

Abstract
Microglia play a prominent role in the brain’s inflammatory response to injury or infection by migrating to affected locations, secreting inflammatory molecules, and phagocytosing damaged tissue. However, because severe or chronic neuroinflammation exacerbates many neurological conditions, controlling microglia actions may provide therapeutic benefits in a diverse array of diseases. Since glycogen synthase kinase-3 (GSK3) promotes inflammatory responses in peripheral immune cells, we investigated if inhibitors of GSK3 attenuated microglia responses to inflammatory stimuli. Treatment of BV-2 microglia with GSK3 inhibitors greatly reduced the migration of microglia in both a scratch assay and in a transwell migration assay. Treatment of BV-2 microglia with lipopolysaccharide (LPS) stimulated the production of interleukin-6 and increased the expression of inducible nitric oxide synthase (iNOS) and NO production. Each of these microglia responses to inflammatory stimulation were greatly attenuated by GSK3 inhibitors. However, GSK3 inhibitors did not cause a general impairment of microglia functions, as the LPS-induced stimulated expression of cylcooxygenase-2 was unaltered. Regulation of microglia functions were also evident in cultured mouse hippocampal slices where GSK3 inhibitors reduced cytokine production and microglial migration, and provided protection from inflammation-induced neuronal toxicity. These findings demonstrate that GSK3 promotes microglial responses to inflammation and that the utilization of GSK3 inhibitors provides a means to limit the inflammatory actions of microglia.
Keywords: neuroinflammation, GSK3, lithium, cytokine, lipopolysaccharide, neuroprotection, microglia motility
1. Introduction
Microglia, the resident immune cells of the brain, constantly survey their microenvironment through process extension and retraction in order to respond to homeostatic disturbances, such as injury, disease, or infection [1, 2]. Resting microglia exhibit a ramified morphology, but upon stimulation, retract their processes and undergo directed chemotaxis along gradients of molecules released by injured cells (e.g., extracellular ATP) or by other immune cells (e.g., CCL2/MCP-1). A commonly used marker of microglial activation is the upregulation of the constitutively expressed CD11b, the αMβ2 integrin and complement receptor 3 [3]. Activated microglia produce an array of inflammatory molecules, act as antigen presenting cells, and phagocytose damaged cells [4]. One of the major products of activated microglia is the proinflammatory cytokine interleukin-6 (IL-6). Exposure to lipopolysaccharide (LPS), a component of the outer membrane of gram-negative bacteria, activates microglia to produce IL-6 as well as other inflammatory molecules, such as nitric oxide (NO). The inflammatory responses of microglia are tightly regulated, but it is thought that excessive or chronic microglial activation can contribute to neurodegenerative processes [5]. Therefore, modulation of microglia responses may provide a therapeutic target for the treatment of severe or chronic neuroinflammatory conditions.
We recently reported [6] that LPS-stimulated production of IL-6 by monocytes was attenuated by inhibitors of glycogen synthase kinase-3 (GSK3), a constitutively active serine-threonine kinase [7]. Therefore, in order to investigate if neuroinflammation is regulated by GSK3, in the present study we tested if GSK3 inhibitors are able to attenuate LPS-stimulated proinflammatory cytokines produced by microglia as well as regulating the induction of iNOS and cyclooxygenase-2 (COX-2). GSK3 also regulates the motility of a number of types of cells [8]. Migration is a critical component of the microglial response to inflammatory stimuli, yet no studies have examined if GSK3 regulates microglial migration. Therefore, we tested if GSK3 regulates microglial migration both in vitro and in situ in acute hippocampal slices. Altogether, the results show that GSK3 inhibitors reduce microglial migration and attenuate the production of inflammatory molecules by activated microglia. Importantly, the results also demonstrate that the attenuation of microglial activity by GSK3 inhibitors provides neuroprotection during neuroinflammatory conditions, indicating that GSK3 is a potential therapeutic target to attenuate neuroinflammation.
2. Material and methods
2.1 Reagents and cells
Reagents were obtained from the following sources: LiCl (Sigma, St. Louis, MO), kenpaullone, indirubin-3’-monoxime (Alexis Biochemicals, San Diego, CA), CHIR99021 (University of Dundee), SB216763 and SB415286 (Tocris, Ellisville, MO), CCL2 (R&D Systems, Minneapolis, MN), SB203580, D4476, and roscovitine (Calbiochem, La Jolla, CA). Protein-free E. coli (K235) LPS was a generous gift from Dr. S. Michalek, and was prepared as previously described [9]. Mouse microglia BV-2 cells (a gift from Dr. E. Benveniste) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in 5% CO2 atmosphere at 37°C. Cells were grown to 80% confluency before experimental treatments, and where indicated were washed two times and incubated in serum-free media overnight before treatments.
2.2 Animals
CX3CR1gfp/gfp (Jackson Laboratory, Bar Harbor, ME) and C57BL/6 (Frederick Cancer Research, Frederick, MD) mice were housed in an animal facility with regulated temperature, humidity, and a 12 hr light cycle. All mice were housed and treated in accordance with National Institutes of Health and the University of Alabama at Birmingham Institutional Animal Care and Use Committee guidelines.
2.3 In vitro Migration Assays
Scratch assays were performed as described [10]. Briefly, confluent BV-2 microglia in 6-well plates were washed with serum-free DMEM three times, and preincubated with GSK3 inhibitors for 30 min. A line down the center of each well was scraped with a p200 pipette tip, followed by a wash to remove debris. Images were taken at 10x magnification, scratch widths were measured, and wound closure was calculated by dividing widths measured after a 6 hr incubation by the initial scraped width. Each experiment was carried out in triplicate and three fields were counted per well by scorers blinded to experimental conditions.
Transwell migration assays were performed in modified Boyden chambers (BD Bioscience, New Bedford, MA) as previously described [11], with slight modifications. BV-2 microglia (4 × 104 cells in 200 μl of DMEM) were added to the upper chamber and allowed to adhere to the polycarbonate filters (8 μm pore) for 30 min at 37°C in a humidified atmosphere of 95% air and 5% CO2. GSK3 inhibitors were placed in the lower chamber, CCL2 was placed in either the lower or both chambers, and the cells were allowed to migrate for an additional 5.5 hr. Cells that did not migrate and remained on the upper surface of the filter were removed, and cells that had migrated to the lower surface were stained with the fluorescent nuclear stain DAPI (Sigma) and counted. In at least three independent experiments, three wells per treatment were counted in nine random fields at 40× magnification per well by scorers blind to experimental conditions.
2.4 Measurement of cytokines and nitric oxide (NO)
IL-6 and TNFα were measured with an enzyme-linked immunosorbent assay (ELISA) kit (eBioscience, San Diego, CA) according to the manufacturer’s instructions. Nitrite, a stable breakdown product of NO, was measured with a Griess Reagent System (Promega, Madison, WI).
2.5 Flow cytometry
Surface expression of CD11b on BV-2 microglia was analyzed by flow cytometry. Cells suspended in cold, phenol red-free, DMEM with 3% BSA were incubated in the dark with Alexa-647 conjugated CD11b antibody and propidium iodide (PI) for 30 min on ice. Following incubation, the cell suspension was centrifuged at 500xg for 5 min, washed twice, and resuspended in phenol red-free DMEM. Cells were gated based on morphological characteristics. Apoptotic and necrotic cells were not accepted for flow cytometry analysis, as indicated by PI positive staining (< 1% of cells).
2.6 Immunoblot analysis
Cells were washed twice with PBS, and were lysed with 100 μl lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin A, 1 nM okadaic acid, and 1% Triton-X-100). The lysates were centrifuged at 10,000xg for 10 min at 4°C. Protein concentrations were determined using the Bradford method [12]. Samples were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for 5 min. Proteins were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with primary antibodies to cyclooxygenase-2 (COX-2; 1:1000), inducible nitric oxide synthase (iNOS; 1:1000), (BD Biosciences, San Jose, CA), or α-tubulin (1:10,000) (Sigma). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence, and the protein bands were quantitated with a densitometer. Quantitative measurements from at least three independent experiments were tested for statistical significance using analysis of variance (ANOVA).
2.7 Hippocampal Slices
Organotypic hippocampal slice cultures were prepared as previously described [13] with slight modifications. Briefly, the hippocampus from C57BL/6 postnatal 6-8 day old mice was dissected in cold dissecting buffer (Earl’s Balanced Salt Solution (EBBS), 36 mM D-glucose, 100 U/ml penicillin, and 100 μg/ml streptomycin). The hippocampus was placed on a bed of polymerized agar and sliced into 400 μm sections using a MT-TS tissue slicer (Siskiyou Inc, Grants Pass, OR). Slices were placed in fresh dissecting buffer, separated, and allowed to recover for 30 min at 4°C. Slices were placed on top of cell culture inserts (0.4 μm pore size, 30 mm in diameter; Millipore, Bedford, MA) with 1 ml pre-warmed media under the insert (Minimum Essential Medium (MEM) with Earl’s Salt, 25% EBBS, 25% heat-inactivated horse serum, 36 mM D-glucose, 1 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). Slices were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2, media was replaced after the first day in culture and every 2-3 days subsequently. For cytokine production measurements, two days prior to treatments, slices were incubated in serum-free media (B-27 supplemented Neurobasal media with 1 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin).
For measurements of microglial migration, acute hippocampal slices from CX3CR1+/gfp mice were prepared as described above and imaging was performed as previously described with slight modifications [14]. Slices were transferred to glass-bottom 35 mm dishes containing 4 ml pre-warmed artificial cerebrospinal fluid (129 mM NaCl, 3 mM KCl, 1.8 mM MgSO4, 1.6 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose) and test agents were added. Slices were placed on a 37°C temperature-controlled microscope stage (Warner Instruments, Hamden, CT) and were imaged at 5 min intervals for 4-6 hr at 10x-magnification, which accurately monitored the movements of over 40 microglial cell bodies. Imaging sessions commenced 60 min after tissue slicing. The migration of 40 cells was analyzed by a person blinded to the treatments with Image Pro Plus 6.0 (Media Cybernetics, Bethesda, MD) using the Correlation Track feature to obtain X and Y coordinates for each cell.
For analysis of neurotoxicity, HSCs (10 DIV) were treated with LPS for 24 or 48 hr with or without GSK3 inhibitors. Propidium iodide (13 μM) was added for 30 min to stain the nuclei of degenerating neurons to evaluate cell death [15]. Images were obtained using a 4x objective, converted to binary images, segmented and measured using integrated optical density in ImageJ 1.38d (National Institutes of Health, Bethesda, MD). Double-labeling of neurons was performed by fixing HSCs after imaging in 4% paraformaldehyde in PBS for 2 hr, permeabilization with 0.25% Triton-X 100 in PBS for 30 min, blocking for 30 min in 0.1% BSA, and staining with primary antibody to NeuN (Chemicon, Temecula, CA) overnight at 4°C. Slices were washed and incubated with Alexa 546-conjugated antibody to rabbit IgG (Molecular Probes). Slices were then viewed and photographed using an inverted Olympus fluorescence microscope.
3. Results
3.1 GSK3 regulates both random and directed migration of microglia in vitro
We tested if GSK3 regulates BV-2 microglial migration using two methods, a scratch assay and a transwell migration assay. During a 6 hr incubation period after implementation of a scratch, BV-2 microglia migrated into the scratched zone sufficiently to reduce the depleted area by 53% (Fig. 1A). To assess the effect of GSK3 on microglial migration, cells were incubated with three structurally distinct selective GSK3 inhibitors, lithium [16], indirubin-3’-monoxime [17], or kenpaullone [18], for 30 min prior to implementing the scratch. All GSK3 inhibitors substantially diminished microglial migration, as in the presence of the GSK3 inhibitors lithium, indirubin-3’-monoxime, or kenpaullone microglial migration only reduced the scratched zone size by 16%, 11%, and 13%, respectively, an average 75% reduction in migration (Fig. 1B). As negative controls, migration was not affected by two kinase inhibitors that do not inhibit GSK3, SB203580, an inhibitor of p38 kinase [19], and D4476, an inhibitor of casein kinase 1 (CK1) [20]. These results are the first to indicate that GSK3 promotes microglial migration.
Figure 1. GSK3 inhibitors reduce migration of microglia in vitro.
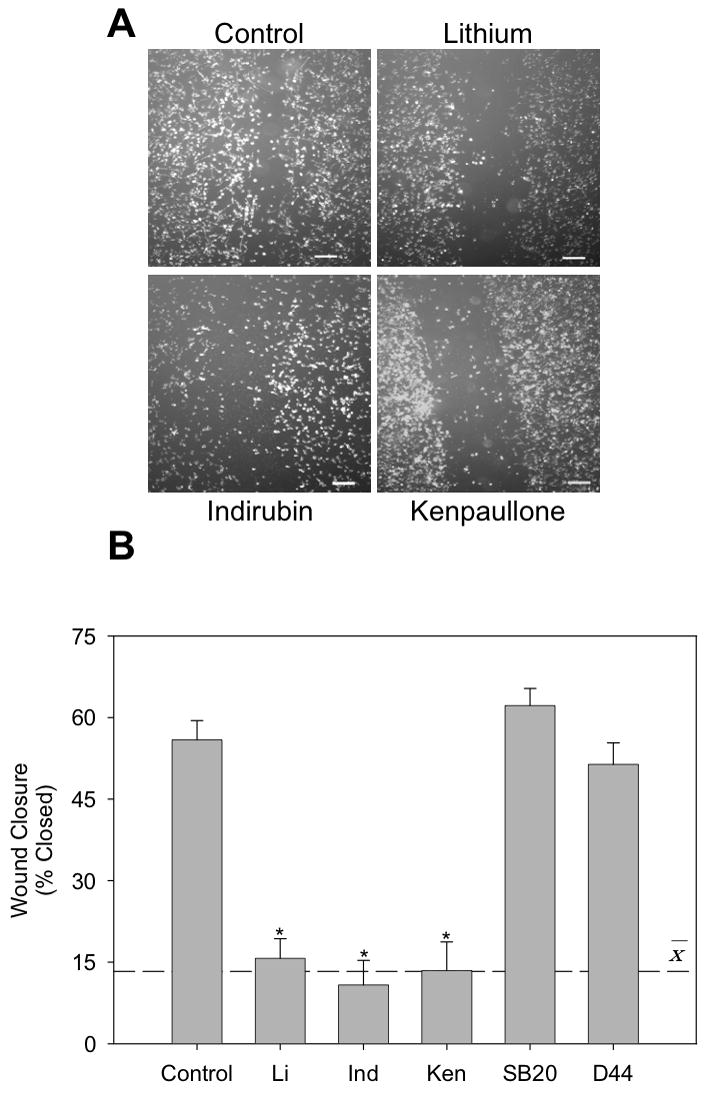
In the scratch migration assay, BV-2 microglia were preincubated for 30 min with GSK3 inhibitors 20 mM lithium (Li), 10 μM indirubin-3’-monoxime (Ind), or 10 μM kenpaullone (Ken), or with non-GSK3 kinase inhibitors 10 μM SB203580 and 10 μM D4476. The scratch was implemented and the cells were imaged immediately and after incubation for 6 hr. (A) Representative images at 6 hr after scratching (scale bar = 200 μm). (B) Wound closure was calculated by dividing widths measured after a 6 hr incubation by the initial scrape width. Values are Means ± SEM; n=3; the dashed line represents the average response with the three GSK3 inhibitors. *p<0.05 compared with control in the absence of GSK3 inhibitor.
The transwell cell migration assay allows assessment of both random and chemotactic migration, and microglia exhibit chemotaxis towards CCL2 in this assay [21-23]. Random microglial migration was measured in the absence or presence of four selective GSK3 inhibitors, lithium, SB216763 [24], kenpaullone, or indirubin-3’-monoxime. Treatment with the GSK3 inhibitors attenuated microglial random migration by 66% (lithium), 41% (SB216763), 58% (kenpaullone), and 84% (indirubin-3’-monoxime), an average inhibition of migration of 62% (Fig. 2A). BV-2 microglial migration was significantly increased in the presence of 50 μM CCL2 in the lower chamber, whereas CCL2 placed in both the upper and lower chambers to eliminate the chemotactic gradient abolished CCL2-induced increased migration (Fig. 2B). Incubation with GSK3 inhibitors prior to CCL2 addition to the upper chamber significantly attenuated chemotactic migration by 60% (lithium) and 81% (indirubin-3’-monoxime) (Fig. 2B). Taken together, these results indicate that GSK3 inhibitors reduce the ability of microglia to migrate both randomly and towards a directed stimulus in vitro.
Figure 2. GSK3 inhibitors attenuate random and directed microglial migration in vitro.

(A) Random migration of BV-2 microglia to the bottom of a transwell migration chamber was measured after treatment with GSK3 inhibitors 20 mM lithium (Li), 10 μM SB216763 (SB21), 10 μM kenpaullone (Ken), or 10 μM indirubin-3’-monoxime (Ind) after a 6 hr incubation. (B) Chemotaxis of BV-2 microglia towards 50 μM CCL2 in the bottom chamber was measured with or without GSK3 inhibitors for 6 hr. The chemotactic gradient was abolished by incubating with CCL2 in the top and bottom (CCL2 t+b) chambers. Control values averaged 34 ± 5 cells per field of view, with 9 high-power fields counted per well. Values are Means ± SEM; n = 3 wells per group in at least 3 independent experiments; the dashed line represents the average response with the GSK3 inhibitors. *p<0.05 compared with untreated control cells; †p<0.05 compared with CCL2-treated cells in the absence of GSK3 inhibitor.
3.2 GSK3 regulates the migration of microglia in situ
We examined if GSK3 regulates migration of microglia in situ in acute hippocampal slices. Using slices from CX3CR1+/GFP heterozygous mice allowed for time-lapse visualization of GFP-labeled microglia without affecting their movement or activation [25]. The preparation of hippocampal slices induces a traumatic injury that results in microglial migration and activation [14]. Monitoring microglial activity for 4 hr after hippocampal slice injury revealed that microglia migrated an average of 58 μm (Fig. 3A and Suppl. Movie 1), and the 10% most active microglia in each slice migrated 121 μm (Fig. 3B). There was a significant reduction of microglial migration in slices in which GSK3 was inhibited with 20 mM lithium (Suppl. Movie 2), as the average distance migrated was reduced by 41% to 34 μm (Fig. 3A) and the migration of the 10% most active microglia was reduced by 38% to 72 μm (Fig. 3B). We also calculated the time-dependence of the maximum migratory velocities of all microglia at each hour to test if inhibition of GSK3 delayed the response (Fig. 3C). However, at all times, the migration velocities of microglia were reduced by inhibition of GSK3, indicating that inhibition of GSK3 did not delay the response of microglia but rather attenuated their migratory response to injury.
Figure 3. Lithium attenuates injury-induced migration of microglia in situ.

Microglial movements in acute hippocampal slices prepared from CX3CR1+/GFP heterozygous mice were measured every 5 min for 4 hr with or without incubation with the GSK3 inhibitor 20 mM lithium. (A) The accumulated migration distance of all microglia counted per slice for each treatment. (B) The accumulated migration distance of the 10% microglia moving the farthest per slice. (C) The maximum velocity calculated at each hour of all microglia counted per treatment. (D) The percentage of microglia per slice migrating less than 25 μm (low mobility) and greater than 75 μm (high mobility) during a 4 hr period after an initial 1 hr equilibration. Values are Means ± SEM; n=3 slices with 40 cells per slice counted; *p<0.05 compared with control in the absence of GSK3 inhibitor.
We also categorized all the counted microglia into low mobility (moving less than 25 μm) and high mobility (moving greater than 75 μm) cells. In control slices, approximately 10% of the microglia exhibited low mobility, and 24% of the microglia exhibited high mobility (Fig. 3D and Suppl. Movie 1). Inhibition of GSK3 with lithium significantly increased the microglia exhibiting low mobility to 38%, a nearly four-fold increase in low mobility or immobile cells, and reduced to only 5% the portion of microglia exhibiting high mobility, an 81% reduction in highly mobile microglia (Fig. 3D and Suppl. Movie 2). Collectively these data demonstrate that GSK3 promotes microglial migration and inhibition of GSK3 attenuates the migration of microglial in response to injury.
3.3 GSK3 promotes IL-6 and NO production
Immortalized BV-2 microglia have been shown to respond to LPS stimulation similar to primary microglia [26, 27], producing IL-6 and up-regulating the expression of iNOS and COX-2 [28-31]. There was a large increase in IL-6 production following stimulation of BV-2 microglia with 100 ng/ml LPS for 6 hr. To test if GSK3 promotes IL-6 production in microglia, BV-2 microglia were preincubated with four structurally distinct selective GSK3 inhibitors, lithium, indirubin-3’-monoxime, kenpaullone or CHIR99021 [32], for 30 min prior to stimulation with LPS (100 ng/ml) for 6 hr. All GSK3 inhibitors significantly attenuated IL-6 production in response to LPS treatment, causing decreases in IL-6 production of 63% (lithium), 65% (indirubin-3’-monoxime), 82% (kenpaullone), and 74% (CHIR99021), an average 71% decrease (Fig. 4A). As negative controls, IL-6 production was not reduced by the p38 inhibitor SB203580 or the CK1 inhibitor D4476. Thus, GSK3 activity is required for full induction of IL-6 production by LPS stimulated microglia.
Figure 4. GSK3 promotes IL-6 and nitric oxide production.

(A) BV-2 microglia were preincubated for 30 min with GSK3 inhibitors 20 mM lithium (Li), 10 μM indirubin-3’-monoxime (Ind), 10 μM kenpaullone (Ken), or 10 μM CHIR99021 (Chir), or with non-GSK3 kinase inhibitors 10 μM SB203580 and 10 μM D4476, followed by treatment with 100 ng/ml LPS for 6 hr and released IL-6 was measured. (B) BV-2 microglia were preincubated for 30 min with GSK3 inhibitors 20 mM Li, 10 μM Ind, 10 μM Ken, or 10 μM Chir, followed by treatment with 1 μg/ml LPS for 24 hr and nitrite, the stable breakdown product of NO, was measured. LPS stimulated the production 227 ± 51 pg/ml of IL-6 and 46 ± 6 μM NO. Values are Means ± SEM; n=3; the dashed line represents the average response with the GSK3 inhibitors present. *p<0.05 compared with LPS treatment in the absence of GSK3 inhibitor.
To test if GSK3 promotes NO production, BV-2 microglia were preincubated with four selective GSK3 inhibitors for 30 min prior to stimulation with LPS (1 μg/ml) for 24 hr followed by measuring nitrite concentration, a marker of NO production. All four GSK3 inhibitors significantly attenuated NO production in response to LPS stimulation; decreasing NO production by 68% (lithium), 80% (indirubin-3’-monoxime), 67% (kenpaullone), and 59% (CHIR99021), an average 69% decrease (Fig. 4B). Therefore, GSK3 promotes both IL-6 and NO production by LPS-stimulated microglia.
3.4 GSK3 promotes NO production through iNOS expression
LPS stimulates NO production by increasing the expression of iNOS, as previously reviewed [33]. Therefore, we assessed whether GSK3 inhibitors limit NO production by decreasing LPS-induced iNOS expression in microglia. Stimulation of BV-2 microglia with LPS (100 ng/ml) for 6 hr caused a large increase in the level of iNOS. Pre-incubation for 30 min with four specific GSK3 inhibitors significantly attenuated LPS-induced iNOS expression by 52% (lithium), 80% (indirubin-3’-monoxime), 92% (kenpaullone), and 65% (CHIR99021), an average of 72% (Figs. 5A,B). As negative controls, LPS-induced iNOS expression was not inhibited by the p38 inhibitor SB203580 or the CK1 inhibitor D4476 (Fig. 5E). These results indicate that GSK3 promotes LPS-induced NO production by facilitating iNOS expression.
Figure 5. GSK3 selectively promotes iNOS, but not COX-2, expression after LPS stimulation.

BV-2 microglia were stimulated with 100 ng/ml LPS for 6 hr after preincubation for 30 min with GSK3 inhibitors 20 mM lithium (Li), 10 μM indirubin-3’-monoxime (Ind), 10 μM kenpaullone (Ken), or 10 μM CHIR99021 (Chir), or with 10 μM roscovitine (Ros), as indicated. Representative immunoblots of (A) iNOS and (C) COX-2 and (B and D) quantified measurements from three independent experiments are shown. (E) iNOS and (F) COX-2 levels were measured after preincubation with the GSK3 inhibitor Ind or with non-GSK3 kinase inhibitors 10 μM SB203580 and 10 μM D4476 for 30 min followed by stimulation with LPS for 6 hr. Values are Means ± SEM; n=3; the dashed line represents the average response with the GSK3 inhibitors. *p<0.05 compared with LPS treatment in the absence of inhibitor.
3.5 GSK3 selectively promotes inflammatory signals
Stimulation of BV-2 microglia with LPS (100 ng/ml) for 6 hr also increased the expression of COX-2 (Figs. 5C,D). Pre-incubation for 30 min with four GSK3 inhibitors resulted in differential effects on COX-2 expression, as lithium, indirubin-3’-monoxime, and CHIR99021 had no effect, but kenpaullone significantly attenuated COX-2 expression. In addition to inhibiting GSK3, kenpaullone also inhibits CDK2 [34]. Therefore, to examine whether the decrease in COX-2 expression caused by kenpaullone was due to CDK2 inhibition, BV-2 microglia were pre-incubated for 30 min with a specific CDK inhibitor, 10 μM roscovitine [35]. Roscovitine significantly attenuated LPS-stimulated COX-2 expression in BV-2 microglia, suggesting that the effect of kenpaullone resulted from inhibition of CDK2 rather than GSK3 (Fig. 5D). LPS-stimulated COX-2 expression also was not inhibited by two non-GSK3 inhibitors the p38 inhibitor SB203580 or the CK1 inhibitor D4476 (Fig. 5F). Taken together, this data indicates that GSK3 is not essential for the upregulation of COX-2 expression stimulated by LPS, demonstrating selectivity in the regulatory effects of GSK3 on inflammatory responses to LPS in microglia.
3.6 GSK3 regulates activation of microglia
CD11b, the αMβ2 integrin, is widely used as a marker of activated microglia [3]. Microglia constitutively express CD11b and it is upregulated upon stimulation with LPS [36]. Stimulation of BV-2 microglia with 100 ng/mL LPS for 24 hr significantly upregulated CD11b surface expression to 145% of untreated control levels as measured by flow cytometry (Figs. 6A,B). Inhibition of GSK3 by lithium pretreatment blocked the LPS-induced increase in CD11b expression (Fig. 6B), indicating that GSK3 is required for the upregulation of CD11b associated with neuroinflammation.
Figure 6. GSK3 regulates LPS-induced upregulation of CD11b.

BV-2 microglia were stimulated with 100 ng/ml LPS for 24 hr after preincubation for 30 min with the GSK3 inhibitor 20 mM lithium, followed by analysis of CD11b surface expression by flow cytometry. (A) Representative flow cytometry profiles. (B) Quantitative values are percent (± SEM) of control geographic mean; n=5; *p<0.05 compared with control untreated cells.
3.7 GSK3 promotes inflammation in organotypic hippocampal slice cultures (HSCs)
The regulatory effects of GSK3 were examined in HSCs to assess neuroinflammatory responses in an environment structurally and morphologically similar to the brain in vivo. HSCs stimulated with LPS exhibited concentration-, time-, and days in vitro (DIV)-dependent increases in IL-6 (Fig. 7A) and TNF-α (Fig. 7B) production. Based on these findings, HSCs were stimulated with 100 ng/ml LPS for 6 hr after 10-14 DIV. Pretreatment of HSCs for 30 min with four selective GSK3 inhibitors significantly attenuated the LPS-induced production of IL-6 by 67% (lithium), 60% (kenpaullone), and 37% (CHIR99021), an average of 55% (Fig. 7C) and inhibited TNF-α production by 61% (lithium), 44% (kenpaullone), and 31% (CHIR99021), an average of 45% (Fig. 7D). These results demonstrate that GSK3 promotes pro-inflammatory cytokine production in situ, as well as in cultured microglia.
Figure 7. GSK3 promotes microglial cytokine production in situ.

Organotypic hippocampal slice cultures (HSCs) exhibited concentration-, time-, and days in vitro (DIV)-dependent increases of (A) IL-6 and (B) TNF-α after LPS stimulation. (C) IL-6 and (D) TNF-α were measured by ELISA after preincubation of HSCs for 30 min with GSK3 inhibitors 20 mM lithium (Li), 10 μM kenpaullone (Ken), or 10 μM CHIR99021 (Chir) followed by treatment with 100 ng/ml LPS for 6 hr. LPS stimulated the production of 575 ± 191 pg/ml IL-6 and 169 ± 56 pg/ml TNF-α. Values are presented as percent of LPS stimulation. Means ± SEM; n=12 (4 slices per well from three different mice in three different experiments); the dashed line represents the average response with the three GSK3 inhibitors. *p<0.05 compared with LPS treatment in the absence of GSK3 inhibitor.
3.8 GSK3 inhibitors protect from inflammation-induced neurotoxicity
Because GSK3 promotes microglial production of inflammatory molecules that can injure neurons and GSK3 inhibitors reduce microglial activation, we tested if inhibition of GSK3 reduces inflammation-induced neurotoxicity. Treatment of HSCs with 100 ng/mL LPS for 24 hr caused a substantial increase in neuronal toxicity as assessed by measuring PI uptake, predominantly in the CA1 region (Fig. 8A). Preincubation of HSCs with three selective GSK3 inhibitors for 30 min significantly reduced LPS-induced neurotoxicity by 48% (kenpaullone), 64% (SB415286), and 66% (CHIR99021), an average 59% decrease in PI uptake compared with LPS treatment alone (Fig. 8B). To verify that the cell damage-mediated PI uptake was predominantly in neurons, HSCs were stained with NeuN to identify the neuronal cell layers [37]. NeuN intensely stained the CA1, CA3, and dentate gyrus of untreated control slices (Fig. 8C). In LPS-treated slices, NeuN staining was selectively lost in the CA1/subiculum region where PI uptake was present (Fig. 8D). These results indicate that GSK3 inhibitors provide protection from LPS-stimulated inflammation-induced neurotoxicity.
Figure 8. GSK3 inhibitors protect from inflammation-induced neurotoxicity in situ.

Organotypic hippocampal slice cultures (HSCs) were pretreated with GSK3 inhibitors, 10 μM kenpaullone (Ken), 10 μM SB415286 (SB41), or 10 μM CHIR99021 (Chir) for 30 min, followed by 100 ng/ml LPS stimulation for 24 hr, then HSCs were incubated with 13 μM propidium iodide (PI) for 20 min and PI uptake was measured. (A) Representative images of PI-stained HSCs with CA1 (arrows) and dentate gyrus (arrowheads) indicated. (B) Quantified PI intensity from HSCs. Values are Means ± SEM; n = 12 (4 slices per well from three different mice in three different experiments); the dashed line represents the average response with the three GSK3 inhibitors. *p<0.05 compared with LPS treatment in the absence of GSK3 inhibitor. HSC neurons stained with NeuN (C) in control conditions and (D) after LPS treatment show loss of NeuN staining in the CA1/subiculum region (top row box) corresponding with an increase in PI-stained cells. Top row shows 5 x magnification (scale bar = 200 μm) and middle row shows inset from boxed region at 10 x magnification (scale bar = 100 μm). Bottom row shows inset from middle row at 20 x magnification (scale bar = 50 μm) indicating the transition between healthy, NeuN-positive neurons and dead, PI-labeled neurons.
4. Discussion
The results of this study reveal that GSK3 is an important regulator of microglia. GSK3 promoted both microglial migration and the production of inflammatory molecules by microglia, and also promoted inflammation-induced neurotoxicity. Specifically, microglial migration assessed either with cultured microglia in two experimental paradigms or in acute hippocampal slices was reduced by GSK3 inhibitors. Moreover, in LPS-stimulated microglia, inhibitors of GSK3 greatly reduced the production of IL-6 and NO, and the upregulated expression of CD11b and iNOS, but not of COX-2. Inhibition of GSK3 also reduced inflammatory cytokine production in hippocampal slice cultures, a more native environment for microglia than cultured monolayers, where GSK3 inhibitors additionally attenuated inflammation-induced neurotoxicity. Thus, GSK3 has important roles in the activation and function of microglia and inhibition of GSK3 attenuates neuroinflammation at least in part by reducing microglial responses to inflammatory stimuli.
Migration of microglia plays a critical role in their response to an injury [4]. Therefore, we tested if GSK3 inhibitors regulated the migration of microglia both in cultured cells and in hippocampal slices. The migration of BV-2 microglia without stimulus allows measurements of both random motility and directed chemotactic migration responses in vitro. Treatment of microglia with GSK3 inhibitors reduced both random and directed migration by nearly 70%. These results demonstrate the importance of GSK3 in supporting microglial migration, extending previous reports that GSK3 regulates the migration of other cell-types, as previously reviewed [8]. The actions of GSK3 on migration, from the initiation of polarization and the extension of processes to the modulation of adhesions, have been studied in a variety of different cell types. For example, GSK3 has been shown to regulate the polarization of astrocytes [38], the anchoring of microtubules at the centrosome in HeLa cells [39], the binding of several microtubule-associated proteins to microtubules in neurons [40-42], and focal adhesion turnover through phosphorylation of focal adhesion kinase [43, 44] and the scaffolding protein, paxillin [45]. Altogether, these results indicate that GSK3 regulates several aspects of cellular functions linked with migration, and inhibition of total cellular GSK3 results in decreased cell migration, such as with epithelial cells [46] and, as reported here, with microglia.
In addition to microglial migration, inflammatory responses of microglia were found to be highly dependent on GSK3 activity. Inhibition of GSK3 attenuated by 70% LPS-induced IL-6 production in microglia, which extends to microglia previous reports that GSK3 inhibitors reduced LPS-stimulated IL-6 production in monocytes [6] and in dendritic cells [47]. GSK3 inhibitors also reduced microglial NO production by ~80%. In activated microglia, NO production is dependent on up-regulation of iNOS expression [48]. Accordingly, GSK3 inhibitors attenuated LPS-induced iNOS expression in microglia. Several previous reports linked GSK3 to the regulation of iNOS expression, as GSK3 inhibitors reduced TNF-α induced iNOS expression in hepatocytes [49] and reduced iNOS expression in inflammatory models of spinal cord trauma [50], arthritis [51], carrageenan-induced pleurisy [52], and acute pancreatitis [53]. In contrast to these reports, lithium was found to potentiate LPS-induced iNOS expression in astrocytes, but not in mouse macrophages [54], and lithium alone slightly increased iNOS expression in hepatocytes and in vivo [55]. These variable findings may signify stimulus- or cell type-selective regulatory effects of GSK3 on iNOS expression. However, the majority of reports indicate that GSK3 predominantly promotes iNOS expression, which was found to be the case in microglia and likely underlies attenuation of NO production by GSK3 inhibitors in activated microglia.
In addition to selectively regulating pro-inflammatory molecule production, GSK3 regulates the expression of CD11b, a marker of microglial activation, as lithium abolished the upregulation of CD11b in microglia after LPS stimulation. GSK3 may promote CD11b expression by regulating transcription factors, such as NF-κB that promotes CD11b expression in neutrophils [56] and is activated by GSK3 [57], or indirectly by promoting the production of inflammatory molecules that can induce CD11b expression, such as IL-6, TNF-α, or NO [36].
GSK3 was not necessary for all microglia inflammatory responses, as GSK3 inhibitors did not alter LPS-induced upregulation of COX-2 in microglia. Previously, a variety of effects of lithium and GSK3 have been reported on COX-2 expression dependent on cell-type and stimulus. Several studies found that GSK3 reduces COX-2 expression, as GSK3 inhibited TNF-α-induced NF-κB activation to decrease COX-2 expression [58] and GSK3 inhibition promoted COX-2 expression by increasing β-catenin [59-61]. In contrast, activation of the Akt signaling pathway, which inhibits GSK3, or direct inhibition of GSK3 enhanced the basal expression of COX-2 and its stimulated expression induced by UVB-irradiation [62] or phorbol myristate acetate [63]. Therefore, it is possible that these two opposing actions within microglia negated any effect of GSK3 inhibitors on COX-2 expression in microglia, or that the regulatory effect reported by others depends on cell-type and stimulus.
GSK3 strongly promotes both microglial migration and inflammatory molecule production, actions that are important in the microglial response to stimuli and are attenuated by GSK3 inhibitors. Understanding the role of GSK3 in microglial activation and migration may provide novel targets for controlling the neuroinflammatory component of neurodegenerative diseases. This suggestion is supported by the finding that GSK3 inhibitors attenuated inflammation-induced neurotoxicity in HSCs. This extends to neuroinflammation previous reports that lithium and other GSK3 inhibitors provide neuroprotection from a variety of neurotoxic insults, as previously reviewed [64]. This neuroprotection by GSK3 inhibitors in an inflammatory condition is in accordance with a recent report that urocortin provided neuroprotection after LPS-stimulated neurotoxicity by reducing GSK3 activity [65]. Altogether, these findings demonstrate that GSK3 inhibitors provide neuronal protection and suggest that at least part of this protection is due to attenuation of microglial functions.
Conclusions
Microglia play an important role in neuroinflammatory conditions. Initial inflammatory responses appear to be beneficial for contributing to the resolution of the inflammation-inducing insult, but chronic inflammation appears be deleterious and to contribute to the progression of neurodegenerative diseases [5]. Understanding the intracellular signaling processes that regulate microglial responses is critical for developing novel therapeutics for neuroinflammatory conditions. The present study identified GSK3 as an important intracellular protein that regulates both microglial migration and activation. Moreover, the ability of GSK3 inhibitors to attenuate microglia actions in response to inflammatory signals could prove beneficial in lessening the secondary destruction caused by inflammation and to provide neuroprotection in neuroinflammatory conditions. Altogether, the results indicate that GSK3 inhibitors attenuate the microglial responses of migration and inflammatory molecule release, and that GSK3 inhibitors may provide a novel therapeutic intervention for some conditions that have a neuroinflammatory component.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. S. Michalek for providing the purified LPS, Dr. E. Benveniste for providing the BV-2 cells, Dr. E. Beurel for critical discussions, Dr. H. Sontheimer for use of his microscope facilities and assistance, Anna Zmijewska and Gordon Meares for experimental assistance, and Dr. K. Roth and the UAB Neuroscience Core Facilities (NS47466, NS57098). This research was supported by a Civitan Emerging Scholars award and grants from the National Institutes of Health (MH38752, NS3768, and AG021045).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. Nat Neurosci. 2005;8:752. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 2.Nimmerjahn A, Kirchhoff F, Helmchen F. Science. 2005;308:1314. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 3.Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Brain Res Rev. 2005;48:196. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Hanisch UK, Kettenmann H. Nat Neurosci. 2007;10:1387. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 5.Kim SU, de Vellis J. J Neurosci Res. 2005;81:302. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 6.Martin M, Rehani K, Jope RS, Michalek SM. Nat Immunol. 2005;6:777. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimes CA, Jope RS. Prog Neurobiol. 2001;65:391. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 8.Jope RS, Yuskaitis CJ, Beurel E. Neurochem Res. 2007;32:577. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. J Immunol. 2000;165:618. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 10.Liang CC, Park AY, Guan JL. Nat Protoc. 2007;2:329. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 11.Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N. J Neurosci. 2003;23:1398. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradford MM. Anal Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 13.De Simoni A, Yu LM. Nat Protoc. 2006;1:1439. doi: 10.1038/nprot.2006.228. [DOI] [PubMed] [Google Scholar]
- 14.Kurpius D, Wilson N, Fuller L, Hoffman A, Dailey ME. Glia. 2006;54:58. doi: 10.1002/glia.20355. [DOI] [PubMed] [Google Scholar]
- 15.Noraberg J, Kristensen BW, Zimmer J. Brain Res Protoc. 1999;3:278. doi: 10.1016/s1385-299x(98)00050-6. [DOI] [PubMed] [Google Scholar]
- 16.Klein PS, Melton DA. Proc Natl Acad Sci U S A. 1996;93:8455. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, Eisenbrand G, Meijer L. J Biol Chem. 2001;276:251. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 18.Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderowicz AM, Sausville EA, Kunick C, Meijer L. Eur J Biochem. 2000;267:5983. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]
- 19.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. FEBS Lett. 1995;364:229. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 20.Rena G, Bain J, Elliott M, Cohen P. EMBO Rep. 2004;5:60. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cross AK, Woodroofe MN. J Neurosci Res. 1999;55:17. doi: 10.1002/(SICI)1097-4547(19990101)55:1<17::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 22.El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. Glia. 2006;53:132. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, Ling EA, Dheen ST. J Neurochem. 2007;102:667. doi: 10.1111/j.1471-4159.2007.04535.x. [DOI] [PubMed] [Google Scholar]
- 24.Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Chem Biol. 2000;7:793. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- 25.Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. Nat Neurosci. 2006;9:1512. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 26.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. J Neuroimmunol. 1990;27:229. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- 27.Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H. J Neurosci Res. 1992;31:616. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- 28.Gibbons H, Sato TA, Dragunow M. Mol Brain Res. 2003;110:63. doi: 10.1016/s0169-328x(02)00585-5. [DOI] [PubMed] [Google Scholar]
- 29.Hu LF, Wong PT, Moore PK, Bian JS. J Neurochem. 2007;100:1121. doi: 10.1111/j.1471-4159.2006.04283.x. [DOI] [PubMed] [Google Scholar]
- 30.Laurenzi MA, Arcuri C, Rossi R, Marconi P, Bocchini V. Neurochem Res. 2001;26:1209. doi: 10.1023/a:1013911205494. [DOI] [PubMed] [Google Scholar]
- 31.Wang MJ, Huang HM, Chen HL, Kuo JS, Jeng KC. J Neurochem. 2001;77:830. doi: 10.1046/j.1471-4159.2001.00295.x. [DOI] [PubMed] [Google Scholar]
- 32.Wagman AS, Johnson KW, Bussiere DE. Curr Pharm Des. 2004;10:1105. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 33.Kleinert H, Pautz A, Linker K, Schwarz PM. Eur J Pharmacol. 2004;500:255. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 34.Davies SP, Reddy H, Caivano M, Cohen P. Biochem J. 2000;351:95. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Eur J Biochem. 1997;243:527. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 36.Roy A, Fung YK, Liu X, Pahan K. J Biol Chem. 2006;281:14971. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mullen RJ, Buck CR, Smith AM. Development. 1992;116:201. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 38.Etienne-Manneville S, Hall A. Nature. 2003;421:753. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 39.Fumoto K, Hoogenraad CC, Kikuchi A. EMBO J. 2006;25:5670. doi: 10.1038/sj.emboj.7601459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cho JH, Johnson GV. J Neurochem. 2004;88:349. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 41.Owen R, Gordon-Weeks PR. Mol Cell Neurosci. 2003;23:626. doi: 10.1016/s1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 42.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. Cell. 2005;120:137. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 43.Bianchi M, De Lucchini S, Marin O, Turner DL, Hanks SK, Villa-Moruzzi E. Biochem J. 2005;391:359. doi: 10.1042/BJ20050282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, Kikuchi A. Mol Cell Biol. 2006;26:898. doi: 10.1128/MCB.26.3.898-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai X, Li M, Vrana J, Schaller MD. Mol Cell Biol. 2006;26:2857. doi: 10.1128/MCB.26.7.2857-2868.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farooqui R, Zhu S, Fenteany G. Exp Cell Res. 2006;312:1514. doi: 10.1016/j.yexcr.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 47.Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, Giese T, Ho AD, Zoller M, Dreger P, Luft T. Blood. 2007;109:1584. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
- 48.Saha RN, Pahan K. Antioxid Redox Signal. 2006;8:929. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwabe RF, Brenner DA. Am J Physiol Gastrointest Liver Physiol. 2002;283:G204. doi: 10.1152/ajpgi.00016.2002. [DOI] [PubMed] [Google Scholar]
- 50.Cuzzocrea S, Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muia C, Collin M, Esposito E, Bramanti P, Thiemermann C. J Pharmacol Exp Ther. 2006;318:79. doi: 10.1124/jpet.106.102863. [DOI] [PubMed] [Google Scholar]
- 51.Cuzzocrea S, Mazzon E, Di Paola R, Muia C, Crisafulli C, Dugo L, Collin M, Britti D, Caputi AP, Thiemermann C. Clin Immunol. 2006;120:57. doi: 10.1016/j.clim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 52.Cuzzocrea S, Crisafulli C, Mazzon E, Esposito E, Muia C, Abdelrahman M, Di Paola R, Thiemermann C. Br J Pharmacol. 2006;149:687. doi: 10.1038/sj.bjp.0706902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cuzzocrea S, Malleo G, Genovese T, Mazzon E, Esposito E, Muia C, Abdelrahman M, Di Paola R, Thiemermann C. Crit Care Med. 2007;35:2811. doi: 10.1097/01.ccm.0000295303.62996.9f. [DOI] [PubMed] [Google Scholar]
- 54.Feinstein DL. J Neurochem. 1998;71:883. doi: 10.1046/j.1471-4159.1998.71020883.x. [DOI] [PubMed] [Google Scholar]
- 55.Du Q, Park KS, Guo Z, He P, Nagashima M, Shao L, Sahai R, Geller DA, Hussain SP. Cancer Res. 2006;66:7024. doi: 10.1158/0008-5472.CAN-05-4110. [DOI] [PubMed] [Google Scholar]
- 56.Zhou X, Gao XP, Fan J, Liu Q, Anwar KN, Frey RS, Malik AB. Am J Physiol Lung Cell Mol Physiol. 2005;288:L655. doi: 10.1152/ajplung.00327.2004. [DOI] [PubMed] [Google Scholar]
- 57.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Nature. 2000;406:86. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 58.Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB. J Biol Chem. 2004;279:39541. doi: 10.1074/jbc.M403449200. [DOI] [PubMed] [Google Scholar]
- 59.Haertel-Wiesmann M, Liang Y, Fantl WJ, Williams LT. J Biol Chem. 2000;275:32046. doi: 10.1074/jbc.M000074200. [DOI] [PubMed] [Google Scholar]
- 60.Kim SJ, Im DS, Kim SH, Ryu JH, Hwang SG, Seong JK, Chun CH, Chun JS. Biochem Biophys Res Commun. 2002;296:221. doi: 10.1016/s0006-291x(02)00824-0. [DOI] [PubMed] [Google Scholar]
- 61.Rao R, Hao CM, Breyer MD. J Biol Chem. 2004;279:3949. doi: 10.1074/jbc.M309325200. [DOI] [PubMed] [Google Scholar]
- 62.Tang Q, Gonzales M, Inoue H, Bowden GT. Cancer Res. 2001;61:4329. [PubMed] [Google Scholar]
- 63.Thiel A, Heinonen M, Rintahaka J, Hallikainen T, Hemmes A, Dixon DA, Haglund C, Ristimaki A. J Biol Chem. 2006;281:4564. doi: 10.1074/jbc.M512722200. [DOI] [PubMed] [Google Scholar]
- 64.Beurel E, Jope RS. Prog Neurobiol. 2006;79:173. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang MJ, Lin SZ, Kuo JS, Huang HY, Tzeng SF, Liao CH, Chen DC, Chen WF. J Immunol. 2007;179:6204. doi: 10.4049/jimmunol.179.9.6204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.