

Abstract
The activation of T cells and the initiation of an immune response is tightly controlled through the crosstalk of both positive and negative regulators. Two adaptors which function as negative regulators of T cell activation are ALX and LAX. Previously, we showed that T cells from mice deficient in ALX and LAX display similar hyperresponsiveness, with increased IL-2 production and proliferation upon TCR/CD28 stimulation, and that these adaptors physically associate. Here, we analyze the nature of the association between ALX and LAX. We demonstrate that this association occurs in the absence of TCR/CD28 signaling via a mechanism independent of both tyrosine phosphorylation of LAX and the SH2 domain of ALX. Cotransfection of ALX with LAX resulted in LAX tyrosine phosphorylation in the absence of TCR/CD28 stimulation. ALX-mediated LAX phosphorylation depends upon the ALX SH2 domain, which functions to recruit Lck to LAX. We also show that LAX, like ALX, can inhibit RE/AP reporter activation. However, in contrast to its inhibition of NFAT, the inhibition of RE/AP by LAX is independent of its tyrosine phosphorylation. Therefore, it can be concluded that inhibition of signaling events involved in T cell activation by LAX occurs through mechanisms both dependent on and independent of its tyrosine phosphorylation.
Introduction
T cell activation occurs via the integration of signals from cell surface receptors. Minimally, two signals are required for T cell activation: an antigen-dependent signal generated through the T cell receptor (TCR), and an antigen-independent costimulatory signal primarily provided by CD28 in naïve T cells (1). If only TCR signals are received in the absence of costimulatory signals, T cells become anergic rather than activated. TCR/CD28 signaling initiates a cascade of events, starting with tyrosine phosphorylation of CD3/ζ chains by Src family kinases and recruitment of Syk family tyrosine kinases (2). Downstream pathways are subsequently triggered, leading to events including activation of mitogen-activated protein (MAP) kinases, and the transcription factors NF-κB, AP-1 and NFAT. One of the outcomes of appropriate T cell activation via TCR/CD28 is the transcriptional activation of interleukin-2 (IL-2) (3). The IL-2 promoter contains several transcription factor binding sites, including those for NFAT and AP-1. However, these elements can be activated by TCR signaling alone in reporter assays. A composite element from the IL-2 promoter designated RE/AP, which requires CD28 signals for activation, is the site at which costimulatory signals act, leading to IL-2 upregulation (4, 5).
Adaptor proteins regulate several critical steps of T cell activation. For example, the transmembrane adaptor LAT and cytoplasmic adaptor SLP-76 organize key regulators of TCR signaling into a ‘signalosome’. In mice deficient for either adaptor, T cells fail to develop, and cell lines deficient in either fail to transduce TCR signals leading to IL-2 (6, 7). Adaptors also can have negative functions in T cells. Previously, we identified an adaptor in T cells designated ALX (also known as HSH2) (8). Overexpression of ALX in the Jurkat T cell line inhibited the activation of RE/AP, but not AP-1 reporters, upon TCR/CD28 stimulation, suggesting that it functioned as a negative regulator of T cell activation. Its negative role was confirmed when ALX-deficient mice were generated and examined; ALX-deficient T cells demonstrated enhanced IL-2 production and proliferation upon TCR/CD28 stimulation (9). Analysis of signaling pathways in ALX-deficient T cells demonstrated no alterations in the level or kinetics of proximal induction of tyrosine phosphorylation, calcium flux, ERK, JNK, IKK or Akt in response to TCR/CD28 stimulation. However, ALX-deficient splenocytes exhibited constitutive activation of the p38 map kinase, through the classical MKK3/6 pathway, that was not further enhanced by TCR/CD28 stimulation.
The T cell phenotype in ALX-deficient mice was similar to that observed in mice lacking the transmembrane adaptor LAX (10), and a physical interaction between ALX and LAX was subsequently discovered (9). Overexpression of LAX has been shown to result in inhibition NFAT and AP-1 upon TCR activation, and this inhibition is dependent on four sites of tyrosine phosphorylation (11). However, ALX overexpression results in inhibition of RE/AP activation, with little effect on either an NFAT or AP-1 reporter (8). If the association between ALX and LAX is important, LAX should also inhibit RE/AP reporter activation as well, which may depend on the association with ALX. Here, we demonstrate that the association between ALX and LAX occurs constitutively, and is unaffected by TCR/CD28 stimulation via a mechanism independent of both tyrosine phosphorylation of LAX and the SH2 domain ALX. Coexpression of ALX and LAX in Jurkat T cells increased tyrosine phosphorylation of LAX. ALX-driven LAX phosphorylation required the ALX SH2 domain and was absent in the Lck-deficient J.CaM1 Jurkat cell line. The ALX SH2 domain associates directly with Lck, and recruits Lck to LAX. We also show that LAX, like ALX, can inhibit RE/AP. However, in contrast to its inhibition of NFAT, the inhibition of RE/AP by LAX is independent of its tyrosine phosphorylation, and correlates with its ability to bind to the ALX C-terminal fragment. Therefore, inhibition of signaling events involved in T cell activation by LAX occurs through mechanisms both dependent on and independent of its tyrosine phosphorylation, and ALX may play a part in both of these activities.
Experimental procedures
Expression plasmids
Full length wild type (wt) LAX and the 4YF mutant have been previously described (11). LAX truncations were generated by PCR cloning. The amino acids contained in the LAX truncations are as follows: LAX D1, aa 1-344; LAX D2, aa 1-293; LAX D3, 1-233; LAX D4, aa 1-169; LAX D5, 1-111; LAX TL, aa 1-67. All LAX expression constructs were subcloned into the pEF6 myc-His A vector (Invitrogen), which introduces an carboxy-terminal myc-epitope tag. Untagged WT ALX and the ALX R/K mutant, in which arginine 59 is replaced by lysine, were described previously (12). All ALX truncation mutants used herein contain a portion of ALX fused to the amino terminus of yellow fluorescent protein (YFP). The ALX ΔC construct contains the residues c-terminal to the ALX SH2 domain, aa136-352. The other ALX truncation mutants have been previously described (12). The NFAT and RE/AP luciferase reporters have been previously described (4, 13).
Jurkat transfections and luciferase assays
Transfections, stimulations and luciferase assays were performed as previously described (4, 13). Briefly, 15 × 106 Jurkat T cells were washed once, and resuspended in 0.4 ml of serum-free RPMI. 10 μg of reporter or various amounts of expression plasmid (figure legend) were added. Electroporation was performed using a Gene Pulser II (BioRad) at 250 volts, 950 μF. Cells were resuspended in 10 mls of RPMI with 5% FCS (Gibco BRL). The following day, live cells were counted by trypan blue exclusion (Bio-Whittaker), and 1 × 105 cells per sample were stimulated as denoted in the figures. Cells were left unstimulated or stimulated with antibodies to TCR (C305, 1:1000 final dilution) and CD28 (1 μg/ml) for 7 hours. Luciferase assays were performed as previously described(13).
Immunoprecipitations and immunoblots
Jurkat cells were transfected by electroporation as above with ALX expression plasmids and/or myc-LAX expression plasmids (15 μg of each plasmid or appropriate vector control were used). After overnight recovery, cells were resuspended at 15 × 106/ml in phosphate buffered saline. Cells were incubated for 30 min. at 37 °C and then, if indicated, stimulated with antibodies to TCR (C305, 1:200 final dilution) and CD28 (5 μg/ml) for two minutes immediately prior to lysis. 293T cells were transfected with YFP-ALX expression plasmids combined with myc-LAX expression plasmids using Fugene6 reagent (Roche Applied Science) according to the manufacturer’s instructions (for each combination, 1 μg of each plasmid and 6 μl of Fugene6 was used to transfect cells in one well of a 6 well dish). Cells were incubated with transfection cocktails overnight prior to lysis. For either cell type, lysates were generated with NP-40 buffer and immunoprecipitation performed with anti-myc antibody followed by elution with myc peptide as previously described (9). Alternatively, immunoprecipitations were performed with monoclonal antibodies to YFP (Roche Applied Science). Cell lysates and immunoprecipitates were loaded on denaturing gels and analyzed by western blotting with rabbit antibodies to ALX (8), LAX (11), Myc (Cell Signaling Technologies), or YFP (Cell Signaling Cell Signaling Technologies). Phosphotyrosine containing proteins were detected with the 4G10 mouse monoclonal antibody (Upstate Biotechnology). Lck and LAT were detected in cell lysates with antibodies from Cell Signaling Cell Signaling Technologies.
Glutathione S-Transferase (GST) precipitations
A fusion protein composed of the SH2 domain of ALX fused to GST (GST-SH2) as well as unfused GST were expressed in bacteria and purified as described previously (12). Jurkat cells were stimulated and lysed with NP-40 buffer as described for immunoprecipitation experiments. Approximately 20 μg of GST or GST-SH2 protein bound to glutathione sepharose beads were added to each lysate, which was then rotated for 2 hr at 4 °C. Beads were washed five times in NP-40 buffer and then loaded on denaturing gels for western blotting with antibodies to Lck or to GST (Amersham).
Results
It was previously shown that four tyrosine residues in the cytoplasmic tail of the transmembrane adaptor LAX are inducibly phosphorylated upon TCR activation (11). Since ALX and LAX associate, and T cells deficient in either adaptor exhibit similar hyperresponsiveness to TCR/CD28 stimulation, we hypothesized that the SH2 domain of ALX may bind to phosphotyrosines in LAX. This model would suggest that TCR induced phosphorylation of LAX would increase its association with ALX and that an ALX mutant unable to bind phosphotyrosines would not associate with LAX. To test these predictions, Jurkat T cells were transfected with wild type (WT) ALX, myc-epitope tagged LAX, or both, along with vector controls. The cells were left unstimulated or treated with antibodies to TCR and CD28, and LAX was subsequently immunoprecipitated with antibodies to the myc tag. Jurkat T cells were stimulated for two minutes, which was previously shown to be the optimal time point for LAX phosphorylation (11). As shown in figure 1A, similar to previous work (9), ALX coprecipitated with LAX. This association was constitutive and not increased upon TCR/CD28 stimulation. Efficacy of stimulation was confirmed by blotting whole cell extracts, as well as LAX immunoprecipitations, with an antibody to phosphotyrosine (4G10, figure 1A, bottom panel). Interestingly, mutating the ALX SH2 domain (ALX R/K), did not alter association with LAX (figure 1A). This R/K mutation was previously shown to abrogate binding of tyrosine phosphorylated proteins from Jurkat cell extracts (12), implying that the association of ALX and LAX occurred via a mechanism that did not involve LAX tyrosine phosphorylation. Taken together, these results suggest that the association of ALX and LAX does not depend on TCR activation or phosphotyrosine recognition by the ALX SH2 domain.
Figure 1.
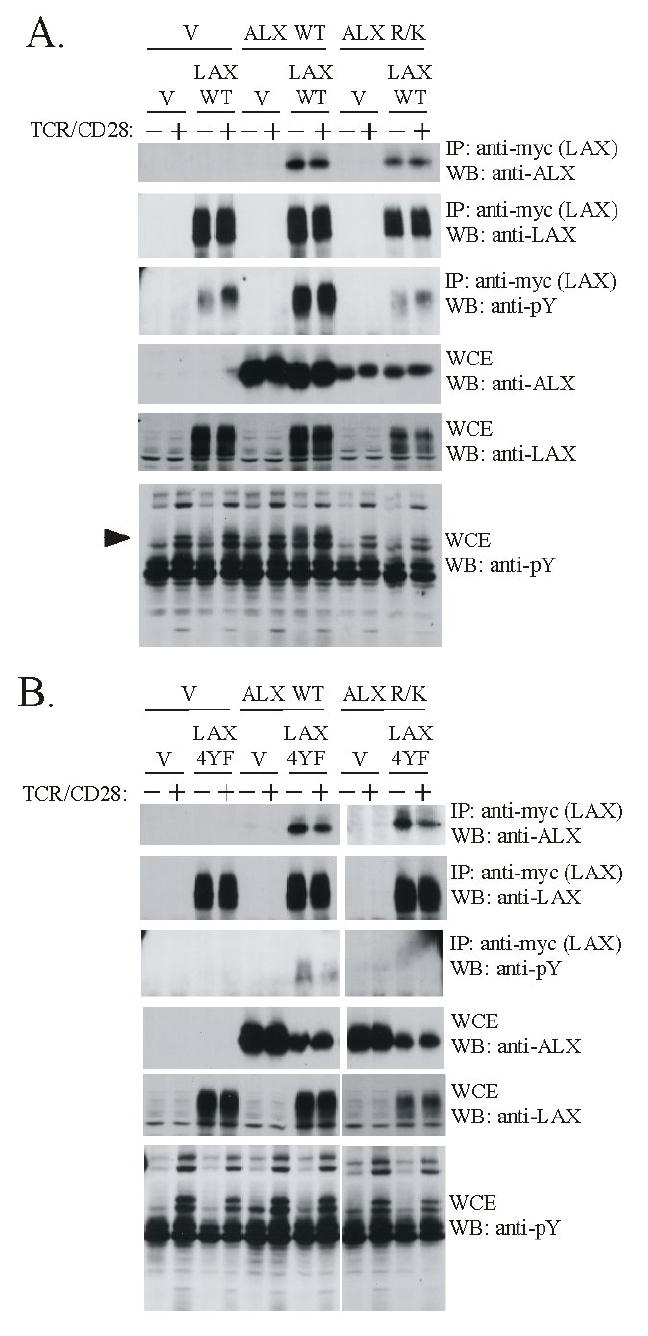
The association of ALX with LAX occurs independently of phosphotyrosine recognition. Jurkat cells were co-transfected with expression plasmids for either wild type ALX or an ALX SH2 domain mutant incapable of phosphotyrosine binding (ALX R/K) along with either (A) wild type LAX or (B) a LAX mutant (LAX 4YF) in which the four sites of tyrosine phosphorylation were replaced. Empty vector (V) was used to standardize total plasmid transfected between samples. The cells were left unstimulated (-) or stimulated for 2 min. (+) with antibodies to TCR and CD28. Whole cell extracts were made (WCE) and subject to immunoprecipitation (IP) with antibodies to a myc-epitope present on LAX. Western blotting (WB) was then performed with the indicated antibodies to analyze expression of LAX and ALX in the WCEs and IPs. Note that LAX appears as a “smear” on immunoblots, which is likely an effect of glycosylation, as is often found in transmembrane proteins. The minor, lower molecular weight bands observed in LAX immunoblots are likely incompletely modified forms of the protein. Anti-phosphotyrosine blotting with 4G10 confirmed TCR/CD28 stimulation, as well as LAX phosphorylation. The arrowhead indicates the position of a tyrosine phosphorylated band present in unstimulated cells only upon transfection with both WT ALX and LAX. This band is likely LAX, based on molecular weight, and consistent with the substantial increase in phosphorylated LAX in the immunoprecipitates observed upon cotransfection with ALX.
To confirm that the association between ALX and LAX is phosphotyrosine independent, experiments were performed with a LAX mutant in which four tyrosine residues (Y193, Y268, Y294 and Y373) are changed to phenylalanine. Previously, these substitutions were shown to eliminate TCR induced phosphorylation of LAX as well as abrogating the ability of LAX to inhibit activation of NFAT, NF-κB or AP-1 reporters (11). As shown in figure 1B, WT ALX coprecipitated with LAX 4YF. In addition, the ALX R/K SH2 domain mutant also associated with the non-phosphorylated LAX 4YF mutant. Similarly, the associations of WT and ALX R/K with LAX 4YF are independent of TCR/CD28 stimulation. Therefore, ALX and LAX associate independent of LAX tyrosine phosphorylation or phosphotyrosine recognition by the SH2 domain of ALX.
Although the ALX SH2 domain does not function to recruit ALX to LAX, it was found to be important for LAX tyrosine phosphorylation. As shown in figure 1A (panel 3), LAX is inducibly tyrosine phosphorylated upon TCR/CD28 stimulation. Surprisingly, the extent of LAX tyrosine phosphorylation in unstimulated cells was substantially increased upon cotransfection of WT ALX, and was not further enhanced by TCR/CD28 stimulation. However, no increase in LAX phosphorylation was observed upon cotransfection of with the ALX SH2 domain mutant (ALX R/K, figure 1A). This increase in LAX tyrosine phosphorylation was significantly lost when ALX was cotransfected with LAX 4YF mutant, demonstrating that the effect of ALX is directed at the physiologically relevant sites in LAX phosphorylated during T cell activation. These results suggest that while the binding of ALX to LAX does not depend on LAX tyrosine phosphorylation, ALX must, through its SH2 domain, recruit additional protein(s) that result in LAX tyrosine phosphorylation. Alternatively, ALX could activate a tyrosine kinase in a general way without coupling it to specific substrates. However, this seems unlikely as ALX expression alone did not grossly alter the pattern of tyrosine phosphorylated proteins in Jurkat whole cell extracts (figures 1A and 1B, bottom panel). In fact, the only increase in tyrosine phosphorylation in the whole cell extracts appears to be LAX itself (figure 1A, marked by arrow in bottom panel), as a band of appropriate molecular weight was only present when WT LAX but not LAX 4YF was cotransfected (compare lane 7 of bottom panels of figures 1A and 1B). Therefore, the phosphotyrosine-independent recognition of LAX by ALX promotes the phosphorylation of LAX, and presumably, phosphotyrosine-dependent interactions between LAX and additional partners involved in regulating T cell activation.
TCR engagement results in the activation of the tyrosine kinase Lck, initiating a cascade of events leading to activation of downstream pathways. Lck has been implicated in LAX tyrosine phosphorylation: Lck overexpression leads to LAX constitutive phosphorylation, and LAX is not inducibly phosphorylated by TCR in the J.Cam1 Jurkat cell line, which is deficient in Lck (11, 14). Thus, ALX-driven LAX phosphorylation may depend on Lck recruitment. To test this model, ALX and LAX cotransfections were repeated using J.Cam1 cells. As shown in figure 2A, cotransfection of ALX with LAX resulted in substantial constitutive LAX phosphorylation in Jurkat, but almost none in JCam1, although association of ALX with LAX was similar in both cell lines. ALX cotransfection stimulated LAX phosphorylation in p116 cells, which lack the tyrosine kinase ZAP-70 (figure 2B, (15)), and in JCam2 cells that lack LAT, an adaptor critical to TCR signaling (data not shown, (16)). Absence Lck, ZAP-70 and LAT in these cell lines was confirmed by Western blot (figures 2A, 2B and data not shown). These results suggest that upon association with LAX, ALX stimulates its phosphorylation in an Lck-dependent manner. To determine if the ALX SH2 domain directly bound to Lck, GST pull down experiments were performed using a fusion protein containing only the SH2 domain of ALX. As shown in figure 2C, Lck was effectively pulled down from Jurkat extracts with a GST ALX SH2 domain fusion protein, but is not with GST alone. J.Cam1 extracts were used as a negative control. The association between the ALX SH2 domain and Lck was constitutive, and was not altered by TCR/CD28 stimulation. We have thus far been unable to demonstrate an interaction between ALX and Lck in coimmunoprecipitation experiments (data not shown), though this does not eliminate the possibility that the association is weak but yet sufficient for transient recruitment of Lck to LAX resulting in LAX tyrosine phosphorylation. Together, the above data suggests that ALX recruits Lck to LAX via its SH2 domain, resulting in LAX tyrosine phosphorylation.
Figure 2.
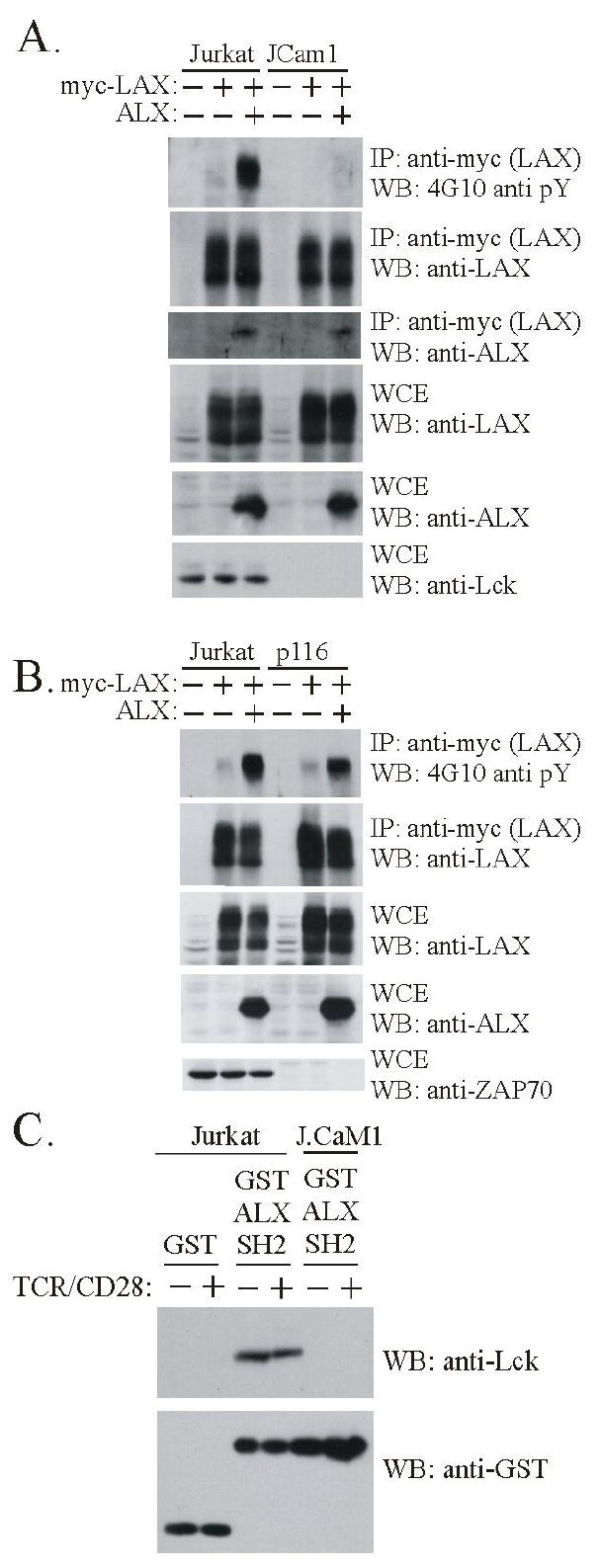
ALX promotes phosphorylation of LAX dependent on the tyrosine kinase Lck. (A) Cells were transfected and subject to immunoprecipitation without TCR stimulation as in Fig. 1 except that, as indicated, Jurkat or J.Cam1 Jurkat mutant (Lck deficient) cell lines were used. Each cell line was transfected with LAX alone or in combination with ALX. Transfection of an expression construct (+) or substitution of empty vector (-) is indicated. Immunoprecipitates and WCE were analyzed by Western blot with antibodies to phosphotyrosine (4G10) to assess LAX phosphorylation, as well as LAX to confirm effective immunoprecipitation, and ALX to confirm association. WCE were analyzed with antibodies to LAX and ALX to assess expression, and against Lck to confirm the deficiencies in JCam1 (B) Examination of the requirement for ZAP-70 in ALX-mediated phosphorylation was examined in p116 ZAP-70 deficient Jurkat T cell line, as was done with JCam1 in part (A) above. (C) Extracts were made from either Jurkat or J.Cam1 cells that were either unstimulated (-) or stimulated for 2 minutes (+) with antibodies to TCR and CD28. Extracts were incubated with either GST or a recombinant protein composed of GST and the SH2 domain of ALX (GST ALX SH2), both bound to beads. The beads were precipitated and washed before being analyzed by western blotting. The anti-Lck blot shows that Lck bound specifically to the SH2 domain of ALX (upper panel). Western blotting with antibodies to GST demonstrates the presence of equivalent amounts of fusion protein in each precipitate (lower panel).
To map the sites within ALX required for the association with LAX, cotransfections were repeated using a series of ALX truncations mutants (shown schematically in figure 3A). 293T cells were utilized for these experiments, rather than Jurkat T cells, since 293T cells lack Lck, and LAX is not phosphorylated in these cells (data not shown, and (11)). This eliminates the potential for indirect associations between ALX and LAX mediated either by Lck or proteins that associate with tyrosine phosphorylation sites within LAX. As shown in figure 3B, the isolated SH2 domain of ALX lacked any ability to interact with LAX. This same ALX truncation has been shown to inhibit RE/AP activation upon overexpression, and a purified fusion protein containing the SH2 domain was able to bind phosphotyrosine containing proteins from Jurkat extracts (12). Hence, it is unlikely that the lack of association of the ALX SH2 domain with LAX is due to a structural change in the SH2 domain when it is expressed in isolation. Thus, combined with the experiments utilizing ALX R/K, it can be concluded the SH2 domain of ALX is neither necessary nor sufficient for the association with LAX. ALX truncations lacking either the N- or C- terminal segments (ALX ΔN and ΔC, respectively) retained the ability to bind to LAX. Since the ALX SH2 domain does not associate with LAX, this implies that ALX contains two separate binding sites for LAX, located both N- and C-terminal to the SH2 domain. The C-terminal segment in isolation (ALX C mutant) was also shown to associate with LAX (figure 3B). In particular, the ALX ΔC and ALX C truncations are mutually exclusive, containing aa 1-134 and 136-352 of ALX. The binding of each of these truncations (ALX ΔC and ALX C) is minimally reduced as compared to WT ALX, indicating that there does not appear to be cooperativity between these two binding sites for LAX.
Figure 3.
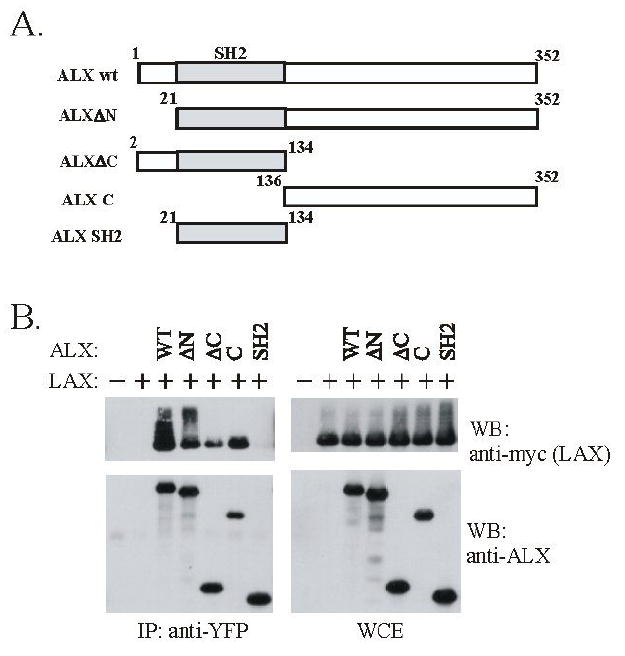
ALX associates with LAX via two distinct regions. (A) Shown is a schematic of the ALX truncations utilized to map the site(s) of LAX interactions. The gray shaded area represents the ALX SH2 domain. The schematic is not shown to scale. (B) Expression plasmids for myc-tagged wt LAX were co-transfected into 293T cells with YFP-tagged wt ALX or ALX truncation mutants along with vector controls (in the first lane, cells were transfected with unfused YFP and empty vector, in the second, with YFP and LAX). Whole cell extracts were generated and subject to immunoprecipitation with antibodies to YFP. Western blotting was then performed with antibodies to ALX, and myc (LAX) to assess protein expression, immunoprecipitation of ALX, and co-precipitation of LAX.
Based upon the association results, if LAX and ALX function together to inhibit T cell activation, then the LAX 4YF mutant may retain some ability to negatively regulate T cell activation. Previously, it has been shown that the LAX 4YF mutation resulted in a loss of ability to inhibit AP-1 and NFAT reporters (11). However, ALX inhibits the activation of the RE/AP composite element from the IL-2 promoter, but does not substantially inhibit TCR-mediated activation of either NFAT or AP-1 (8). Therefore, we examined the ability of the both wt LAX and the 4YF mutant to inhibit activation of RE/AP. As shown in figure 4A, Jurkat T cells were transfected with an RE/AP luciferase reporter, and either vector or increasing amounts of WT LAX or LAX 4YF. TCR/CD28 stimulation activated the RE/AP reporter approximately 30-fold when cotransfected with vector control. Overexpression of LAX, as previously observed with ALX, caused a decrease in the activation of the RE/AP element of approximately 85% (p<0.01, by student’s t test). Interestingly, overexpression of LAX 4YF mutant inhibited the activation of RE/AP by approximately 70% (p<0.01, by student’s t test), nearly as well as WT LAX. WT LAX and the LAX 4YF mutant were expressed at similar levels (figure 4C). The small, but significant (p<0.01, by student’s t test), difference between inhibition of RE/AP by WT LAX and LAX 4YF indicates that LAX phosphorylation may also contribute towards RE/AP inhibition. As a control, the effect of LAX on an NFAT reporter was also examined (figure 4B). Consistent with previous results (11), overexpression of WT LAX blocked activation of NFAT upon TCR/CD28 stimulation (by approximately 80%, p<0.02, by student’s t test), and this ability was almost entirely abrogated by the 4YF mutation. Therefore, these results demonstrate that LAX can inhibit different signaling pathways in T cells in distinct ways: a phosphotyrosine dependent pathway primarily responsible for NFAT inhibition, and a separate pathway independent of LAX tyrosine phosphorylation largely responsible for inhibition of RE/AP.
Figure 4.
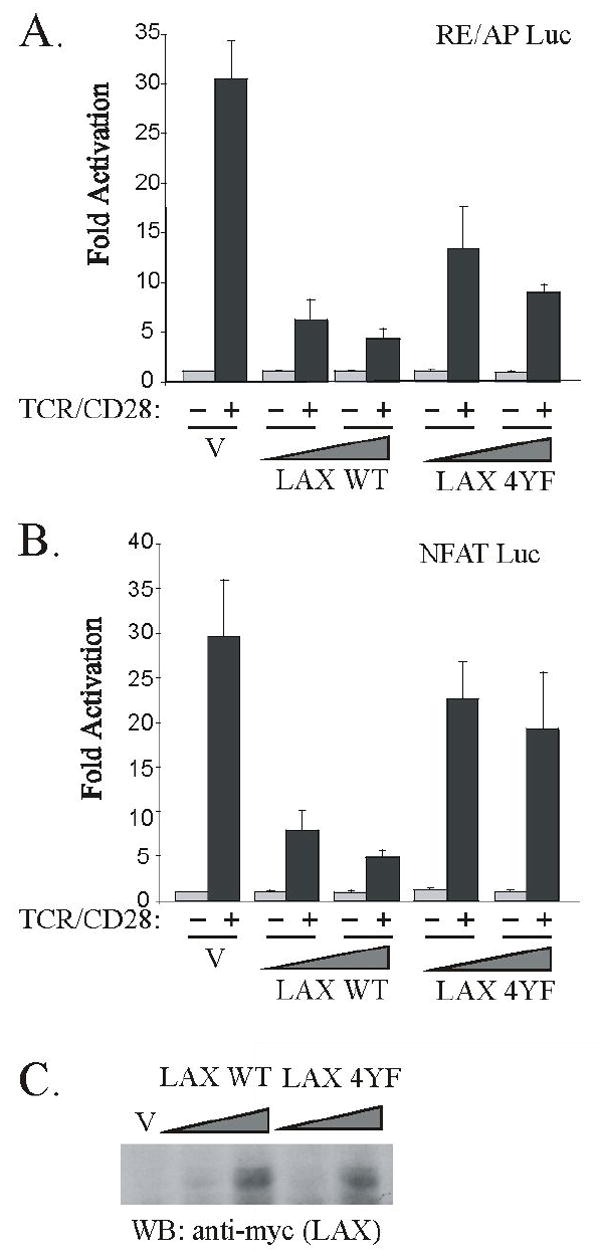
A non-phosphorylatable form of LAX retains the ability to inhibit TCR/CD28-induced activation of RE/AP. Jurkat T cells were transfected with 10 μg of either an (A) RE/AP or (B) NFAT luciferase reporter along with 3μg of either wt LAX or LAX 4YF expression plasmids (and 7 μg empty vector), or with 10μg of either LAX construct, or with 10 μg vector only (V). The following day, cells were left unstimulated or stimulated with anti-TCR/CD28 antibodies for 7 hours prior to assaying luciferase expression. Data is shown as fold activation relative to unstimulated samples. Error bars reflect SEM from two independent experiments. (C) An anti-myc western blot is also presented to show equivalent expression of LAX WT and LAX 4YF in the Jurkat lysates used for luciferase assays above.
To map the sites on LAX which are important for the association with ALX and inhibition of RE/AP, we generated a series of LAX truncation mutants, in which the c-terminal tail is progressively shortened in approximately 50 aa segments: LAX D1, D2, D3, D4 and TL containing aa 1-344, 1-293, 1-233, 1-169, 1-111 and 1-67, respectively. The truncations are shown schematically in figure 5A, along with the locations of the four tyrosines substituted in the 4YF mutant. 293T cells were transfected and coimmunoprecipitations performed with WT LAX and the truncation series combined with either WT ALX, ALX ΔC or ALX C. As shown in figure 5B, wt ALX associated with all the LAX constructs except LAX TL, which contains almost no cytoplasmic tail. ALX ΔC exhibited a similar pattern of association with the LAX truncation mutants (figure 5D). In contrast, ALX C associated only with wt LAX, LAX D1, D2 and D3 (figure 5C). These results suggest that ALX and LAX associate via two distinct sites; the C-terminal segment of ALX binds to the residues between the D3 and D4 truncations (aa 169-233) and the N-terminal segment between the D5 truncation and the start of the cytoplasmic tail (aa 67-11).
Figure 5.
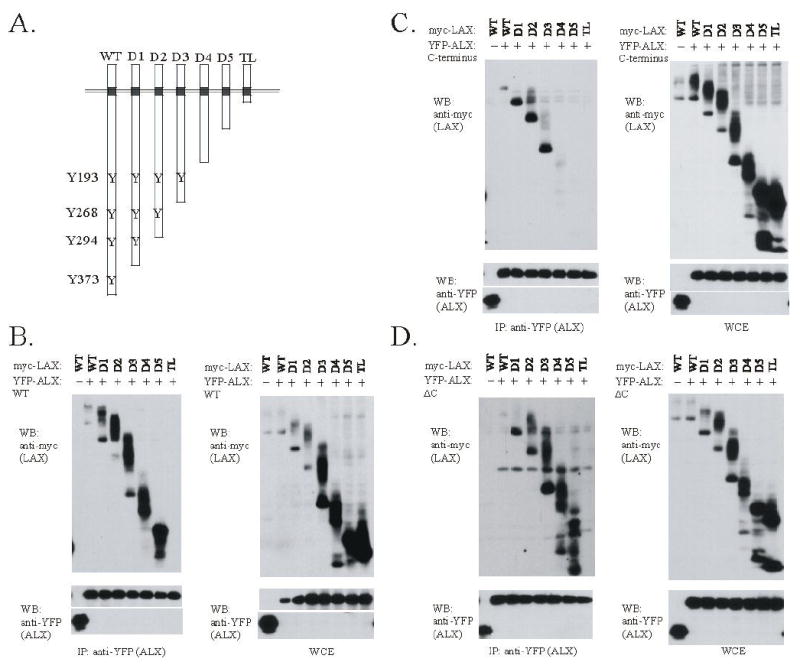
The association of ALX with LAX maps to two distinct site within the LAX cytoplasmic tail. (A) C-terminal truncation mutants of LAX are shown, with the four tyrosines in the cytoplasmic tail of LAX that are sites of TCR-induces tyrosine phosphorylation denoted. (B-D) Expression plasmids for myc-tagged wt LAX or the LAX truncations shown in fig. 5A were co-transfected into 293T cells with YFP-tagged ALX constructs. WT ALX (B), ALX C (C), or ALX ΔC (D) proteins were used. Each experiment was otherwise performed as in Fig. 3. Note that the LAX truncations were expressed at a higher level than WT LAX as shown in the whole cell extract controls, and any increase in association with ALX appears proportional to the increased expression.
If an association with ALX is required for LAX to inhibit RE/AP, it may occur in one of two ways. First, if the site of association or orientation between the proteins is not critical, all LAX truncations that can associate with wt ALX should inhibit RE/AP and only LAX TL would lack an inhibitory effect. Alternatively, if the orientation between ALX and LAX is critical, then the LAX D4, D5 and TL truncations, which fail to bind ALX C, should not inhibit RE/AP activation. As shown in figure 6A, Jurkat T cells were transfected with an RE/AP luciferase reporter, along with expression constructs for wt LAX and all the LAX truncations. WT LAX, LAX D1, D2, and D3 inhibited TCR/CD28-induced activation of the RE/AP reporter to similar extents. However, LAX D4, D5 and TL had no inhibitory effect on RE/AP. Therefore, the inhibition of RE/AP by LAX does not simply correlate with an ability to bind to ALX, but the orientation assumed by ALX with the cytoplasmic tail of LAX may be important.
Figure 6.
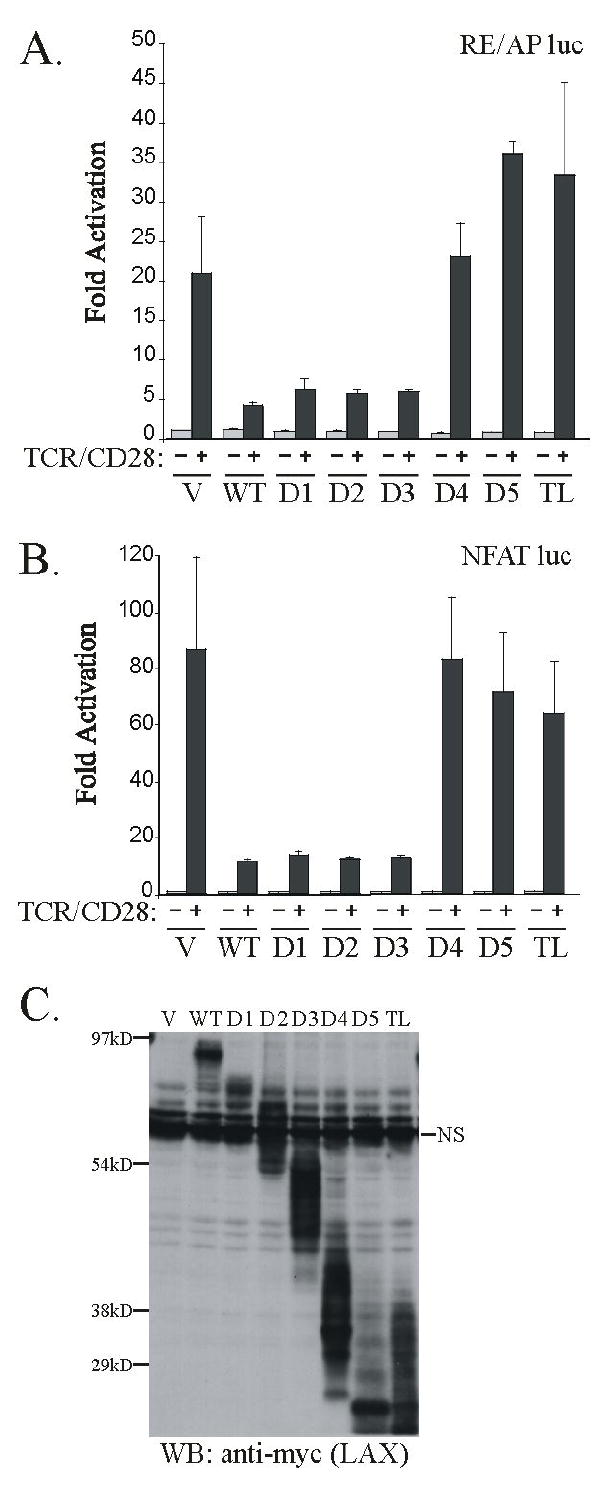
The region of LAX between the D3 and D4 truncations is critical for inhibition of both (A) RE/AP and (B) NFAT reporters. Jurkat T cells were transfected as in Fig. 4, using LAX truncations shown in Fig. 5A. Error bars reflect SEM from three independent experiments. (C) An anti-myc western blot is also presented to show expression of LAX in the Jurkat lysates used for luciferase assays above.
The inhibition of NFAT by LAX is abrogated in the 4YF mutant, indicating that at least one of the four tyrosines is crucial for this inhibition. We utilized the truncation mutants described above to determine which tyrosine is important for the inhition of NFAT by LAX. As diagramed in figure 5A, the D1, D2, D3, and D4 LAX truncations serve to eliminate, respectively, one, two, three, and all four tyrosines from LAX. As shown in figure 6B, Jurkat T cells were transfected with an NFAT luciferase reporter, along with expression constructs for WT LAX and all of the LAX truncations. NFAT activation was inhibited by wt LAX, as well as LAX D1, D2 and D3. However LAX D4, as well as the further truncated constructs, lacked all ability to inhibit NFAT. Since LAX D3 contains only one of the four tyrosines found to be critical for inhibition of NFAT, Y193, we can conclude that the presence of at least this tyrosine residue is necessary for LAX-mediated inhibition of NFAT. LAX Y193 is not sufficient, however, since a mutant of LAX in which only Y193 was mutated to phenylalanine inhibited NFAT activation to a similar extent as wildtype LAX (data not shown), most likely due to the presence of a similar and presumably redundant motifs at Y294 and Y393.
Discussion
The mechanism by which adaptors can negatively regulate signaling pathways is not well defined at present. LAX was originally identified as a transmembrane adaptor that contained similar tyrosine-containing motifs as the transmembrane adaptor LAT (11). LAT is absolutely required for TCR signaling (6). In LAT-deficient mice, T cells do not develop, and no TCR signaling occurs in a Jurkat T cell line deficient in LAT (16). T cell activation results in the tyrosine phosphorylation of LAT and subsequent recruitment of SH2 containing proteins including Grb2 and GADS, which nucleate a complex that transmits TCR signals to downstream pathways. LAX has also been shown to undergo tyrosine phosphorylation upon TCR signaling, leading to the recruitment of Grb2 and GADS. However, while LAT localizes to lipid rafts, LAX is excluded from them (11). In the LAX 4YF mutant, substitutions at four tyrosine residues were shown to eliminate both LAX phosphorylation and the protein associations triggerered by TCR signaling, as well as the inhibition of NFAT activation (11). Thus, one mechanism by which LAX may negatively regulate TCR signaling is to function as a “sink” for SH2 containing proteins that bind to its phosphotyrosines, thus sequestrating these proteins away from LAT and lipid rafts.
The results presented here illustrate a second mechanisms by which LAX can function in which it may nucleate a distinct complex that inhibits TCR signals. LAX was shown to inhibit activation of the RE/AP composite element, which integrates TCR and costimulatory signals leading to IL2 upregulation (4, 5). However, unlike the inhibition of NFAT, RE/AP activation was not abrogated in LAX 4YF, suggesting that a mechanism of inhibition independent of LAX tyrosine phosphorylation must exist. The cytoplasmic adaptor ALX was previously shown to bind to LAX, but not to LAT (9). ALX deficient mice display a phenotype similar to LAX deficient mice (9, 10), and ALX also inhibits RE/AP upon overexpression (8). Here, we show that the interaction between ALX and LAX is independent of LAX tyrosine phosphorylation as the association is observed in unstimulated cells and is not abrogated by mutations in ALX that impair phosphotyrosine binding or mutations in LAX that impair its phosphorylation. Taken together, these results illustrate how LAX can inhibit T cell signaling by a mechanism distinct from simply functioning as a molecular sink for signaling molecules that might otherwise positively regulate T cell activation.
The data presented here also clarify the structural features of ALX and LAX involved in their interaction and their inhibitory effects on signaling. The association of ALX and LAX involves two distinct sites, each interaction occuring independent of tyrosine phosphorylation: ALX ΔC associates with LAX between aa 67-111, while the C-terminal portion of ALX associates with LAX between aa 169-233. ALX contains a single SH2 domain and no other known structural motifs. Four conserved PxxP polyproline sequences, three of which are located outside of the SH2 domain, are present in ALX. However, LAX does not contain any modular binding motifs, such as SH3/WW domains, that associate directly with polyproline sequences. In fact, the polyproline sequences in ALX are not required for LAX association, as LAX associates with an ALX mutant in which these sequences are mutated (data not shown). Thus, the association of ALX and LAX must occur via mechanisms independent of SH2/phosphotyrosine as well as polyproline based interactions, and additional motifs that could mediate an interaction are not obvious. Future characterization of the interaction will require more refined truncations or scanning mutagenesis.
Finally, the truncation analyses presented here also defines the structural features of LAX required for inhibition of RE/AP and NFAT activation. Previously, four tyrosine containing motifs in the cytoplasmic tail of LAX were shown to be critical for its inhibition of NFAT (Y193, Y268, Y294 and Y373) as their combined mutation abrogates this inhibition (11). LAX D3, which retains Y193 but not Y268, Y294 or Y373, inhibits NFAT activation similar to wild type, while LAX D4, which lacks all four critical tyrosines, does not. Hence, inhibition of NFAT by LAX depend on the presence of at least one tyrosine phosphorylation site, and Y193 can serve this function. The truncation analysis also shows that inhibition of RE/AP by LAX depends on sequences contained within aa 169-233 (between D3 and D4 truncations), correlating with the sequences required for binding to ALX. The importance of this segment of LAX for NFAT inhibition may, in fact, reflect a dependence on ALX for LAX tyrosine phosphorylation.
The results presented here lead to a model of LAX function, shown in figure 7. In the absence of stimulation, non-phosphorylated LAX associates with ALX via two independent interactions. LAX is able to inhibit RE/AP activation without undergoing phosphorylation. ALX, via its SH2 domain, associates with Lck and recruits it to the complex with LAX. This results in LAX tyrosine phosphorylatiopn, enabling LAX to associate with additional signaling molecules, including Gads, Grb2 and PLCγ1. These associations allow LAX to inhibit activation of NFAT. Thus, LAX regulates T cell activation by two distinct but interconnected mechanisms.
Figure 7.
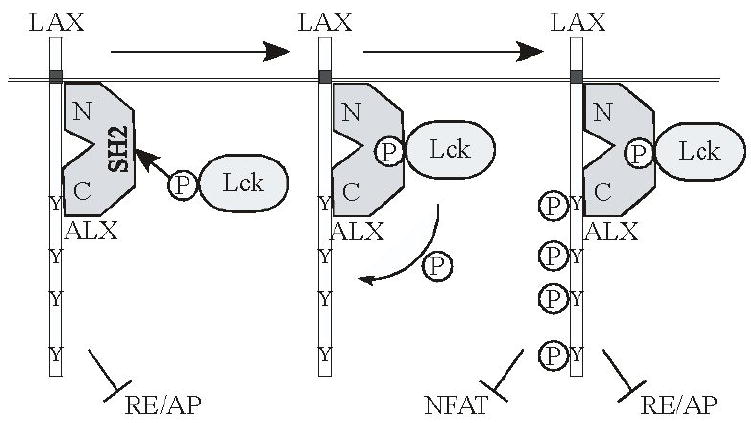
Model for LAX-mediated inhibition of RE/AP and NFAT.
Acknowledgments
We thank Art Weiss for C305 antibody, and Yen-Yu Tina Chen for generating the ALX c-terminal expression construct. This work was supported by NIH R01 AI054974 to V.S.S.
Abbreviations used in this paper
- ALX
adaptor in lymphocytes of unknown function X
- HSH2
hematopoietic SH2 protein
- LAX
linker for activation of X cells
- LAT
linker for activation of T cells
- PI3K
phosphatidyl-inositol-3-kinase
- YFP
yellow fluorescent protein
- WT
wild type
- GST
Glutathione S-Transferase
Footnotes
The authors declare that they have no competing/financial conflict of interest.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Op Imm. 1997;9:396–404. doi: 10.1016/s0952-7915(97)80087-8. [DOI] [PubMed] [Google Scholar]
- 2.Kane LP, Lin J, Weiss A. It’s all Rel-ative: NF-kappaB and CD28 costimulation of T-cell activation. Trends Immunol. 2002;23:413–420. doi: 10.1016/s1471-4906(02)02264-0. [DOI] [PubMed] [Google Scholar]
- 3.Jain J, Loh C, Rao A. Transcriptional Regulation of the IL-2 gene. Curr Op Immunol. 1995;7:333–342. doi: 10.1016/0952-7915(95)80107-3. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro VS, Truitt KE, Imboden JB, Weiss A. CD28 mediates transcriptional upregualtion of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol Cell Biol. 1997;17:4051–4058. doi: 10.1128/mcb.17.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro VS, Mollenauer MN, Weiss A. Nuclear factor of activated T cells and AP-1 are insufficient for IL-2 promoter activation: requirement for CD28 up-regulation of RE/AP. J Immunol. 1998;161:4051–4058. [PubMed] [Google Scholar]
- 6.Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, Grinberg A, Tsay HC, Jacobs HM, Kessler CM, Long EO, Love PE, Samelson LE. Essential role of LAT in T cell development. Immunity. 1999;10:323–332. doi: 10.1016/s1074-7613(00)80032-1. [DOI] [PubMed] [Google Scholar]
- 7.Clements JL, Yang B, Ross-Barta SE, Eliason SL, Hrstka RF, Williamson RA, Koretzky GA. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science. 1998;281:416–419. doi: 10.1126/science.281.5375.416. [DOI] [PubMed] [Google Scholar]
- 8.Greene TA, Powell P, Nzerem C, Shapiro MJ, Shapiro VS. Cloning and characterization of ALX, an adaptor downstream of CD28. J Biol Chem. 2003;278:45128–45134. doi: 10.1074/jbc.M306283200. [DOI] [PubMed] [Google Scholar]
- 9.Perchonock CE, Fernando MC, Quinn WJI, Nguyen CT, Sun J, Shapiro MJ, Shapiro VS. Negative regulation of interleukin-2 and p38 mitogen-activated protein kinase during T cell activation by the adaptor ALX. Mol Cell Biol. 2006;26:6005–6015. doi: 10.1128/MCB.02067-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu M, Granillo O, Wen R, Yang K, Dai K, Wang D, Zhang W. Negative regulation of lymphocyte activation by the adaptor protein LAX. J Immunol. 2005;174:5612–5619. doi: 10.4049/jimmunol.174.9.5612. [DOI] [PubMed] [Google Scholar]
- 11.Zhu M, Janssen E, Leung K, Zhang W. Molecular cloning of a novel gene encoding a membrane-associated adaptor protein (LAX) in lymphocyte signaling. J Biol Chem. 2002;277:46151–46158. doi: 10.1074/jbc.M208946200. [DOI] [PubMed] [Google Scholar]
- 12.Shapiro MJ, Powell P, Ndubuizu A, Nzerem C, Shapiro VS. The ALX Src homology 2 domain is both necessary and sufficient to inhibit T cell receptor/CD28-mediated upregulation of RE/AP. J Biol Chem. 2004;279:40647–40652. doi: 10.1074/jbc.M404198200. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro VS, Mollenauer MN, Greene WC, Weiss A. c-Rel Regulation of IL-2 Gene Expression may be Mediated through Activation of AP-1. J Exp Med. 1996;184:1663–1670. doi: 10.1084/jem.184.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straus DB, Weiss A. Genetic evidence for the involvement of the ick tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70:585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 15.Williams BL, Schreiber KL, Zhang W, Wange RL, Samelson LE, Leibson PJ, Abraham RT. Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70 deficient Jurkat T-cell line. Mol Cell Biol. 1998;18:1388–1399. doi: 10.1128/mcb.18.3.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A. LAT is required for TCR-mediated activation of PLCgamma1 and the Ras pathway. Immunity. 1998;9:617–626. doi: 10.1016/s1074-7613(00)80659-7. [DOI] [PubMed] [Google Scholar]