006;45:5284. doi: 10.1021/ic052059i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The following abbreviations are used in this paper: Por, a generalized porphyrin dianion; OEP, dianion of octaethylporphyrin; TPP, dianion of meso-tetraphenyl-porphyrin; Tp-OCH 3PP, dianion of meso-tetra-p-methoxyphenylporphyrin; TTP, dianion of meso-tetratolylporphyrin, TpivPP, dianion of α, α, α, α-tetrakis(o-pivalamidophenyl)-porphyrin; Piv2C8P, dianion of α,α ,5,15-[2,2’-(octanediamido)diphenyl]-α,α,10-20-bis(o-pivalamidophenyl)porphyrin; Im, generalized imidazole; RIm, generalized hindered imidazole; HIm, imidazole; 1-MeIm, 1-methylimidazole; 2-MeHIm, 2-methylimidazole; 1,2-Me2Im, 1,2-dimethylimidazole; phen, phenanthroline; DBU, 1,8-Diazabicyclo[5.4.0]undec-7-ene; Np, porphyrinato nitrogen; Ct, the center of four porphyrinato nitrogen atoms.
- 19.Debrunner P. In: Iron Porphyrins Part 3. Lever ABP, Gray HB, editors. Chapter 2 VCH Publishers Inc.; New York: 1983. [Google Scholar]
- 20.Champion PM, Chiang R, Müunck E, Debrunner P, Hager LP. Biochemistry. 1975;14:4159. doi: 10.1021/bi00690a002. [DOI] [PubMed] [Google Scholar]
- 21.Hu C, Noll BC, Piccoli PMB, Schultz AJ, Schulz CE, Scheidt WR. J Am Chem Soc. 2008;130:3127. doi: 10.1021/ja078222l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balch AL, Watkins JJ, Doonan DJ. Inorg Chem. 1979;18:1228. [Google Scholar]
- 23.The increase in imidazole binding constants upon the addition of 1,10-phenanthroline had been noted previously by Abbott and Rafson, but the origin of the effect not established. Abbott EH, Rafson PA, editors. J Am Chem Soc. Vol. 96. 1974. p. 7378.
- 24.Adler AD, Longo FR, Finarelli JD, Goldmacher J, Assour J, Korsakoff L. J Org Chem. 1967;32:476. [Google Scholar]
- 25.Milgram BC, Eskildsen K, Richter SM, Scheidt WR, Scheidt KA. J Org Chem. 2007;72:3941. doi: 10.1021/jo070389+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Adler AD, Longo FR, Kampus F, Kim J. J Inorg Nucl Chem. 1970;32:2443. [Google Scholar]; (b) Buchler JW. In: Porphyrins and Metalloporphyrins. Smith KM, editor. Chapter 5 Elsevier Scientific Publishing; Amsterdam, The Netherlands: 1975. [Google Scholar]
- 27.(a) Fleischer EB, Srivastava TS. J Am Chem Soc. 1969;91:2403. [Google Scholar]; (b) Hoffman AB, Collins DM, Day VW, Fleischer EB, Srivastava TS, Hoard JL. J Am Chem Soc. 1972;94:3620. doi: 10.1021/ja00765a060. [DOI] [PubMed] [Google Scholar]
- 28.Sheldrick GM. Acta Crystallogr Sect A. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 29.R1 = Σ ∥ Fo∣ − ∣Fc ∥ /Σ∣Fo∣ , wR2 = {Σ[w (Fo2− Fc2)2]/Σ[w(F02)2}1/2 and R(F2) = Σ ∥ Fo∣2 − ∣Fc∣2∣/Σ∣Fo∣2. The conventional R-factors R1 are based on F, with F set to zero for negative F2. The criterion of F2 > 2σ(F2) was used only for calculating R1. R-factors based on F2 (wR2) are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger.
- 30.Sheldrick GM. SADABS. Universität Göttingen; Gttingen, Göermany: 2006. [Google Scholar]
- 31.Spek AL. J Appl Cryst. 2003;36:7. [Google Scholar]
- 32.Many of these failed crystallization experiments yielded oils suggesting the presence of several species.
- 33.Quinn R, Mercer-Smith J, Burstyn JN, Valentine JS. J Am Chem Soc. 1984;106:4136. [Google Scholar]
- 34.Lemoine P, Viossat V, Dayan E, Dung N-H, Viossat B. Inorg Chim Acta. 2006;356:4274. [Google Scholar]
- 35.Allen FH. Acta Crystallogr. 2002;B58:380. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]; Bruno IJ, Cole JC, Edgington PR, Kessler MC, Macrae F, McCabe P, Pearson J, Taylor R. Acta Crystallogr. 2002;B58:389. doi: 10.1107/s0108768102003324. [DOI] [PubMed] [Google Scholar]
- 36.Scheidt WR, Reed CA. Chem Rev. 1981;81:543. [Google Scholar]
- 37.Scheidt WR, Gouterman M. In: Iron Porphyrins, Part One. Lever ABP, Gray HB, editors. Addison-Wesley; Reading, MA: 1983. p. 89. [Google Scholar]
- 38.Collman JP, Kim N, Hoard JL, Lang G, Radonovich LJ, Reed CA. Abstracts of Papers; 167th National Meeting of the American Chemical Society; Los Angeles, CA. April 1974; Washington, D. C.: American Chemical Society; p. INOR 29. [Google Scholar]
- 39.Mandon D, Ott-Woelfel F, Fischer J, Weiss R, Bill E, Trautwein AX. Inorg Chem. 1990;29:2442. [Google Scholar]
- 40.Sun L, Shelnutt JA. Program is available via the internet at http://jasheln.unm.edu.
- 41.Kent TA, Spartalian K, Lang G, Yonetani T, Reed CA, Collman JP. Biochem Biophys Acta. 1979;580:245. doi: 10.1016/0005-2795(79)90137-5. [DOI] [PubMed] [Google Scholar]
- 42.Debrunner PG. In: Iron Porphyrins Part 3. Lever ABP, Gray HB, editors. Chapter 2 VCH Publishers Inc.; New York: 1983. [Google Scholar]
- 43.Kent TA, Spartalian K, Lang G. J Chem Phys. 1979;71:4899. [Google Scholar]
- 44.Kent TA, Spartalian K, Lang G, Yonetani T. Biochem Biophys Acta. 1977;490:331. doi: 10.1016/0005-2795(77)90008-3. [DOI] [PubMed] [Google Scholar]
- 45.Bade D, Parak F. Biophys Struct Mechanism. 1976;2:219. doi: 10.1007/BF00535368. [DOI] [PubMed] [Google Scholar]
- 46.Eicher H, Bade D, Parak F. J Chem Phys. 1976;64:1446. [Google Scholar]
- 47.Huynh BH, Paraefthymiou GC, Yen CS, Wu CS. J Chem Phys. 1974;61:3750. [Google Scholar]
- 48.Eicher H, Trautwein A. J Chem Phys. 1970;52:932. doi: 10.1063/1.1673077. [DOI] [PubMed] [Google Scholar]
- 49.Eicher H, Trautwein A. J Chem Phys. 1969;50:2540. doi: 10.1063/1.1671413. [DOI] [PubMed] [Google Scholar]
- 50.Momenteau M, Scheidt WR, Eigenbrot CW, Reed CA. J Am Chem Soc. 1988;110:1207. [Google Scholar]
- 51.Shaevitz BA, Lang G, Reed CA. Inorg Chem. 1988;27:4607. [Google Scholar]
- 52.Bominaar EL, Ding X, Gismelseed A, Bill E, Winkler H, Trautwein AX, Nasri H, Fisher J, Weiss R. Inorg Chem. 1992;31:1845. [Google Scholar]
- 53.Nasri H, Fischer J, Weiss R, Bill E, Trautwein AX. J Am Chem Soc. 1987;109:2549. [Google Scholar]
- 54.Schappacher M, Ricard L, Fisher J, Weiss R, Montiel-Montoya R, Bill E. Inorg Chem. 1989;28:4639. doi: 10.1111/j.1432-1033.1987.tb13436.x. [DOI] [PubMed] [Google Scholar]
- 55.Schappacher M, Ricard L, Weiss R, Montiel-Montoya R, Gonser U, Bill E, Trautwein AX. Inorg Chim Acta. 1983;78:L9. [Google Scholar]
- 56.Hu C, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2005;44:4346. doi: 10.1021/ic050320p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.(a) Wagner GC, Kassner RJ. J Am Chem Soc. 1974;96:5593. doi: 10.1021/ja00824a059. [DOI] [PubMed] [Google Scholar]; (b) Wagner GC, Kassner RJ. Biochim Biophys Acta. 1975;392:319. doi: 10.1016/0304-4165(75)90013-6. [DOI] [PubMed] [Google Scholar]
- 58.Collman JP, Brauman JI, Doxsee KM, Halbert TR, Bumenberg E, Linder RE, LaMar GN, Del Gaudio J, Lang G, Spartalian K. J Am Chem Soc. 1980;102:4182. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S4 show ORTEP diagrams of [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form B molecule 2), [Fe(OEP)(2-MeHIm)]·(1,10-phen) (molecule 1 and 2), and [Fe(TPP)(2-MeHIm)]·(1,10-phen) (form A). Figure S5 shows atomic displacements from the mean plane of the 24-atom core. Table S1 shows analysis results of the core conformations by the normal structural decomposition method. Table S2 gives full details on the fits for applied field Mössbauer measurements of [Fe(OEP)(2-MeHIm)]·(1,10-phen). Table S3 shows variable temperature Mössbauer Parameters for crashed out solid sample of TPP derivative. Tables S4–S21 give complete crystallographic details, atomic coordinates, bond distances and angles, anisotropic temperature factors, and fixed hydrogen atom positions. This information is available as a PDF file. The crystallographic information files (CIF) are also available. This material is available free of charge via the Internet at http://pubs.acs.org.