

Abstract
CYP2C9 is a significant P450 protein responsible for drug metabolism. With the increased use of heterocyclic compounds in drug design, a rapid and efficient pre-drug screening of these potential type II binding compounds is essential to avoid adverse drug reactions. To understand binding modes, we use quinoline-4-carboxamide analogs to study the factors that determine the structure-activity relationships. The results of this study suggest that the more accessible pyridine with the nitrogen para to the linkage can coordinate directly with the ferric heme iron, but this is not seen for the meta or ortho isomers. The π-cation interaction of the naphthalene moiety and Arg 108 residue may also assist in stabilizing substrate binding within the active-site cavity. The type II substrate binding affinity is determined by the combination of steric, electrostatic, and hydrophobicity factors; meanwhile, it is enhanced by the strength of lone pair electrons coordination with the heme iron.
INTRODUCTION
Cytochrome P450 enzymes (P450s) constitute a superfamily of heme-containing monooxygenases that are responsible for a large range of metabolic reactions on endogenous and exogenous substrates. Three of the major P450 isoforms involved in drug metabolism are CYP2C9, CYP2D6, and CYP3A4, which typically metabolize anions, cations and large lipophilic molecules, respectively. Given the structural diversity of drugs, broad substrate selectivity is required for these isoforms to mediate metabolism. This makes the metabolic P450 enzymes different than other enzymes, including P450 enzymes that catalyze endogenous reactions, in that multiple metabolites are often formed from very different binding orientations. 1, 2 Even very tight binding inhibitors have been shown to bind with multiple orientations in the catalytic sites of metabolic P450 enzymes. 3 Given this broad substrate selectivity, it is not surprising that potentially severe drug-drug interactions can occur due to P450s binding multiple compounds with high affinity. 4 5, 6 Additionally, given that the physiological concentrations of administered drugs are normally near 1μM, it is generally accepted that compounds that bind with a Ki less than 10 μM may cause drug-drug interactions.
To minimize these interactions, it is important to understand the mechanisms involved in substrate binding to P450s, which are described by two binding modes called Type I and Type II. 7 Type I and Type II substrate binding modes have been extensively studied using UV spectroscopy 8-12, which characterizes spectral changes in the heme-Soret absorption band. In Type I binding, the substrate binds in a hydrophobic pocket located near the catalytic site of P450. This binding displaces a water molecule that was coordinated to the catalytic face of the heme iron. The resulting five-coordinated heme iron shifts from a low-spin to high-spin state with an accompanying blue shift in the Soret band of the absolute UV spectrum. Type I binding makes the heme more easily reduced by P450 reductase, starting the catalytic cycle of oxidative metabolism. A Type II binding mode describes a situation where a substrate binds as a sixth ligand to the heme iron. In addition to the displacement of the coordinated water, the compound itself serves as a ligand. The heme iron remains in low-spin state, which makes reduction by P450 reductase more difficult. Type II substrates typically contain a sp2 hybridized nitrogen atom as part of a heteroaromatic system where the nitrogen lone pair coordinates with the heme iron. Type II substrates show an increase in binding affinity and results in an overall rate decrease in the catalytic cycle of oxidative metabolism. Even though binding modes are divided into two categories, the existence of mixed or multiple binding modes is inevitable. There could be an equilibrium between Type I and Type II binding. 10
In addition to UV spectroscopy, electron paramagnetic resonance (EPR) spectroscopy has been used to differentiate between Type I and Type II binding modes by directly probing spin state changes in the heme iron for both bacterial and mammalian P450 enzymes. 8, 12-14 This technique provides information regarding the spin state of ferric iron, which has as many as five unpaired electrons. Depending on substrate binding, different spin state heme signals will appear on the spectra as a result of ligand binding. A high-spin state ferric iron (S=5/2) gives EPR signals as g = 7.95, 3.78, and 1.74; a low-spin state ferric iron (S=1/2) gives signals as g = 2.42, 2.25, and 1.92.
In order to improve metabolic stability and inhibitory potency, current practices in drug design incorporate nitrogen heterocycles (pyridines, imidazoles, and triazoles) moieties into drug molecules. 11, 15 Nitrogen incorporation has the potential of creating a Type II binder that, in addition to affecting metabolic rate, could potentially inhibit co-administered drugs and lead to toxicity. Therefore, to facilitate rational drug design, it is necessary to understand and be able to predict the binding mode of a given compound as well as to balance the inhibitory potency with the metabolic stability.
Finally, some P450 enzymes are therapeutic targets. Examples include; aromatase for breast cancer 16, CYP2A6 for smoking cessation 17, 18 and CYP51 for high cholesterol 19, 20, yeast infections, and fungal infections (tinea pedis, tinea cruris, and candida in HIV-AIDS. 21 CYP51 is also a target for parasitic diseases including American trypanosomiasis (Chagas disease) and African trypanosomiasis (sleeping sickness). Many of the currently available therapeutic agents used to treat these diseases are high affinity Type II binders 21. While Type II binding enhances affinities for P450 enzymes relative to other non-P450 targets, the non-specific inhibition of P450 enzymes using these therapeutics leads to toxicity. For example, Azole antifungal agents that target CYP51 can cause birth defects similar to those observed with a phenotypic variation in the P450 reductase function that is related to a decrease in steroid metabolism. 22 In order to design a viable therapeutic, the compound must specifically target P450 disease related isoforms while not inhibiting drug metabolizing P450 isoforms. Since Type II binding potentially enhances binding to all P450 isoforms, it is particularly important to understand and be able to attenuate this binding interaction.
CYP2C9 (2C9) is one of the most significant drug metabolizing P450 isoforms, metabolizing many non-steroidal anti-inflammatory drugs including ibuprofen, flurbiprofen, diclofenac, the antidiabetic sulfonylureas, and the anticoagulant (S)-warfarin 4, 23, 24. During routine screening of diverse inhibitors of 2C9 we found a quinoline-4-carboxamide compound that bound with a low Ki value. In an effort to elucidate the role of nitrogen coordination to heme plays in Type II substrate binding interactions with 2C9, we have synthesized a series of quinoline-4-carboxamide analogs (Figure 1 and Table 1). In addition to other structural changes, we have intentionally varied the position of the sp2 nitrogen atom in the ring system to study heme coordination in Type II binding interactions. Binding affinity, spectroscopy, computational models, and docking experiments to the known crystal structure of 2C9 are used to determine the strength and nature of these interactions. Our analogs were designed to probe binding interactions as a result of both apoprotein interactions and Type II coordination while maintaining the core binding motif in order to decrease differences in binding not related to coordination to the heme iron.
Figure 1.
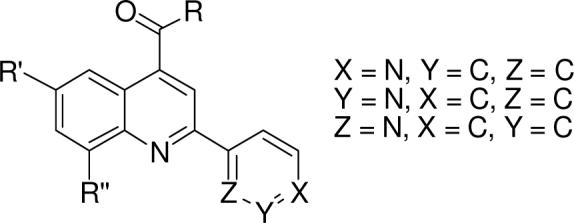
Quinoline-4-carboxamide analogs.
Table 1.
Ki values of Type II binding analogs
| Group | Compd | Structure (para) | Ki +/− SC (μM) | Compd | Structure (meta) | Ki +/− SD (μM) | Compd | Structure (ortho) | Ki +/− SD (μM) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 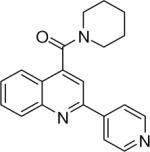 |
9.94 +/− 0.053 | 2 | 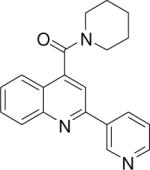 |
9.74 +/− 0.031 | 3 | 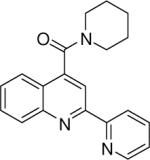 |
23.61 +/− 6.08 |
| 2 | 4 | 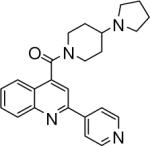 |
3.67+/−0.082 | 5 | 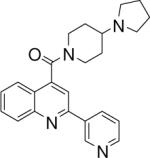 |
24.08+/−1.03 | 6 | 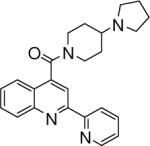 |
76.19 +/− 11.78 |
| 3 | 7 | 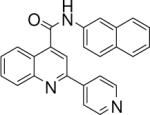 |
0.033 +/− 0.002 | 8 |  |
1.15 +/− 0.002 | 9 |  |
10.67 +/− 1.32 |
| 4 | 10 |  |
0.125 +/− 0.014 | 11 |  |
0.152 +/− 0.004 | 12 | 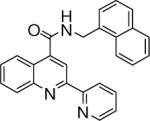 |
>43.5 |
| 5 | 13 | 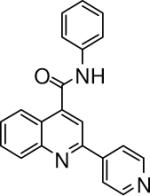 |
0.10 +/− 0.008 | 14 | 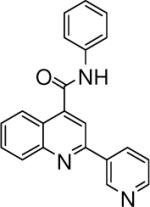 |
4.06 +/− 1.53 | 15 | 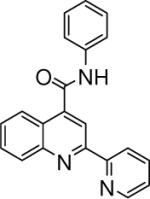 |
3.47 +/− 1.40 |
| 6 | 16 | 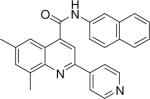 |
0.011 +/− 0.001 | 17 | 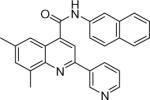 |
8.55 +/− 1.02 | 18 | 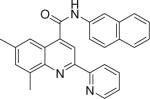 |
>20.6 |
| 7 | 19 | 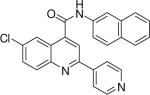 |
0.012 +/− 0.0007 | 20 | 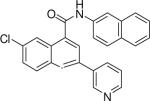 |
1.89 +/− 0.20 | N/Aa | ||
| 8 | 21 | 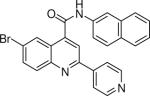 |
0.009 +/− 0.001 | 22 | 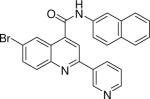 |
>37.85 | N/Aa | ||
| 9 | N/Aa | 23 | 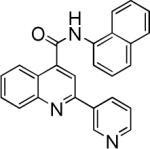 |
1.25 +/− 0.43 | 24 |  |
1.32+/− 0.10 |
compounds not available
EXPERIMENTAL METHODS
Enzymes, chemicals, and instruments
2C9 Baculosomes were prepared as described previously 25. Diclofenac and β-NADPH were purchased from Sigma-Aldrich (St. Louis, MO). HPLC-grade solvents were purchased from EMD Chemicals Inc. (Gibbstown, NJ) and used without purification. All other chemicals were purchased from commercial sources and used without purification. The 1H and 13C NMR spectra were gathered on a Varian Mercury Vx 300 MHz spectrometer using TMS as internal standard except the 13C NMR spectra of compound 19, 20, 21, 22, which were collected by Varian Inova 500 MHz. A Hewlett Packard 8453 single-beam spectrophotometer with HP 845x UV-visible system (Hewlett Packard, Palo Alto, CA) was used to determine substrate concentrations. HPLC system was from Agilent 1100 Series with an Alltima C18 5 μm 3.2 × 150 mm column from Alltech. Values of pKa were estimated using MarvinSketch (ChemAxon Kft. Budapest, Hungary).
Baculosomes incubation conditions
Diclofenac was used as a 2C9 substrate and Ki values were determined from the formation rates of 4-hydroxydiclofenac. Four different final concentrations of diclofenac (5, 10, 20, and 30 μM) in 100 mM potassium phosphate buffer at pH 7.4 were used. Three different inhibitor concentrations were added to each different concentration of diclofenac. 2C9 Baculosomes (1 pmole) were added to the incubation mixtures containing the substrate and selected inhibitor and followed by preincubation for 5 minutes at 37°C. NADPH was added to a final concentration of 1 mM to initiate the reaction. The final volume of all incubations was 500 μL. Reactions were quenched after 5 minutes by adding 200μL of 6% acetic acid in acetonitrile containing an internal standard (0.05 mM phenacetin). The enzyme was removed by centrifugation and product formation was analyzed using HPLC separation with UV detection. The HPLC system used reverse phase chromatography beginning with 30% of mobile phase A: MeOH and 70% of mobile phase B: (30% acetonitrile: 70% water containing 1mM perchloric acid) with a linear gradient to 95% of MeOH over 6 minutes 26. Each compound's Ki was determined from Eq. 1 using GraphPad Prism4 (San Diego, CA) graphing software. The low enzyme concentration of 1 pM means that substrate depletion does not need to be corrected for even for the tightest binding inhibitors.
| (eq. 1) |
Ki determination from IC50 measurements
Measurements were performed using diclofenac at Km concentrations. Inhibitors at three different concentrations were added across to the reaction mixtures containing the same substrate concentration and P450 enzyme (1 pmole). The same incubation procedure and HPLC analysis (detailed above) were used to measure product formation rates. IC50 and Ki values were calculated by GraphPad Prism4 graphing software using equation 2
| (eq. 2) |
Reconstituted 2C9 system
2C9 protein premix was prepared as described previously 27 using purified 2C9 protein 28. The incubation conditions were same as baculosomes incubation assay except 20 pmol of purified 2C9 protein was used in each sample with an incubation time of 10 minutes.
Synthesis of proposed Type II binders based on quinoline-4-carboxamide
All compounds were synthesized as outlined in Scheme 1 with slight modifications for each analog.
Scheme 1.

The general procedure for quinoline-4-carboxamide analogs is given with compound 7: to a 100 mL round bottom flask equipped with a water condenser and stir bar were combined potassium hydroxide (1.71 g, 0.030 mol) and ethanol (8 mL). The reaction mixture was stirred at 80 °C until the the potassium hydroxide was completely dissolved. At this time isatin (1.61 g, 0.011 mol) was added followed by a dropwise addition of 4-acetylpyridine (1.57 g, 0.013 mol). The mixture was refluxed with stirring at 80 °C for 3 days. At the end of this time, the reaction solvent was evaporated using a rotary evaporator and the residue was dissolved in 50 mL water and neutralized with 1N HCl to pH ∼ 6. The resulting solid was collected by vacuum filtration and washed with ethanol (25 mL × 2), acetone (25 mL × 2) and pentane (25 mL × 2) to give crude 2-(pyridin-4-yl)quinoline-4-carboxylic acid, which was used directly to generate the acyl chloride in the next step. The acyl chlorides were formed by combining the crude 2-(pyridin-4-yl)quinoline-4-carboxylic acid (formed above, 500 mg, 2.00 mmol) with neat thionyl chloride (10 mL) in a 100 mL round bottom flask fitted with an air condenser under reflux for two hours. At the end of this time, the condenser was removed and the excess thionyl chloride was evaporated using a stream of argon to give 2-(pyridin-4-yl)quinoline-4-carbonyl chloride. The crude 2-(pyridin-4-yl)quinoline-4-carbonyl chloride (200 mg, 0.744 mmol) was dissolved in a 1:1:1 mixture of acetone:dichloromethane:triethylamine (15 mL) in a 100 mL round bottom flask. To this flask, 2-naphthylamine (117.2 mg, 0.818 mmol) was added and the reaction mixture refluxed for two hours. Following solvent evaporation, the crude compound was purified by chromatography (30 g, silica gel 60, 0.063−0.200 mm) using ethyl acetate. Crystallization from diisopropyl ether gave pure N-(naphthalen-2-yl)-2-(pyridin-4-yl)quinoline-4-carboxamide in 14.67 % yield overall.
Piperidin-1-yl(2-(pyridin-4-yl)quinolin-4-yl)methanone (1)
The reaction conditions were the same as above except piperidine was used instead of 2-naphthylamine to form the amide to give an overall yield of 69.5 %. mp 136−137 °C 1H NMR (CDCl3) δ 8.79 (dd, J = 1.5, 4.8 Hz, 2H), 8.23 (d, J = 8.4 Hz, 1H), 8.06 (dd, J = 1.5, 4.5 Hz, 2H), 7.87−7.78 (m, 3H), 7.63 (m, 1H), 3.90 (m, 2H), 3.17 (t, J = 5.7 Hz, 2H), 1.83−1.68 (m, 4H), 1.49−1.43 (m, 2H); 13C NMR (CDCl3) δ 166.78, 154.51, 150.84, 148.64, 146.27, 144.61, 130.95, 130.77, 128.37, 124.86, 124.12, 121.75, 115.38, 48.54, 43.00, 26.91, 25.97, 24.61. ESI-MS m/z 318.5 (M + 1).
Piperidin-1-yl(2-(pyridin-3-yl)quinolin-4-yl)methanone (2)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and piperidine was used instead of 2-naphthylamine to form the amide. The Overall yield was 71.4%. mp 146−147 °C 1H NMR (CDCl3) δ 9.36 (dd, J = 0.9, 2.1 Hz, 1H), 8.72 (dd, J = 1.8, 4.8 Hz, 1H), 8.50 (m, 1H), 8.21 (d, J = 8.1 Hz, 1H), 7.86−7.76 (m, 3H), 7.60 (m, 1H), 7.47 (m, 1H), 3.90 (m, 2H), 3.18 (t, J = 5.4 Hz, 2H), 1.82−1.68 (m, 4H), 1.49−1.44 (m, 2H); 13C NMR (CDCl3) δ 166.86, 154.67, 150.79, 149.03, 148.73, 144.48, 135.13, 134.81, 130.84, 130.54, 127.93, 124.88, 123.96, 123.67, 115.37, 48.54, 42.99, 26.91, 25.98, 24.63. ESI-MS m/z 318.5 (M + 1).
Piperidin-1-yl(2-(pyridin-2-yl)quinolin-4-yl)methanone (3)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine and piperidine was used instead of 2-naphthylamine to form the amide. Overall yield was 33.9%. mp 157−158 °C 1H NMR (CDCl3) δ 8.73 (m, 1H), 8.66 (m, 1H), 8.51 (s, 1H), 8.21 (d, J = 8.7 Hz, 1H), 7.91−7.85 (m, 2H), 7.76 (m, 1H), 7.58 (m, 1H), 7.37 (m, 1H), 3.89 (m, 2H), 3.21 (t, J = 5.7 Hz, 2H), 1.82−1.66 (m, 4H), 1.56−1.39 (m, 2H); 13C NMR (CDCl3) δ 167.20, 156.10, 155.93, 149.44, 148.32, 143.88, 137.22, 130.54, 130.33, 127.84, 125.02, 124.62, 124.54, 121.99, 115.91, 48.56, 42.90, 26.90, 26.00, 24.70. ESI-MS m/z 318.3 (M + 1), 233.1 (M – 85), 207.2 (M – 111).
(2-(pyridin-4-yl)quinolin-4-yl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (4)
The reaction conditions were the same as above except 4-(1-pyrrolidinyl)piperidine was used instead of 2-naphthylamine to form the amide. The crude compound was purified with chromatography using 10% MeOH in CH2Cl2. Overall yield was 39.4%. mp 164−165 °C 1H NMR (CDCl3) δ 8.79 (s, 2H), 8.23 (d, J = 8.7 Hz, 1H), 8.06 (m, 2H), 7.91−7.78 (m, 3H), 7.67−7.59 (m, 1H), 4.85 (m, 1H), 3.42 (d, J = 13.8 Hz, 1H), 3.16−2.99 (m, 2H), 2.69 (m, 4H), 2.45−2.38 (m, 1H), 2.19−2.13 (m, 1H), 1.86−1.76 (m, 6H), 1.60−1.49 (m, 1H); 13C NMR (CDCl3) δ 166.72, 154.41, 150.80, 148.50, 146.09, 144.11, 130.96, 130.65, 128.80, 124.83, 123.97, 121.70, 115.37, 61.84, 51.75, 45.93, 40.46, 31.79, 30.52, 23.33. ESI-MS m/z 387.5 (M + 1).
(2-(pyridin-3-yl)quinolin-4-yl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (5)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and 4-(1-pyrrolidinyl)piperidine was used instead of 2-naphthylamine to form the amide. The crude compound was purified by chromatography using 10% MeOH in CH2Cl2. Overall yield was 50.2%. mp 164−165 °C 1H NMR (CDCl3) δ 9.37 (dd, J = 1.8, 18.6 Hz, 1H), 8.73−8.71 (m, 1H), 8.53−8.47 (m, 1H), 8.23−8.19 (m, 1H), 7.92−7.77 (m, 3H), 7.67−7.56 (m, 1H), 7.49−7.45 (m, 1H), 4.90 (m, 1H), 3.46 (d, J = 14.4 Hz, 1H), 3.14−2.98 (m, 2H), 2.75 (m, 4H), 2.17 (m, 1H), 2.03−1.49 (m, 7H); 13C NMR (CDCl3) δ 166.72, 154.59, 150.73, 148.99, 148.90, 144.17, 135.04, 134.99, 130.89, 130.48, 128.09, 124.78, 123.91, 123.57, 115.37, 61.78, 51.84, 46.06, 40.54, 32.56, 31.63, 23.37. ESI-MS m/z 387.4 (M + 1).
(2-(pyridin-2-yl)quinolin-4-yl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (6)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine and 4-(1-pyrrolidinyl)piperidine was used instead of 2-naphthylamine to form the amide. The crude compound was purified by chromatography using 10% MeOH in CH2Cl2. Overall yield was 60.3%. mp 81−82 °C 1H NMR (CDCl3) δ 8.73 (m, 1H), 8.65 (m, 1H), 8.51 (dd, J = 18.9 Hz, 1H), 8.21 (dd, J = 8.4 Hz, 1H), 7.92−7.75 (m, 3H), 7.65−7.58 (m, 1H), 7.40−7.35 (m, 1H), 4.92 (m, 1H), 3.49 (d, J = 12.9 Hz, 1H), 3.07−2.93 (m, 2H), 2.79 (m, 4H), 2.62−2.53 (m, 1H), 2.20−2.13 (m, 1H), 1.88−1.77 (m, 6H), 1.68−1.55 (m, 1H); 13C NMR (CDCl3) δ 167.22, 156.22, 155.84, 149.51, 148.26, 143.59, 137.24, 130.49, 130.32, 128.07, 127.85, 124.96, 124.56, 121.97, 115.96, 61.98, 51.86, 46.18, 40.53, 32.67, 31.67, 23.44. ESI-MS m/z 387.4 (M + 1).
N-(naphthalen-2-yl)-2-(pyridin-4-yl)quinoline-4-carboxamide (7)
The reaction conditions were described as above. mp 251−252 °C 1H NMR (DMSO) δ 11.07 (s, 1H), 8.79 (dd, J = 1.8, 4.8 Hz, 2H), 8.56−8.54 (m, 2H), 8.33 (dd, J = 1.5, 4.5 Hz, 2H), 8.24 (d, J = 8.4 Hz, 2H), 7.96−7.88 (m, 4H), 7.78−7.71 (m, 2H), 7.54−7.43 (m, 2H); 13C NMR (DMSO) δ 165.98, 154.22, 151.25, 148.58, 145.67, 144.12, 137.04, 134.01, 131.45, 130.91, 130.61, 129.22, 129.08, 128.24, 127.32, 125.89, 125.78, 124.60, 122.06, 121.08, 117.66, 117.09. ESI-MS m/z 376.3 (M + 1), 348.4 (M – 28), 297.4 (M – 79), 233.1 (M – 143), 205.3 (M – 171).
N-(naphthalen-2-yl)-2-(pyridin-3-yl)quinoline-4-carboxamide (8)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine. Overall yield was 19.8%. mp 215−216 °C 1H NMR (CDCl3) δ 9.19 (d, J = 1.8 Hz, 1H), 9.06 (s, 1H), 8.52−8.50 (m, 2H), 8.37 (m, 1H), 8.28 (dd, J = 0.6, 8.1 Hz, 1H), 8.16 (d, J = 8.4, 1H), 7.97 (s, 1H), 7.91−7.76 (m, 4H), 7.72 (dd, J = 2.1, 8.7 Hz, 1H), 7.62 (m, 1H), 7.50 (m, 2H), 7.33−7.29 (m, 1H); 13C NMR (CDCl3) δ 165.81, 154.05, 150.37, 148.98, 148.55, 143.53, 135.46, 135.04, 134.41, 134.05, 131.27, 130.94, 130.40, 129.37, 128.34, 128.11, 127.92, 127.04, 125.75, 125.26, 123.95, 123.71, 120.00, 117.52, 116.29. ESI-MS m/z 376.5 (M + 1).
N-(naphthalen-2-yl)-2-(pyridin-2-yl)quinoline-4-carboxamide (9)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (2:1). Overall yield was 23.9%. mp 261−262 °C 1H NMR (CDCl3) δ 8.80 (s, 1H), 8.74−8.68 (m, 2H), 8.48−8.42 (m, 2H), 8.26−8.20 (m, 2H), 7.95−7.78 (m, 5H), 7.66−7.61 (m, 2H), 7.55−7.39 (m, 3H); 13C NMR (CDCl3) δ 165.65, 155.64, 155.47, 149.16, 148.62, 142.34, 137.25, 135.03, 133.81, 131.03, 130.36, 130.25, 129.06, 128.13, 127.88, 127.66, 126.73, 125.44, 124.57, 124.46, 121.87, 119.89, 117.31, 116.18. ESI-MS m/z 376.5 (M + 1).
N-(naphthalen-1-ylmethyl)-2-(pyridin-4-yl)quinoline-4-carboxamide (10)
The reaction conditions were the same as above except 1-naphthylmethylamine was used instead of 2-naphthylamine to form the amide. Overall yield was 72.3%. mp 188−189 °C 1H NMR (CDCl3) δ 8.71 (d, J = 6 Hz, 2H), 8.25−8.15 (m, 3H), 7.96−7.86 (m, 5H), 7.77 (m, 1H), 7.67−7.54 (m, 4H), 7.50−7.45 (m, 1H), 6.42 (m, 1H), 5.24 (d, J = 5.4 Hz, 2H); 13C NMR (CDCl3) δ 167.06, 154.15, 150.76, 148.93, 145.93, 143.35, 134.26, 132.88, 131.60, 130.89, 130.64, 129.45, 129.29, 128.57, 127.61, 127.18, 126.53, 125.71, 125.24, 124.23, 123.58, 121.62, 116.23, 42.81. ESI-MS m/z 390.5 (M + 1).
N-(naphthalen-1-ylmethyl)-2-(pyridin-3-yl)quinoline-4-carboxamide (11)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and 1-naphthylmethylamine was used instead of 2-naphthylamine to form the amide. Overall yield was 10.3%. mp 175−176 °C 1H NMR (CDCl3) δ 9.15 (s, 1H), 8.59 (d, J = 3.3 Hz, 1H), 8.38 (m, 1H), 8.21 (d, J = 8.1 Hz, 2H), 8.11 (d, J = 8.7 Hz, 1H), 7.93−7.82 (m, 3H), 7.74 (m, 1H), 7.66−7.53 (m, 4H), 7.49−7.44 (m, 1H), 7.39−7.35 (m, 1H), 6.61 (m, 1H), 5.23 (d, J = 5.1 Hz, 2H); 13C NMR (CDCl3) δ 167.17, 154.04, 150.53, 148.86, 148.53, 143.27, 134.96, 134.34, 134.23, 133.04, 131.63, 130.72, 130.24, 129.35, 129.25, 128.06, 127.56, 127.11, 126.50, 125.70, 125.29, 123.86, 123.76, 123.66, 116.16, 42.76. ESI-MS m/z 390.3 (M + 1), 262.1 (M – 128), 233.0 (M – 157), 207.2 (M – 183), 141.1 (M – 249).
N-(naphthalen-1-ylmethyl)-2-(pyridin-2-yl)quinoline-4-carboxamide (12)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine and 1-naphthylmethylamine was used instead of 2-naphthylamine to form the amide. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (2:1). Overall yield was 23%. mp 198−199 °C 1H NMR (CDCl3) δ 8.66−8.57 (m, 3H), 8.37 (d, J = 7.8 Hz, 1H), 8.24−8.14 (m, 2H), 7.91−7.82 (m, 3H), 7.75 (m, 1H), 7.64−7.44 (m, 5H), 7.37−7.32 (m, 1H), 6.54 (m, 1H), 5.22 (d, J = 5.4 Hz, 2H); 13C NMR (CDCl3) δ 167.40, 155.76, 155.69, 149.29 148.65, 142.55, 137.33, 134.18, 133.07, 131.65, 130.36, 129.24, 129.13, 128.12, 127.41, 127.09, 126.40, 125.67, 124.79, 124.63, 123.73, 122.01, 116.35, 42.60. ESI-MS m/z 390.5 (M + 1).
N-phenyl-2-(pyridin-4-yl)quinoline-4-carboxamide (13)
The reaction conditions were the same as above except aniline was used instead of 2-naphthylamine to form the amide. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (2:1). Overall yield was 67.8%. mp 202−203 °C 1H NMR (CDCl3) δ 8.86 (s, 1H), 8.57 (d, J = 4.2 Hz, 2H), 8.24−8.16 (m, 2H), 7.88 (d, J = 6 Hz, 2H), 7.81−7.76 (m, 4H), 7.60 (m, 1H), 7.44 (t, J = 8.1 Hz, 2H), 7.24 (t, J = 7.8 Hz, 1H); 13C NMR (CDCl3) δ 165.55, 153.70, 150.48, 148.89, 145.93, 143.33, 138.01, 131.02, 130.61, 129.56, 128.75, 125.50, 125.25, 124.12, 121.64, 120.39, 116.25. ESI-MS m/z 326.5 (M + 1).
N-phenyl-2-(pyridin-3-yl)quinoline-4-carboxamide (14)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and aniline was used instead of 2-naphthylamine to form the amide. Overall yield was 41.3%. mp 215−216 °C 1H NMR (CDCl3) δ 9.16 (d, J = 1.8 Hz, 1H), 8.82 (s, 1H), 8.52 (dd, J = 1.5, 4.5 Hz, 1H), 8.36 (m, 1H), 8.23 (d, J = 7.5 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.92 (s, 1H), 7.81−7.75 (m, 3H), 7.60 (m, 1H), 7.44 (t, J = 8.1 Hz, 2H), 7.34−7.30 (m, 1H), 7.24 (t, J = 7.8 Hz, 1H); 13C NMR (CDCl3) δ 165.64, 154.04, 150.37, 148.95, 148.53, 143.56, 138.01, 135.02, 134.39, 130.91, 130.37, 129.55, 128.29, 125.45, 125.23, 123.94, 123.67, 120.40, 116.25. ESI-MS m/z 326.5 (M + 1).
N-phenyl-2-(pyridin-2-yl)quinoline-4-carboxamide (15)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine and aniline was used instead of 2-naphthylamine to form the amide. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (2:1). Overall yield was 32.4%. mp 207−208 °C 1H NMR (CDCl3) δ 8.75−8.66 (m, 3H), 8.38 (d, J = 7.8 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.02 (s, 1H), 7.91 (m, 1H), 7.82−7.73 (m, 3H), 7.62 (m, 1H), 7.46−7.38 (m, 3H), 7.22 (t, J = 7.5 Hz, 1H); 13C NMR (CDCl3) δ 165.74, 155.83, 155.67, 149.37, 148.79, 142.62, 137.81, 137.46, 130.56, 130.44, 129.48, 128.31, 125.62, 125.37, 124.78, 124.65, 122.08, 120.49, 116.37. ESI-MS m/z 326.3 (M + 1).
6,8-dimethyl-N-(naphthalen-2-yl)-2-(pyridin-4-yl)quinoline-4-carboxamide (16)
The reaction conditions were the same as above except isatin was substituted with 5,7-dimethylisatin. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (1:2). Overall yield was 35.6%. mp 233−234 °C 1H NMR (CDCl3) δ 8.74 (s, 1H), 8.61 (dd, J = 1.5, 4.8 Hz, 2H), 8.51 (d, J = 2.1 Hz, 1H), 7.98 (dd, J = 1.8, 4.8 Hz, 2H), 7.90−7.84 (m, 5H), 7.70 (dd, J = 2.1, 9 Hz, 1H), 7.55−7.45 (m, 3H), 2.84 (s, 3H), 2.48 (s, 3H); 13C NMR (CDCl3) δ 166.35, 150.96, 150.46, 146.60, 146.27, 142.61, 138.88, 138.29, 135.46, 134.05, 133.42, 131.25, 129.35, 128.09, 127.91, 127.04, 125.74, 124.20, 121.72, 121.42, 119.99, 117.46, 115.59, 22.24, 18.31. ESI-MS m/z 404.5 (M + 1).
6,8-dimethyl-N-(naphthalen-2-yl)-2-(pyridin-3-yl)quinoline-4-carboxamide (17)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and 5,7-dimethylisatin was used instead of isatin. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (1:2). Overall yield was 13.9%. mp 224−225 °C 1H NMR (CDCl3) δ 9.23 (d, J = 1.8 Hz, 1H), 9.11 (s, 1H), 8.52−8.46 (m, 2H), 8.35 (m, 1H), 7.90−7.83 (m, 5H), 7.72 (dd, J = 2.1, 8.7 Hz, 1H), 7.54−7.43 (m, 3H), 7.29−7.25 (m, 1H), 2.75 (s, 3H), 2.46 (s, 3H); 13C NMR (CDCl3) δ 166.53, 151.11, 149.90, 148.45, 146.54, 142.74, 138.20, 137.99, 135.63, 134.63, 134.06, 133.23, 131.20, 129.29, 128.10, 127.88, 126.97, 125.65, 123.80, 123.67, 121.70, 120.08, 117.46, 115.50, 22.17, 18.29. ESI-MS m/z 404.4 (M + 1).
6,8-dimethyl-N-(naphthalen-2-yl)-2-(pyridin-2-yl)quinoline-4-carboxamide (18)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine and 5,7-dimethylisatin was used instead of isatin. The crude compound was purified by chromatography using mixture of hexanes and ethyl acetate (1:2). Overall yield was 25.1%. mp 226−227 °C 1H NMR (CDCl3) δ 8.74−8.70 (m, 3H), 8.46 (d, J = 2.1 Hz, 1H), 8.16 (s, 1H), 8.00 (s, 1H), 7.92−7.82 (m, 4H), 7.63 (dd, J = 2.4, 8.7 Hz, 1H), 7.48 (m, 3H), 7.39−7.35 (m, 1H), 2.89 (s, 3H), 2.50 (s, 3H); 13C NMR (CDCl3) δ 166.28, 155.94, 152.95, 148.91, 146.08, 141.72, 137.97, 137.58, 137.02, 135.18, 133.81, 132.69, 130.95, 128.97, 127.86, 127.62, 126.63, 125.32, 124.37, 124.14, 121.91, 121.65, 119.96, 117.25, 115.57, 21.95, 18.13. ESI-MS m/z 404.3 (M + 1).
6-chloro-N-(naphthalen-2-yl)-2-(pyridin-4-yl)quinoline-4-carboxamide (19)
The reaction conditions were the same as above except isatin was substituted with 5-chloroisatin. Overall yield was 8.3%. mp 225−226 °C 1H NMR (DMSO) δ 11.10 (s, 1H), 8.81 (d, J = 5.1 Hz, 2H), 8.64 (s, 1H), 8.56 (s, 1H), 8.34−8.31 (m, 3H), 8.26 (d, J = 9 Hz, 1H), 7.97−7.88 (m, 4H), 7.77 (d, J = 8.7 Hz, 1H), 7.54−7.45 (m, 2H); 13C NMR (DMSO) δ 164.6, 154.0, 150.5, 146.4, 144.6, 142.0, 136.1, 133.2, 132.8, 132.0, 131.2, 130.2, 128.4, 127.5, 126.5, 125.1, 124.6, 123.9, 121.3, 120.4, 118.2, 116.6. ESI-MS m/z 410.3 (M + 1), 382.4 (M – 28), 267.0 (M – 143), 239.2 (M – 171).
6-chloro-N-(naphthalen-2-yl)-2-(pyridin-3-yl)quinoline-4-carboxamide (20)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and 5-chloroisatin was used instead of isatin. Overall yield was 3.2 %. mp 228−229 °C 1H NMR (DMSO) δ 11.07 (s, 1H), 9.54 (s, 1H), 8.73 (d, J = 7.5 Hz, 2H), 8.61 (s, 1H), 8.56 (s, 1H), 8.30 (d, J = 2.7 Hz, 1H), 8.23 (d, J = 9 Hz, 1H), 7.97−7.88 (m, 4H), 7.78 (d, J = 8.4 Hz, 1H), 7.64−7.60 (m, 1H), 7.54−7.43 (m, 2H); 13C NMR (DMSO) δ 164.8, 154.5, 150.9, 148.6, 146.6, 141.9, 136.2, 134.9, 133.3, 132.4, 131.9, 131.2, 130.3, 128.6, 127.6, 126.7, 125.2, 124.2, 124.0, 120.6, 118.4, 116.7. ESI-MS m/z 410.2 (M + 1), 382.4 (M – 28), 267.0 (M – 143), 239.2 (M – 171).
6-bromo-N-(naphthalen-2-yl)-2-(pyridin-4-yl)quinoline-4-carboxamide (21)
The reaction conditions were the same as above except isatin was substituted with 5-bromoisatin. Overall yield was 5.6%. mp 208−209 °C 1H NMR (DMSO) δ 11.10 (s, 1H), 8.81 (d, J = 3.9 Hz, 2H), 8.63 (s, 1H), 8.56 (s, 1H), 8.47 (d, J = 2.1 Hz, 1H), 8.33 (d, J = 5.7 Hz, 2H), 8.18 (d, J = 8.7 Hz, 1H), 8.04 (dd, J = 2.1, 9.3 Hz, 1H), 7.92 (m, 3H), 7.77(dd, J = 2.1, 8.7 Hz, 1H), 7.54−7.43 (m, 2H); 13C NMR (DMSO) δ 164.7, 154.1, 150.3, 146.6, 144.9, 141.9, 136.2, 133.9, 133.3, 132.1, 130.3, 128.5, 127.6, 127.5, 127.3, 126.6, 125.2, 125.1, 121.7, 121.5, 120.5, 118.4, 116.6. ESI-MS m/z 454.2 (M + 1), 426.3 (M – 28), 311.0 (M – 143), 283.2 (M – 171).
6-bromo-N-(naphthalen-2-yl)-2-(pyridin-3-yl)quinoline-4-carboxamide (22)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and 5-bromoisatin was used instead of isatin. Overall yield was 6.1%. mp 240−241 °C 1H NMR (DMSO) δ 11.07 (s, 1H), 9.54 (s, 1H), 8.74−8.71 (m, 2H), 8.61 (s, 1H), 8.56 (s, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 9 Hz, 1H), 8.03−7.88 (m, 4H), 7.77(dd, J = 1.5, 8.7 Hz, 1H), 7.64−7.60 (m, 1H), 7.54−7.43 (m, 2H); 13C NMR (DMSO) δ 164.7, 154.5, 150.9, 148.6, 146.7, 141.7, 136.2, 134.8, 133.7, 133.3, 131.9, 130.3, 128.5, 127.6, 127.2, 126.6, 125.2, 124.6, 124.0, 121.1, 120.5, 118.3, 116.7. ESI-MS m/z 454.2 (M + 1), 426.3 (M – 28), 311.0 (M – 143), 283.1 (M – 171).
N-(naphthalen-1-yl)-2-(pyridin-3-yl)quinoline-4-carboxamide (23)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 3-acetylpyridine and 1-naphthylamine was used instead of 2-naphthylamine to form the amide. Overall yield was 35.8%. mp 201−202 °C 1H NMR (CDCl3) δ 9.27 (s, 1H), 8.67−8.61 (m, 2H), 8.47 (d, J = 7.8 Hz, 1H), 8.36 (d, J = 8.1 Hz, 1H), 8.21 (t, J = 7.8 Hz, 2H), 8.11 (s, 1H), 7.95−7.91 (m, 2H), 7.85−7.80 (m, 2H), 7.67−7.52 (m, 4H), 7.42−7.38 (m, 1H); 13C NMR (CDCl3) δ 166.05, 154.16, 150.44, 148.93, 148.57, 143.31, 134.86, 134.27, 134.21, 131.71, 130.76, 130.35, 128.98, 128.19, 127.11, 126.75, 126.30, 125.77, 124.98, 123.74, 123.59, 121.33, 120.53, 116.19. ESI-MS m/z 376.5 (M + 1).
N-(naphthalen-1-yl)-2-(pyridin-2-yl)quinoline-4-carboxamide (24)
The reaction conditions were the same as above except 4-acetylpyridine was substituted with 2-acetylpyridine and 1-naphthylamine was used instead of 2-naphthylamine to form the amide. Overall yield was 3.8%. mp 207−208 °C 1H NMR (DMSO) δ 10.99 (s, 1H), 8.87−8.81 (m, 2H), 8.68 (d, J = 7.8 Hz, 1H), 8.31−8.23 (m, 2H), 8.13−7.99 (m, 3H), 7.92−7.86 (m, 3H), 7.75 (t, J = 7.2 Hz, 1H), 7.64−7.57 (m, 4H); 13C NMR (DMSO) δ 167.15, 155.84, 155.32, 150.18, 148.39, 143.90, 138.35, 134.51, 133.63, 131.20, 130.51, 129.20, 128.93, 128.80, 127.21, 126.97, 126.92, 126.35, 126.02, 125.79, 125.04, 124.11, 123.53, 121.92, 117.14. ESI-MS m/z 376.5 (M + 1).
Difference spectroscopy of binding and substrate titrations
Difference spectra were collected using a double-beam Olis upgraded Aminco DW-2000 spectrophotometer (Olis, Inc., Bogart, GA). The wavelength range encompassed 350 to 500 nm. Both sample and reference cuvettes contained purified 2C9 in 100 mM KPi buffer with 20% glycerol, pH 7.4 10. To obtain difference spectra, a baseline was recorded with 2C9 enzyme and buffer in both sample and reference cuvettes followed by substrate titratation into the sample cuvette, and the same volume of solvent was added to the reference cuvette. The difference spectra of samples containing purified 2C9 0.49 μM and substrates were scanned at 30°C. Substrate concentrations were between 0.107 and 109.9 μM. Substrate binding curves were fit by nonlinear regression with GraphPad Prism4 using equation ΔA = (Bmax × [S]) / (Ks + [S]), where A is the absorbance difference, Bmax is the maximum absorbance difference, S is the substrate concentration. The types of substrate-induced binding spectra with P450 heme were determined by the positions of wavelengths for peaks (λmax) and minima (λmin) on the spectra 29.
EPR studies on binding modes
EPR spectra were recorded on a Bruker ESP-300e X-band spectrometer fitted with a liquid helium flow cryostat (ESR-9, Oxford Instruments). The ferric 2C9 samples were prepared in KPi buffer (100 mM, pH 7.4, 20% glycerol) and frozen in liquid nitrogen prior to spectral acquisition. The protein concentrations of samples were 18.6 μM (substrate-free form) and 18.5 μM (substrate-bound form), respectively with a substrate concentration of 1 mM. Spectra were recorded at 20 K with microwave frequency of 9.614 GHz, microwave power of 10 mW, modulation amplitude of 16 gauss, modulation frequency of 100 kHz, receiver gain of 1e4, conversion time of 168 ms, and time constant of 20 ms.
pKa measurement
Data were collected on a diode array HP 845 UV/Vis spectrophotometer. Samples were prepared by adding 500 μL of 0.1 mM substrate stock solution in DMSO to 500 μL of 0.1 M KPi buffer prepared at pH values between 1.04 and 10.03. As the pyridine nitrogen is protonated at lower pH, peaks emerged with λmax at 287 nm. Absorbance was plotted versus pH and fit to a sigmoidal function using GraphPad Prism 4. Results agreed with MarvinSketch calculated values.
Automated Docking
Substrates were docked into the crystal structure of 2C9 1r9o 30 using MOE 2006.08 (Chemical Computing Group Inc. Montreal, Quebec, Canada). Compounds were sketched and underwent energy minimization as entries. Selected substrates were then fit into the active site cavity using the automatic docking program, exploring the conformational space associated with each rotatable bond. The parameters were as follows: receptor – receptor atoms; site – selected atoms (glycerol + flurbiprofen); ligand – sketched substrates; placement – alpha triangle; scoring – London dG; retain – 100. The parameters for inclusion of the residues by ligand interactions calculations in MOE were fields of H-bond, ionic, and solvent with minimum score 10% prospectively. Only the substrate and amino acid residues within 4.5 Å were allowed to move during the minimization. The protein was frozen when the autodocking was performed. The water molecules were modeled within the crystal structure before the docking process was initiated. The force field for both docking and energy minimization were Hamiltonian – MMFF94x in MOE.
DFT calculations
Density functional calculations were performed using Gaussian 03 31. The TPSS functional 32 was used with the cc-pVDZ basis set with effective core potential on iron 33, the aug-cc-pVDZ on sulfur 34 and nitrogen 35 with diffuse d functions removed, and the cc-VDZ basis set on carbon and hydrogen 36. The basis set and optimized geometries are available as supplemental material. Solvation energies were calculated for the gas phase optimized geometries using the polarized continuum model self consistent reaction field with the solvent set to water's dielectric constant. The heme model was the abbreviated heme with an S-H fifth ligand used by Shaik and coworkers. 37
RESULTS
All quinoline-4-carboxamide analogs were synthesized by the route shown in Scheme 1. Results of the inhibition assays are shown in Table 1. From the experimental results, we saw a trend in binding affinity related to the positions of the Type II accessible nitrogen in the pyridine moiety (Figure 1). Compounds with pyridine nitrogen in the para position (relative to the linkage) generally exhibit higher binding affinity than those in meta and ortho. In addition, compounds with an aromatic ring system at the carboxyamide moiety bind more tightly than those that lack a ring system (Figure 1, R=aromatic).
UV difference spectra were used to determine substrate binding modes and spectral dissociation constants Ks for the quinoline-4-carboxamide analogs shown in Table 1 whose absorbance did not interfere with the UV absorption of the titration. Of those analogs, 10 and 13 gave clear Type II binding spectra (representative titrations results are shown in Figure 2). Since the Ki values reported in Table 1 were obtained using baculosomes, we used the reconstituted 2C9 system to determine if purified enzyme gave different Ki values relative to the baculosomes preparations. The results gave Ki values for compound 10 of 113 +/− 6.85 nM in the purified system and 125 nM in the baculosomes. For compound 13 the Ki was 79.8 +/− 5.91 nM in the purified system and 100 nM in the baculosome system. The results indicate that for these substrates the purified reconstituted system and the baculosomes system gave similar results. Compound 14 showed a peak at ∼ 415 nm, but no obvious trough in difference spectra, which implies that 14 exhibits mixed Type II and Type I binding modes.
Figure 2.

Binding spectrum and titration curves of compound 13, Ks = 0.325 μM.
Since EPR measurements are not hindered by the substrate absorbing in the Soret band range, additional compounds could be examined for binding mode. Compounds 7, 8, 9, 16, 17,18, 19, 21 were studied by EPR to probe binding modes (Figure 3 and 4). From these results, analogs with the pyridine nitrogen in the para position to the linkage (7, 16, 19, 21), EPR spectra showed a low-spin shift which was absent with compounds having nitrogen in the meta or ortho positions. EPR spectrum of substrate-free 2C9 had g-values at 2.42/2.38, 2.24, and 1.92 (data not shown) and the Type II binding substrate bound 2C9 spectrum had g-values at 2.51, 2.26, and 1.88. This result suggests that the para nitrogen is coordinated to the heme iron, while the meta and ortho compounds are not.
Figure 3.

EPR spectra of 2C9 in the present of substrates 7 (para), 8 (meta), 9 (ortho), 16 (para), 17 (meta), and 18 (ortho).
Figure 4.

EPR spectra of 2C9 in the present of substrates 7 (para), 16 (para), 19 (para), and 21 (para).
To determine which portion of the total binding energy depends on pure Type II interaction (as opposed to apoprotein interactions), the density functional method TPSS was used for the core structure of the three pyridinylquinolines (ortho, meta, and para pyridines, Figure 5), and the differences in energies for nitrogen substitution ortho, meta, and para from the linkage to quinoline were determined. The results indicate that the ortho nitrogen linkage has a much longer N-Fe bond and significantly distorts the heme from planarity. In fact, this structure was so distorted that the geometry could not be minimized. The meta and para linkage both have 2.1 Å N-Fe bond lengths with only a slight distortion in the planarity of the heme. Electronically, the para linkage has the strongest interaction energy by 0.6 kcal/mol, however, this is not a significant difference given the level of computation.
Figure 5.

Core structures used to determine nitrogen-iron coordination energies and solvation energies.
To determine if differences in solvation energy might influence the overall binding energy, a series of ortho, meta, para linkages were used to calculate the solvation energies for each core structure using a polarized continuum self consistent reaction field model as implemented in Gaussian 03. Results indicate insignificant differences in solvation energy between the meta and para isomer, while the ortho isomer was around 2 kcal/mol less stabilized by solvent. This difference in solvent interaction is also reflected in the measured differences in pKa values for the ortho compound 18 of 3.05 versus nearly identical values of 4.27 and 4.30 for para (16) and meta (17) respectively. These results indicate that difference in binding affinity for the compounds with ortho linkage may be complicated by differences in solvation.
DISCUSSION
Understanding the mechanism of Type I and Type II substrate binding modes in P450 drug metabolizing isoforms is critical to optimizing drug design and reducing drug toxicity. Metabolic stability, enzyme inhibition, metabolite regioselectivity, and formation of reactive metabolites are all functions of binding modes. It has been shown that substrates which are capable of Type II binding are more potent inhibitors than those capable of only Type I binding 38. In addition, potent inhibition is possible when coordination between the heteroatom (nitrogen atom) lone pair electrons and the ferric heme iron is combined with apoprotein interactions (lipophilic and electrostatic) 11. Type II binding slows the rate of P450-mediated oxidation, increasing substrate metabolic stability as well as the potency of inhibition. Therefore, elucidating how Type II interactions can be used to optimize drugs metabolic properties without increasing drug toxicity is necessary to optimize drug design.
In this study we investigated the potential increase in binding affinity that can be attributed to Type II binding in the absence of other binding features. To this end, we synthesized a series of quinoline-4-carboxamide analogs that differ in the placement of nitrogen capable of forming a Type II complex with the heme iron of 2C9 (Table 1). Each set of three compounds differed only in the position of their linkage and are assumed to have almost identical solvation energies allowing for the determination of the nitrogen coordination binding. One caveat is that solvation energy calculations and pKa measurements show that interpretation of changes in binding affinity for the ortho compounds may be complicated by differences in solvation relative to the meta and para linkages.
The effects of varying the position of the nitrogen atom in the pyridine ring on the binding affinity of quinoline-4-carboxamide analogs to 2C9 are shown in Table 1 and Figure 6. In general, compounds with para nitrogen in the pyridine ring bind more tightly than meta or ortho compounds; compounds with meta nitrogens are better binders than ortho compounds. Para compounds generally bind to 2C9 with Ki between 9 − 125 nM except for compounds 1 and 4, which have Ki values of 9.94 and 3.67 μM, respectively. The binding affinity of para compounds are increased by a factor of up to 4200-fold compared with meta compounds (such as 21 and 22) and a factor of up to 1900-fold compared with ortho compounds (such as 16 and 18). Meta compounds overall have a larger range of binding affinities to 2C9, having Ki values from 0.15 μM to 37.85 μM and these compounds can be 300-fold tighter binders than ortho compounds. In general, ortho compounds bind less tightly than those of their para and meta counterparts. Ki values of ortho compounds are ranged from 76.1 − 1.32 μM. The most likely explanation for these results is that the para compounds are capable of stronger Type II binding interaction than the meta compounds, which are capable of stronger Type II interaction than the ortho compounds.
Figure 6.

Ki versus nitrogen atom in para, meta and ortho position toward linkage of nine different groups of compounds indicate that in general the order of tightness of binding is para>meta>ortho (Table 1).
To determine if Type II binding is responsible for the differences in binding affinity, we used both UV and EPR spectroscopy (Table 2) to determine substrate binding modes of several compounds from Table 1. Binding mode analysis using UV spectroscopy was limited to only compounds that did not absorb in the region on interested 350−500 nm. UV difference spectra results for para compounds (1, 10, and 13) show typical Type II binding spectra, with peaks at ca. 430 nm and trough at ca. 410 nm. The meta compounds (2 and 14), however, show what appears to be mixed Type I and Type II binding in the UV difference spectra where peaks are at ca. 418 nm, but not an obvious trough. The ortho compound (15) does not give a either a Type I or Type II binding spectra.
Table 2.
B3LYP/cc-pVDZ Energies Substrates and Heme-Substrate Complexes for Core Structures Shown in Figure 5.
| Linkage | Substrate Energya) | Type II Energyb) | Solvation Energyc) |
|---|---|---|---|
| Para | −649.207 | −2135.127 | −10.23 |
| Meta | −649.208 | −2135.126 | −9.87 |
| Ortho | −649.344 | Not stable. | −8.03 |
Energies in Hartrees for the para, meta and ortho linked core structures in Figure 5.
Energies in Hartrees for each core structure coordinated to the Heme.
PCM solvation energies on kcal/mol for the different linkages with the dielectric set to that of water.
EPR studies on para compounds 7, 16, 19, and 21 suggest that the para nitrogen atom on the pyridine moiety is coordinated to the P450 iron. The low-spin g value shifts of 1.88 to 1.91 and 2.45 to 2.51 observed for these compounds are typical of the low-spin shift commonly seen in Type II binding substrates (normal nitrogen donors) in other P450 enzymes 8, 39. However, compounds 16, 19, and 21 also show a low spin signal at 2.38 that is characteristic of a non substrate bound EPR spectrum (Figure 4). The coexistence of both the shifts and the unbound signal at 2.38 for compounds 16, 19, and 21 suggests compound 7 is able to bind pure Type II while the others bind in mixed mode. The difference in binding modes of these para compounds may be attributed to the increase in steric bulk (and hydrophobicity) of compounds 16, 19, and 21 relative to compound 7. While all are tight binders, increasing steric bulk may interfere with formation of a nitrogen-ferric complex allowing for the mixed binding mode seen in the EPR. Overall, UV and EPR studies support the hypothesis that the pyridine rings with the para-substituted nitrogen have strong Type II binding interactions. The meta compounds appear to be able to coordinate to the heme to a lesser extent, while the ortho compounds do not appear to coordinate to the heme.
Interestingly, the UV difference spectra appear to provide a more sensitive test of substrate binding modes than EPR spectra. UV difference spectrum gathered on meta compound 2 showed what may be interpreted as mixed Type I and Type II binding that was not apparent in the EPR spectrum. The disagreement between UV and EPR spectra of meta compounds could be a sensitivity issue or other artifacts requiring additional investigation.
The spectral dissociation constants (Ks) for compounds 10 and 13 (1.28 and 0.325 μM) were higher than the inhibitor dissociation constant, Ki, (0.125 and 0.1μM) determined by IC50 experiments These discrepancies are not unusual 40, 41 and may be attributed to the different methods used to measure each value. Ks was measured as the Type II binding substrate dissociation constant when the heme iron was in the +3 oxidation state and the amount of spin state change between high and low was depending on the equilibrium of the nature non-substrate bound protein. On the other hand Ki was determined as the overall inhibitor dissociation constant where the catalytic cycle was completed and the iron is in equilibrium between +3 and +2 oxidation states, allowing the inhibitor to bind in either Type I or Type II modes. Mixed substrate binding and heme iron oxidation state equilibria further complicate the elucidation of the substrate binding modes within the active site. Another possible reason that the Ks was higher than Ki was that the substrate heme coordination was incomplete. Incomplete heme coordination has been documented by Locuson et al 2007 10.
One potential complication of this interpretation would arise if the para, meta, and ortho compounds had different pKa values. To determine if the nitrogen atom in the pyridine ring was differentially protonated for the ortho, meta and para substituted pyridines in the physiological pH, the pKa values of all analogs were predicted using by MarvinSketch. To ensure that these predictions were reliable, pKa values were experimentally determined for compounds 16, 17, and 18. The results from computational prediction/experimental determination were 4.15/4.27, 3.44/4.30, and 3.36/3.05 for para, meta, and ortho respectively. All the compounds have predicted pKa less than 4.3 for the pyridine nitrogen, which indicates that at physiological pH, the analogs are free to coordinate with the heme iron. In addition, lower pKa corresponds to greater delocalization of the lone pair in the aromatic system. Delocalization decreases the ability of an sp2 nitrogen to Type II bind. The piperidine nitrogen on compounds 4, 5, and 6, which are predicted to have pKa values of ca. 9.1. Since these values are well above physiological pH, they would exist in their protonated forms. In general, positively charged compounds do not bind strongly to 2C9, which may explain the overall low affinity of compounds 4, 5, and 6 for the enzyme.
To determine if differential interactions of the para, meta and ortho compounds result from different heme interactions, quantum calculations of binding energies the different compounds were determined (Table 2). The results indicate that the ortho substituted pyridines do not strongly interact with the heme. However, the para and meta compounds interact with essentially the same energy. These results suggest the meta compounds may have less favorable apoprotein interactions than the para substituted compounds. Further, these computational results suggest that active site residues play an important role in determining affinity for the meta and para compounds’ ability to form Type II complexes. For meta and ortho compounds, interactions with the apoprotein rather than just the electrostatic interaction with the heme appear to have the most significant influence on binding affinity versus para compounds.
Crystal structures that show Type II binding interactions include P450cam with nicotine, CYP51 with 4-phenylimidazole (4-PI) and fluconazole (FLU), P450eryF with ketoconazole, and CYP2B4 with bifonazole 42-45. Generally, these structures show the heterocyclic substrates Type II bound to the heme iron with the remainder of the molecule bent away from the I-helix in the active site pocket and the essential conformational changes of apoprotein are necessary for substrates binding. With these substrate-enzyme binding interactions in mind, we applied computational modeling methods to further visualize overall substrate binding interactions.
In an attempt to ascertain specific substrate-protein interactions, we used automated docking on two of our tight binders, quinoline-4-carboxamide analog 7 and 8 (Figures 7 and 8). Both of these compounds contain naphthalene functionalized amides and differ only in the position of the sp2 nitrogen in the pyridine moiety. Indeed, all compounds from Table 1 that contain aromatic amides (especially naphthalene) bind more tightly than substrates with non-aromatic amides. This suggests that in addition to Type II coordination, the aromatic amide systems may interact favorably with active site residues. Docking of Type II binder 7 into the crystal structure of 2C9 is relatively straight-forward since the nitrogen-heme coordination is known. This important feature of nitrogen position on pyridine is one of the major characteristics on substrate structures that determine overall binding. For compound 7, a direct coordination between the heme and the para pyridine nitrogen is shown and this pyridine ring system is tilted away from the I-helix. In this orientation, the naphthalene ring faces Phe 114, allowing an edge-to-face π-interaction. In addition, the positively charged side chain of Arg 108 is oriented in such a way to allow interaction with the naphthalene ring through parallel stacking geometry. Presumably, a π-cation interaction from the positively charged Arg 108 side chain with the aromatic ring of amide allows for favorable enthalpic contributions of van der Waals and electrostatic interactions. This type of energetically favorable π -cation interaction has been described in various biological systems and recognition process 46, 47. In addition, the quinoline ring portion of 7 located in a hydrophobic pocket surrounded by Phe 476, Leu 208, Ile 205 and Leu 362. This conformation allows for potential π - π interactions between the quinoline ring and Phe 476.
Figure 7.
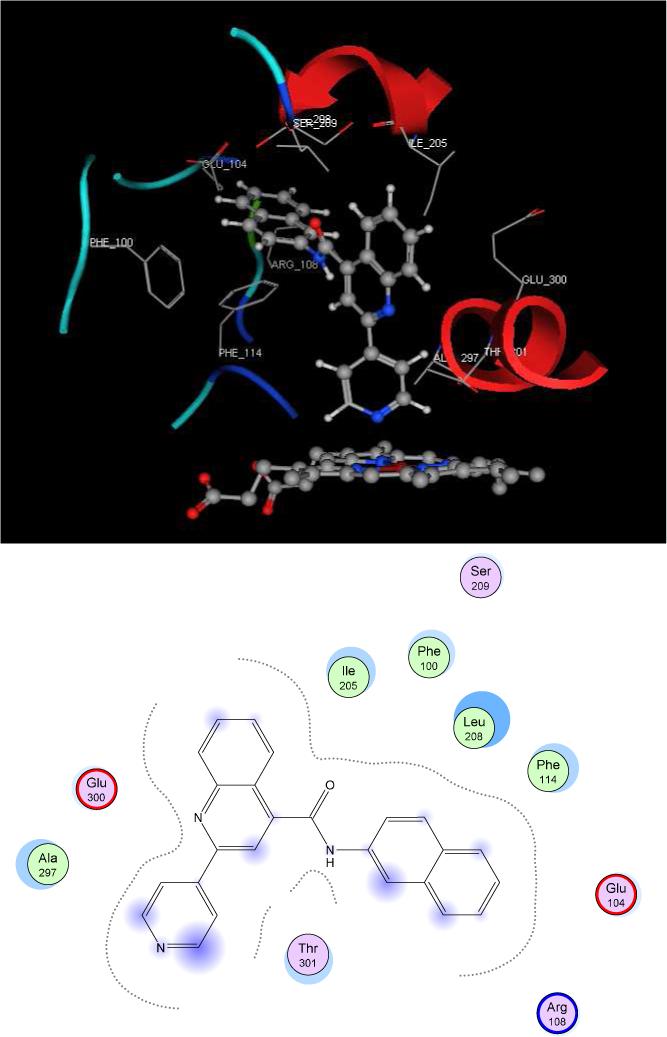
Proposed type II binding mode with compound 7 using the flurbiprofen complex of 2C9dH (Protein Data Bank code 1r9o), on the top. 2D-diagram of ligand-residues interactions is presented on the bottom.
Figure 8.
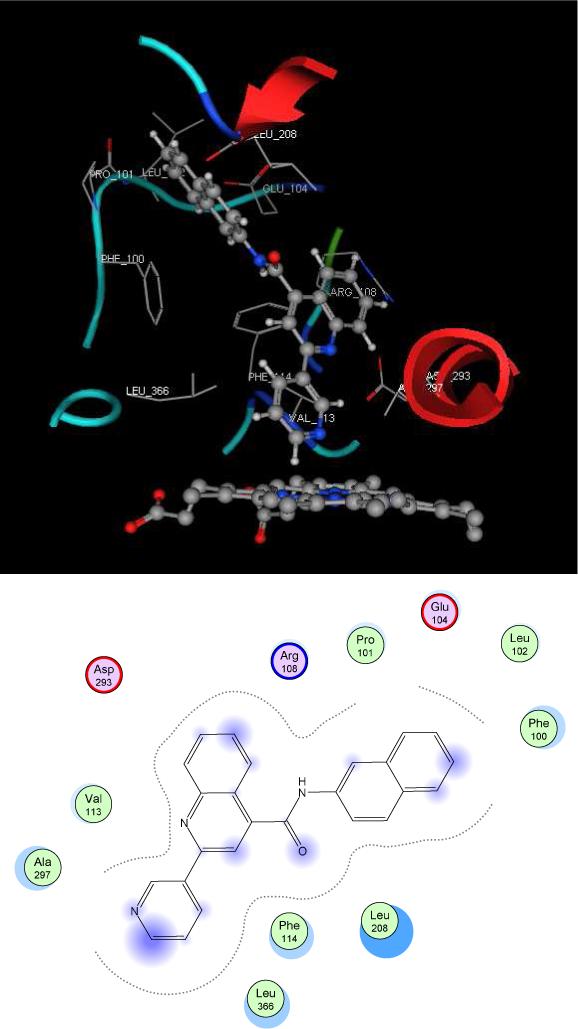
Proposed implausible type II binding mode with compound 8 using the flurbiprofen complex of 2C9dH (Protein Data Bank code 1r9o).
The proposed binding orientation for compound 8 does not have the nitrogen-atom coordinated to the heme iron. This may be a result of steric interference with the I-helix, even though all the other ligand-residue interactions are similar to those for compound 7. These docking results suggest the primary difference in binding affinity between meta and para compounds is a result of steric interaction with the I helix resulting in diminished or nonexistent Type II binding.
CONCLUSION
Type II interactions involving coordination of a pyridine nitrogen to the heme results in an up to 3500 fold increase in binding affinity. These significant increases in affinity can be modulated by changing the position of the nitrogen relative to substituents and could be used to dial-in affinities for P450 enzymes to minimize drug-drug interactions, while maintaining very similar reactivity and solubility in a potential drug.
Supplementary Material
Table 3.
List of UV and EPR Spectral Results for Selected Compounds.
| Compound | Ki (μM) | Binding Type | |
|---|---|---|---|
| UV min (nm)/max (nm) | EPR | ||
| 1 | 9.94 | Type II 410/429 | Type II |
| 2 | 9.74 | Mixed −/413 | Type I |
| 7 | 0.033 | N.Db | Type II |
| 8 | 1.15 | N.Da | Type I |
| 9 | 10.67 | N.Da | Type I |
| 10 | 0.125 | Type II 413/428 | N.Da |
| 13 | 0.10 | Type II 410/431 | N.Da |
| 14 | 4.06 | Mixed −/417 | N.Da |
| 16 | 0.011 | N.Dc | Type II/Type I |
| 17 | 8.55 | N.Dc | Type I |
| 18 | >20.6 | N.Dc | Type I |
| 19 | 0.012 | N.Db | Type II/Type I |
| 21 | 0.009 | N.Db | Type II/Type I |
Data not collected.
Substrate absorbance prevented characterization.
Spectrum is not clear.
Acknowledgement
This work was supported by National Institutes of Health Grants GM032165 and GM084546. We would like to thank Professor Kirk Peterson for providing the basis set for iron.
Glossary
- P450s
Cytochrome P450 enzymes
- EPR
electron paramagnetic resonance
- 2C9
CYP2C9
- 4-PI
4-phenylimidazole
- FLU
fluconazole
- PDB
Protein Data Bank
Footnotes
Supporting Information Available: 1) The cc-aug-pVDZ basis set and effective core potential used for calculations and tables of Cartesian coordinates for geometries used for Table 2; 2) The chromatographic data for target compounds. These materials are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Locuson CW, Wahlstrom JL, Rock DA, Rock DA, Jones JP. A new class of CYP2C9 inhibitors: Probing 2C9 specificity with high-affinity benzbromarone derivatives. Drug Metab. Dispos. 2003;31:967–971. doi: 10.1124/dmd.31.7.967. [DOI] [PubMed] [Google Scholar]
- 2.Locuson CW, Suzuki H, Rettie AE, Jones JP. Charge and substituent effects on affinity and metabolism of benzbromarone-based CYP2C19 inhibitors. J. Med. Chem. 2004;47:6768–6776. doi: 10.1021/jm049605m. [DOI] [PubMed] [Google Scholar]
- 3.Locuson CW, Rock DA, Jones JP. Quantitative binding models for CYP2C9 based on benzbromarone analogues. Biochemistry. 2004;43:6948–6958. doi: 10.1021/bi049651o. [DOI] [PubMed] [Google Scholar]
- 4.Rettie AE, Jones JP. Clinical and toxicological relevance of CYP2C9: Drug-drug interactions and pharmacogenetics. Annual Review of Pharmacology and Toxicology. 2005;45:477–494. doi: 10.1146/annurev.pharmtox.45.120403.095821. [DOI] [PubMed] [Google Scholar]
- 5.Hudelson MG, Jones JP. Line-walking method for predicting the inhibition of P450 drug metabolism. J. Med. Chem. 2006;49:4367–73. doi: 10.1021/jm0601553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hudelson MG, Ketkar NS, Holder LB, Carlson TJ, Peng CC, Waldher BJ, Jones JP. High confidence predictions of drug-drug interactions: predicting affinities for cytochrome P450 2C9 with multiple computational methods. J. Med. Chem. 2008;51:648–54. doi: 10.1021/jm701130z. [DOI] [PubMed] [Google Scholar]
- 7.Schenkman JB. Studies on the nature of the type I and type II spectral changes in liver microsomes. Biochemistry. 1970;9:2081–91. doi: 10.1021/bi00812a009. [DOI] [PubMed] [Google Scholar]
- 8.Kumaki K, Sato M, Kon H, Nebert DW. Correlation of type I, type II, and reverse type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J Biol Chem. 1978;253:1048–58. [PubMed] [Google Scholar]
- 9.Schenkman JB, Remmer H, Estabrook RW. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 10.Locuson CW, Hutzler JM, Tracy TS. Visible spectra of type II cytochrome P450-drug complexes: evidence that “incomplete” heme coordination is common. Drug Metab. Dispos. 2007;35:614–22. doi: 10.1124/dmd.106.012609. [DOI] [PubMed] [Google Scholar]
- 11.Chiba M, Jin L, Neway W, Vacca JP, Tata JR, Chapman K, Lin JH. P450 interaction with HIV protease inhibitors: relationship between metabolic stability, inhibitory potency, and P450 binding spectra. Drug Metab. Dispos. 2001;29:1–3. [PubMed] [Google Scholar]
- 12.Roberts AG, Campbell AP, Atkins WM. The thermodynamic landscape of testosterone binding to cytochrome P 450 3A4: Ligand binding and spin state equilibria. Biochemistry. 2005;44:1353–1366. doi: 10.1021/bi0481390. [DOI] [PubMed] [Google Scholar]
- 13.LeLean JE, Moon N, Dunham WR, Coon MJ. EPR Spectrometry of Cytochrome P450 2B4: Effects of Mutations and Substrate Binding. Biochem. Biophys. Res. Commun. 2000;276:762–766. doi: 10.1006/bbrc.2000.3539. [DOI] [PubMed] [Google Scholar]
- 14.Tsai R, Yu CA, Gunsalus IC, Peisach J, Blumberg W, Orme-Johnson WH, Beinert H. Spin-state changes in cytochrome P-450cam on binding of specific substrates. Proc. Natl. Acad. Sci. U. S. A. 1970;66:1157–63. doi: 10.1073/pnas.66.4.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiba M, Tang C, Neway WE, Williams TM, Desolms SJ, Dinsmore CJ, Wai JS, Lin JH. P450 interaction with farnesyl-protein transferase inhibitors. Metabolic stability, inhibitory potency, and P450 binding spectra in human liver microsomes. Biochem. Pharmacol. 2001;62:773–776. doi: 10.1016/s0006-2952(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 16.Johnston SR, Martin LA, Head J, Smith I, Dowsett M. Aromatase inhibitors: combinations with fulvestrant or signal transduction inhibitors as a strategy to overcome endocrine resistance. J. Steroid Biochem. Mol. Biol. 2005;95:173–81. doi: 10.1016/j.jsbmb.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Denton TT, Zhang X, Cashman JR. Nicotine-related alkaloids and metabolites as inhibitors of human cytochrome P-450 2A6. Biochem. Pharmacol. 2004;67:751–6. doi: 10.1016/j.bcp.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 18.Sellers EM, Kaplan HL, Tyndale RF. Inhibition of cytochrome P450 2A6 increases nicotine's oral bioavailability and decreases smoking. Clin Pharmacol Ther. 2000;68:35–43. doi: 10.1067/mcp.2000.107651. [DOI] [PubMed] [Google Scholar]
- 19.Ekins S, Mankowski DC, Hoover DJ, Lawton MP, Treadway JL, Harwood HJ., Jr. Three-dimensional quantitative structure-activity relationship analysis of human CYP51 inhibitors. Drug Metab. Dispos. 2007;35:493–500. doi: 10.1124/dmd.106.013888. [DOI] [PubMed] [Google Scholar]
- 20.Gibbons GF. The role of cytochrome P450 in the regulation of cholesterol biosynthesis. Lipids. 2002;37:1163–70. doi: 10.1007/s11745-002-1016-x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos. 2002;30:314–8. doi: 10.1124/dmd.30.3.314. [DOI] [PubMed] [Google Scholar]
- 22.Zarn JA, Bruschweiler BJ, Schlatter JR. Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14 alpha-demethylase and aromatase. Environ. Health Perspect. 2003;111:255–261. doi: 10.1289/ehp.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 24.Rettie AE, Jones JP. CLINICAL AND TOXICOLOGICAL RELEVANCE OF CYP2C9: Drug-Drug Interactions and Pharmacogenetics. Annual Review of Pharmacology and Toxicology. 2005;45:477–494. doi: 10.1146/annurev.pharmtox.45.120403.095821. [DOI] [PubMed] [Google Scholar]
- 25.Rushmore TH, Reider PJ, Slaughter D, Assang C, Shou M. Bioreactor systems in drug metabolism: synthesis of cytochrome P450-generated metabolites. Metabolic engineering. 2000;2:115–125. doi: 10.1006/mben.2000.0147. [DOI] [PubMed] [Google Scholar]
- 26.BD . 4‘-Hydroxydiclofenac Standard. BD Biosciences; San Jose, CA USA: 2005. [Google Scholar]
- 27.Shaw PM, Hosea NA, Thompson DV, Lenius JM, Guengerich FP. Reconstitution premixes for assays using purified recombinant human cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b5. Arch. Biochem. Biophys. 1997;348:107–115. doi: 10.1006/abbi.1997.0378. [DOI] [PubMed] [Google Scholar]
- 28.Locuson CW, Gannett PM, Tracy TS. Heteroactivator effects on the coupling and spin state equilibrium of CYP2C9. Arch. Biochem. Biophys. 2006;449:115–129. doi: 10.1016/j.abb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Jefcoate CR. Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 1978:52. doi: 10.1016/s0076-6879(78)52029-6. [DOI] [PubMed] [Google Scholar]
- 30.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P4502C9 complexed with flurbiprofen at 2.0-angstrom resolution. J. Biol. Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J, J. A, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 32.Staroverov VN, Scuseria GE, Tao JM, Perdew JP. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003;119:12129–12137. [Google Scholar]
- 33.Peterson KA, Dolg M. Unpublished results.
- 34.Woon DE, Dunning TH. Gaussian-Basis Sets for Use in Correlated Molecular Calculations .3. The Atoms Aluminum through Argon. J. Chem. Phys. 1993;98:1358–1371. [Google Scholar]
- 35.Kendall RA, Dunning TH, Harrison RJ. Electron-Affinities of the 1st-Row Atoms Revisited - Systematic Basis-Sets and Wave-Functions. J. Chem. Phys. 1992;96:6796–6806. [Google Scholar]
- 36.Dunning TH. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989;90:1007–1023. [Google Scholar]
- 37.Cho KB, Moreau Y, Kumar D, Rock DA, Jones JP, Shaik S. Formation of the active species of cytochrome P450 by using iodosylbenzene: A case for spin-selective reactivity. Chemistry-a European Journal. 2007;13:4103–4115. doi: 10.1002/chem.200601704. [DOI] [PubMed] [Google Scholar]
- 38.Schenkman JB, Sligar SG, Cinti DL. Substrate interaction with cytochrome P-450. Pharmacol. Ther. 1981;12:43–71. doi: 10.1016/0163-7258(81)90075-9. [DOI] [PubMed] [Google Scholar]
- 39.Dawson JH, Andersson LA, Sono M. Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J. Biol. Chem. 1982;257:3606–3617. [PubMed] [Google Scholar]
- 40.Verras A, Kuntz ID, Ortiz de Montellano PR. Computer-assisted design of selective imidazole inhibitors for cytochrome p450 enzymes. J. Med. Chem. 2004;47:3572–9. doi: 10.1021/jm030608t. [DOI] [PubMed] [Google Scholar]
- 41.Ahlstroem MM, Zamora I. Characterization of Type II Ligands in CYP2C9 and CYP3A4. J. Med. Chem. 2008;51:1755–1763. doi: 10.1021/jm701121y. [DOI] [PubMed] [Google Scholar]
- 42.Strickler M, Goldstein BM, Maxfield K, Shireman L, Kim G, Matteson DS, Jones JP. Crystallographic studies on the complex behavior of nicotine binding to P450cam (CYP101). Biochemistry. 2003;42:11943–50. doi: 10.1021/bi034833o. [DOI] [PubMed] [Google Scholar]
- 43.Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14alpha -sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cupp-Vickery JR, Garcia C, Hofacre A, McGee-Estrada K. Ketoconazole-induced conformational changes in the active site of cytochrome P450eryF. J. Mol. Biol. 2001;311:101–110. doi: 10.1006/jmbi.2001.4803. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, White MA, Muralidhara BK, Sun L, Halpert JR, Stout CD. Structure of Microsomal Cytochrome P450 2B4 Complexed with the Antifungal Drug Bifonazole: insight into P450 conformational plasticity and membrane interaction. J. Biol. Chem. 2006;281:5973–5981. doi: 10.1074/jbc.M511464200. [DOI] [PubMed] [Google Scholar]
- 46.Pellequer J-L, Zhao B, Kao H-I, Bell CW, Li K, Li QX, Karu AE, Roberts VA. Stabilization of Bound Polycyclic Aromatic Hydrocarbons by a p-Cation Interaction. J. Mol. Biol. 2000;302:691–699. doi: 10.1006/jmbi.2000.4033. [DOI] [PubMed] [Google Scholar]
- 47.Dougherty DA, Stauffer DA. Acetylcholine binding by a synthetic receptor: implications for biological recognition. Science (Washington, DC, United States) 1990;250:1558–1560. doi: 10.1126/science.2274786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.