

Abstract
Background
The mammalian FoxO transcription factors function to regulate diverse physiological processes. Emerging evidence that both BDNF and lithium suppress FoxO activity suggests a potential role of FoxOs in regulating mood-relevant behavior. Here, we investigated whether brain FoxO1 and FoxO3a can be regulated by serotonin and antidepressant, and whether their genetic deletion affects behaviors.
Methods
C57BL/6 mice were treated with d-fenfluramine to increase brain serotonergic activity, or with the antidepressant imipramine. The functional status of brain FoxO1 and FoxO3a was audited by immunoblot analysis for phosphorylation and subcellular localization. The behavioral manifestations in FoxO1 and FoxO3a deficient mice were assessed via the Elevated Plus Maze Test, Forced Swim Test, Tail Suspension Test, and Open Field Test.
Results
Increasing serotonergic activity by d-fenfluramine strongly increased phosphorylation of FoxO1 and FoxO3a in several brain regions, and reduced nuclear FoxO1 and FoxO3a. The effect of d-fenfluramine was mediated by the PI3K/Akt signaling pathway. Chronic, but not acute, treatment with the antidepressant imipramine also increased the phosphorylation of brain FoxO1 and FoxO3a. When FoxO1 was selectively deleted from brain, mice displayed reduced anxiety. In contrast, FoxO3a-deficient mice presented with a significant antidepressant-like behavior.
Conclusion
FoxOs may be a transcriptional target for anxiety and mood disorder treatment. Despite their physical and functional relatedness, FoxO1 and FoxO3a influence distinct behavioral processes linked to anxiety and depression. Findings in this study reveal important new roles of FoxOs in brain and provide a molecular framework for further investigation of how FoxOs may govern mood and anxiety disorders.
Keywords: FoxOs, serotonin, Akt, antidepressant, depression, anxiety
Introduction
FoxO (forkhead box, class O) belongs to the large family of forkhead transcription factors that are characterized by a conserved “forkhead box” DNA-binding domain (1-3). Four mammalian FoxO transcription factors, FoxO1, FoxO3a, FoxO4, and FoxO6 (4-8), have been identified as orthologs of DAF16, an insulin-responsive transcription factor involved in regulating longevity of worms (9) and flies (10).
The activity of FoxO transcription factors is largely controlled by posttranslational regulation. Activation of growth factor receptors activates phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway which results in phosphorylation of FoxO proteins at three serine/threonine residues – threonine 24, serine 256 and serine 316 for FoxO1, and threonine 32, serine 253 and serine 315 for FoxO3a (11-16). Phosphorylation of FoxOs results in redistribution from the nucleus to the cytosol and therefore prevents their binding to DNA (11, 15-18). Besides inhibition by Akt-induced phosphorylation, phosphorylation by other protein kinases, nuclear acetylation, and proteasomal degradation also contribute to the multiple levels of posttranslational regulation of FoxO transcriptional activity (19-23).
Studies in mammalian cells have shown that activation of FoxO transcription factors can induce the expression of genes involved in apoptosis (11, 16, 24-26), cell survival and differentiation (27-29), cell cycle arrest (30-33), and resistance to oxidative stress (34-37). Consistent with their capacity to regulate diverse networks of genes, the FoxOs have been linked to a number of human diseases, such as cancer (4-7, 21, 38-41) and diabetes (42-44). FoxOs also have diverse roles in developmental and reproductive biology, in which FoxO1-null mice dying on embryonic day 10 from impaired vasculogenesis (45, 46), whereas FoxO3a-null mice exhibiting premature ovarian failure (46, 47).
Fewer studies have explored the function of FoxOs in the brain and its link to neuropsychiatric diseases. In neuronal tissues, FoxO3a induces the expression of the proapoptotic Bcl-2 interactive mediator (Bim) (26, 48-52). Ischemia in the brain has been shown to elevate the level of active FoxO3a (49, 53, 54). Neurotrophins, such as BDNF, activate signaling pathways leading to phosphorylation and inactivation of FoxOs (48, 55, 56). More recently, a study in C. elegans (57) revealed that activation of serotonin receptors led to inhibition of FoxO transcriptional activity via activation of Akt, which appeared to be an important mechanism in stress modulation in worms. Additionally, we have also reported that FoxO3a transcriptional activity in mouse brain is inhibited by the mood stabilizer lithium (58). We therefore hypothesized that FoxO is an important mediator of behaviors normally regulated by neuromodulators. In this study, we sought to test this hypothesis by examining the regulation of mouse brain FoxO1 and FoxO3a by serotonin and antidepressant and by testing anxiety- and mood-relevant behaviors in mice lacking FoxO1 or FoxO3a.
Materials and Methods
Animals and treatments
The Institutional Animal Care and Use Committee at the University of Alabama at Birmingham approved the experimental protocol using mice. Adult male C57BL/6 mice (8-10 weeks of age, Frederick Cancer Research, MD) were used for pharmacological treatments. After a one-week accommodation in the university animal facility, mice were treated with d-fenfluramine or imipramine dissolved in saline and injected intraperitoneally (i.p.). For intra-cerebroventricular (i.c.v.) injection, mice were anesthetized with ketamine and xylazine (100 mg/kg:10 mg/kg) to placed a guide cannula (2.2 mm) stereotaxically (posterior 0.8 mm and left 1.6 mm to Bregma). Five days later, the PI3K inhibitor LY294002 (5 nM) or vehicle (10% DMSO) was infused into the left ventricle via an internal cannula. Animals were given d-fenfluramine (i.p.) 90 min after LY294002. At the end of treatment, mice were rapidly decapitated and brain regions (cerebral cortex, hippocampus, and striatum) were immediately dissected in ice-cold saline and used for immunoassays.
FoxO1 and FoxO3a mouse strains
See details in Supplemental Materials
RT-PCR
Brain RNA was extracted with Trizol Reagent (Invitrogen Life Technologies, Carlsbad, CA) and chloroform, precipitated with isopropanol, washed with 75% ethanol, and stored at -80°C in DEPC-treated water. RT-PCR was performed using the SuperScript III One-step RT-PCR system (Invitrogen Life Technologies, Carlsbad, CA) with the forward and reverse primers of FoxO1 (5′-CCTGTCGTACGCCGACCTCATCAC-3′ and 5′-GTCCATGGACGCAGCTCTTCTCCG-3′). PCR products (15 μl) were separated on a 2% agarose gel, visualized by ethidium bromide staining, and photographed with Fluor-S MultiImager (Bio-Rad, Hercules, CA).
Protein preparation and immunoblotting
Proteins from brain homogenate or cortical nuclear extracts were prepared as previously described (48, 59). Proteins were resolved in 7.5-10% SDS-polyacrylamide gels, and immunoblotted with antibodies to phospho-Ser256-FoxO1, FoxO1, phospho-Ser253-FoxO3a, phospho-Thr308-Akt, phospho-Ser473-Akt, Akt (Cell Signaling Technologies, Danvers, MA), phospho-Thr32-FoxO3a, FoxO3a, CREB (Upstate Biotech, Lake Placid, NY), and β-tubulin (Sigma-Aldrich, St. Louis, MS). Following a reaction with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG, the immunoreactions were detected by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ), and protein bands were quantified with a densitometer.
Immunohistochemistry
The immunohistochemistry method was as previous described (59). Briefly, brains were immersion-fixed in Bouin's fixative overnight at 4°C, processed in paraffin, and 4 μm brain sections were prepared on a microtome. Deparaffinized sections were incubated with anti-FoxO1 or anti-FoxO3a, labeled with horseradish peroxidase-conjugated anti-rabbit IgG, and counter-stained with Hoechst 33,258. Immuno-fluorescence in brain sections was viewed with a digital confocal microscope and photographed using a 100× objective. Negative controls of FoxO1 and FoxO3a immunostains were obtained from Brain FoxO1 knockout mouse and FoxO3a-deficient mouse, respectively.
Behavior tests
Male and female mice, 10-14 weeks old, were subjected to the Forced Swim Test (FST), Tail Suspension Test (TST), Elevated Plus Maze Test (EPMT), and Open Field Test (OFT) (see details in Supplemental Materials). No more than two behavior tests were conducted within one week and mice were kept in their home cage with food and water between tests.
Statistical analysis
All data are presented as mean ± SEM. Statistical analyses were conducted using unpaired Student's t-test or one-way analysis of variance (ANOVA) for significant differences (p≤0.05). Any unexplained outlier value greater than ±2 standard deviations from the mean of the group was excluded. Any significant difference by ANOVA was followed by a post hoc test.
Results
To assess whether FoxOs can be regulated by serotonin in the mammalian brain, we first measured phosphorylation of brain FoxO1 and FoxO3a after serotonergic activity was increased by administration of d-fenfluramine, a drug that selectively stimulates serotonin release and blocks serotonin reuptake. C57BL/6 mice received an acute treatment with d-fenfluramine (50 mg/kg, i.p.) for 0.5, 1, and 2 hr. Following the d-fenfluramine injection, the Akt sensitive phosphorylation and the total levels of FoxO1 and FoxO3a in homogenates from cerebral cortex, hippocampus, and striatum were detected by immunoblots (Figure 1).
Figure 1.
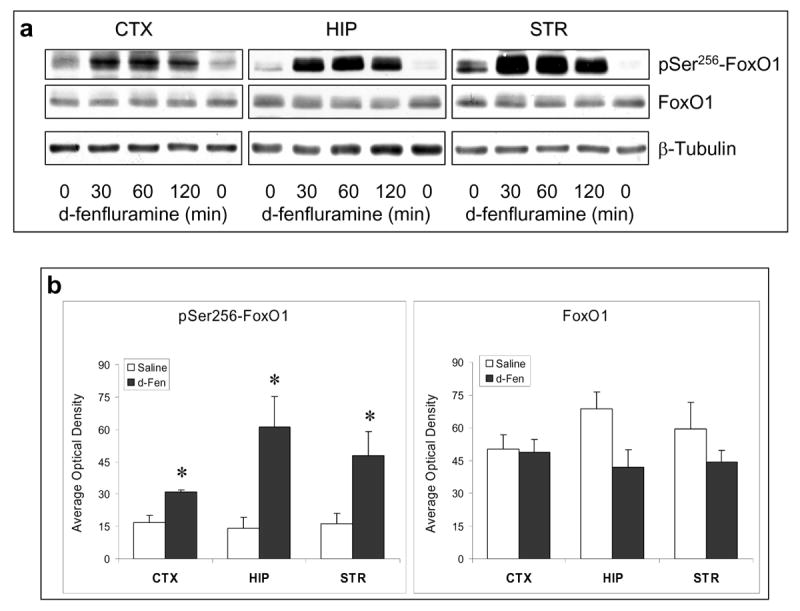
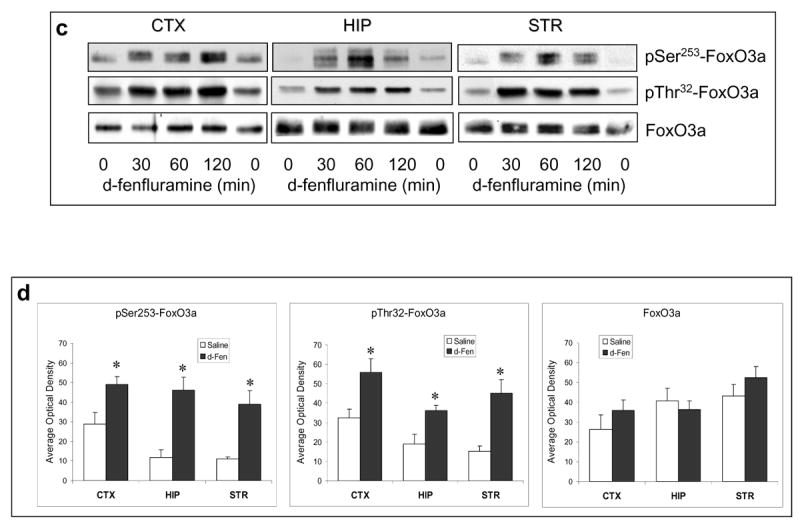
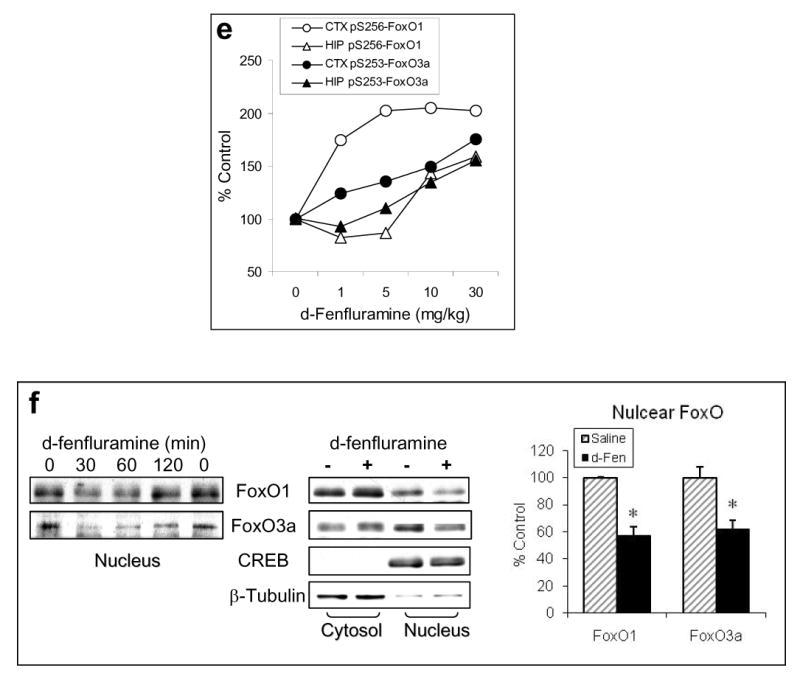
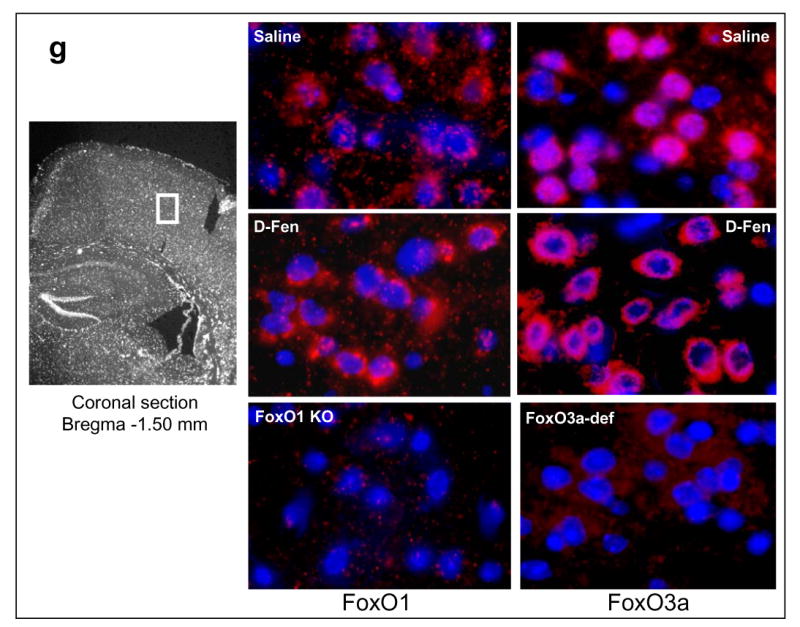
Regulation of FoxOs by d-fenfluramine. C57BL/6 mice were treated with d-fenfluramine before FoxO proteins from homogenates of the cerebral cortex (CTX), hippocampus (HIP), and striatum (STR) were examined by immunoblots. (a) Immunoblots of brain phospho-Ser256-FoxO1 and total FoxO1 after saline (0 min) or d-fenfluramine treatment (50 mg/kg, i.p.). (b) Quantification of phospho-Ser256-FoxO1 (t-test; CTX: t=-4.324, p=0.003; HIP: t=-3.205, p=0.013; STR: t=-2.559, p=0.043; n=4-5 per group) and total FoxO1 after saline or d-fenfluramine (d-Fen) treatment for 2 hr. (c) Immunoblots of brain phospho-Ser253-FoxO3a, phospho-Thr32-FoxO3a, and total FoxO3a after d-fenfluramine treatment. (d) Quantification of phospho-Ser253-FoxO3a (t-test; CTX: t=-2.805, p=0.023; HIP: t=-4.356, p=0.005; STR: t=-4.660, p=0.003; n=4-5 per group), phospho-Thr32-FoxO3a (t-test; CTX: t=-2.842, p=0.030; HIP: t=-2.993, p=0.040; STR: t=-4.110, p=0.015; n=3-4 per group), and total FoxO3a after saline or d-fenfluramine treatment for 2 hr. Data are mean ± SEM. *p<0.05 when values are compared to saline treatment in Student's t-test. (e) Dose-dependent response of FoxO1 and FoxO3a to d-fenfluramine in the cerebral cortex and hippocampus. Values are expressed as % saline treatment (Control). (f) Immunoblots of nuclear and cytosolic FoxO1 and FoxO3a from the cerebral cortex of mice treated with saline (0 min) or d-fenfluramine for indicated times (left panel) or 1 hr (middle panel). Data from nuclear FoxO1 and FoxO3a were quantified and calculated as % saline treatment. *p<0.01 when values are compared to saline treatment (t-test; FoxO1: t=5.765, p=0.001; FoxO3a: t=3.911, p=0.006; n=5 per group). (g) Immunohistochemical detection of brain FoxO1 and FoxO3a. C57BL/6 mice were treated with saline- or d-fenfluramine. Coronal section of mouse brain at Bregma -1.5 mm position was photographed under a 10× objective (left) with the mid cortical region squared for capturing Immuno-fluorescent images of FoxO1 and FoxO3a under a 100× objective. In the colored fluorescent photographs, red color shows FoxO1 and FoxO3a, respectively, and blue color shows nuclear stain. For negative controls, brain sections from the FoxO1 knockout mouse and the FoxO3a-deficient mouse were immuno-stained with anti-FoxO1 and anti-FoxO3a antibodies, respectively.
Acute d-fenfluramine treatment greatly increased the level of phospho-Ser256-FoxO1 in the cerebral cortex, hippocampus, and striatum (Figure 1a). The effect of d-fenfluramine was statistically significant in all tested brain regions when data from the 2-hr d-fenfluramine treatment were compared to saline controls (Figure 1b). D-fenfluramine did not significantly change the level of total FoxO1, although a trend towards a decrease was observed in the hippocampus and striatum. Thus, the initial data suggest that a surge of synaptic serotonin can rapidly increase brain FoxO1 phosphorylation.
Similar to the effect on FoxO1, d-fenfluramine treatment greatly increased the levels of phospho-Ser253-FoxO3a and phospho-Thr32-FoxO3a in the cerebral cortex, hippocampus, and striatum (Figure 1c). The level of phosphorylated FoxO3a after 2-hr d-fenfluramine treatment was significantly higher than saline controls, whereas the level of total FoxO3a was not changed by d-fenflurmaine in the tested brain regions (Figure 1d).
The above experiments were conducted using a high dose of d-fenfluramine (50 mg/kg) that is comparable with our previously published results (60). Mice tolerated this dose of d-fenfluramine well with 100% survival rate. To examine if the response of FoxOs to d-fenfluramine is dose-dependent, mice were also treated with different doses of d-fenfluramine (0, 1, 5, 10, 30 mg/kg) for 2 hr (Figure 1e). Except FoxO1 in the cerebral cortex, which was more sensitive to a lower dose of d-fenfluramine (1 mg/kg), a dose range of 10-30 mg/kg was sufficient to induce a 50% increase in FoxO1 phosphorylation in the hippocampus and FoxO3a phosphorylation in both cerebral cortex and hippocampus. To assure maximal effect in all experiments, we then used 50 mg/kg d-fenfluramine for all experiments and data analysis in this study.
Phosphorylation of FoxOs at the Akt-sensitive serine/threonine residues is known to cause translocation of FoxOs from the nucleus to the cytosol, resulting in transcriptional inactivation (11, 14-17). We therefore measured the nuclear and cytosolic FoxO1 and FoxO3a in the cerebral cortex after d-fenfluramine treatment. Both immunoblots and immunostained images indicated that d-fenfluramine treatment reduced nuclear FoxO1 and FoxO3a (Figure 1f, 1g), and the effect of d-fenfluramine was statistically significant when compared to saline treatment (Figure 1f). These results suggest that d-fenfluramine can inactivate FoxO1 and FoxO3a in the brain, likely through an increase in the inhibitory phosphorylation.
Since phosphorylation of FoxOs is primarily regulated by the growth factor-activated PI3K/Akt signaling pathway, we measured Akt phosphorylation, an indicator of Akt activation, after d-fenfluramine treatment. Similar to the effects on FoxOs, d-fenfluramine caused a rapid increase in phospho-Thr308-Akt and phospho-Ser473-Akt in the cerebral cortex, hippocampus, and striatum (Figure 2a). The level of Akt phosphorylation after 2-hr d-fenfluramine treatment was significantly higher than saline controls, but d-fenfluramine had no effect on the level of total Akt in the tested brain regions (Figure 2b).
Figure 2.
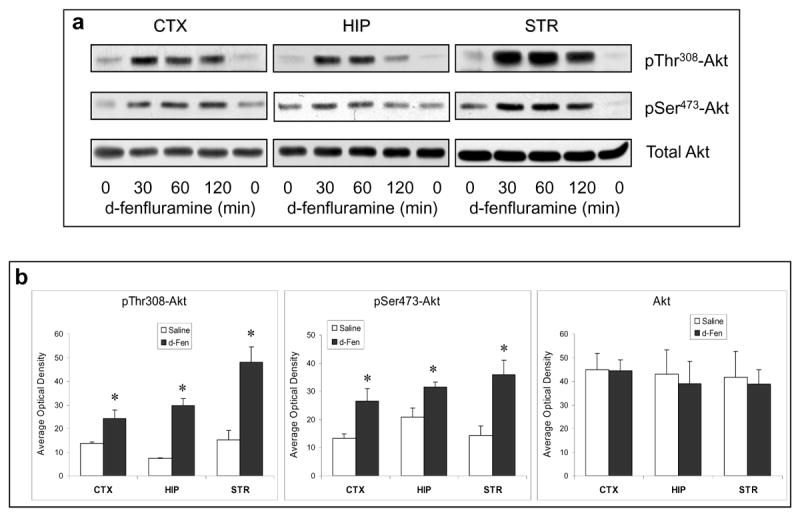
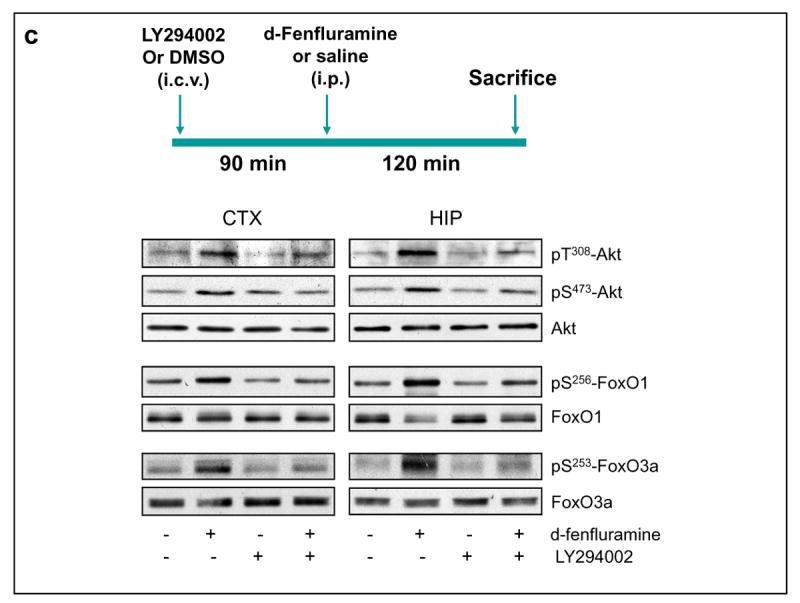
Regulation of Akt signaling pathway by d-fenfluramine. (a) C57BL/6 mice were treated with d-fenfluramine (50 mg/kg, i.p.) before phospho-Thr308-Akt, phospho-Ser473-Akt and total Akt were examined in brain homogenates. (b) Quantification of phospho-Thr308-Akt (t-test; CTX: t=-2.873, p=0.021; HIP: t=-7.842, p=0.001; STR: t=-4.208, p=0.014; n=3-5 per group), phospho-Ser473-Akt (t-test; CTX: t=-2.887, p=0.020; HIP: t=-2.848, p=0.047; STR: t=-3.588, p=0.023; n=3-5 per group), and total Akt after saline and d-fenfluramine (d-Fen) treatment for 2 hr. Data are mean ± SEM. *p<0.05 when values are compared to saline treatment in Student's t-test. (c) Mice received unilateral intra-cerebroventricular (i.c.v.) injection of either LY294002 (5 nM in 1 μl) or DMSO (vehicle, 1 μl) 1.5 hr prior to receiving saline or d-fenfluramine treatment (2 hr). Immunoblots of phosphorylated and total Akt, FoxO1, and FoxO3a are representatives of five separate experiments.
To determine if the above changes resulted from PI3K activation, mice were treated with the PI3K inhibitor LY294002 (5 nM in 1 μL i.c.v.) 90 min prior to d-fenfluramine treatment. LY294002 moderately reduced the d-fenfluramine-induced increases in phospho-Thr308-Akt and phospho-Ser473-Akt in both cerebral cortex and hippocampus. It also reduced d-fenfluramine-induced increases in phospho-Ser256-FoxO1 and phospho-Ser253-FoxO3a, with the effect significant in the cerebral cortex and moderate in the hippocampus (Figure 2c and Table 1). The above results suggest that increasing brain serotonergic activity by d-fenfluramine activates Akt at least partially via PI3K to phosphorylate and inactivate FoxO transcription factors in the mouse brain.
Table 1.
The Effect of PI3K Inhibitor LY294002 on d-Fenfluramine-Induced Phosphorylation of Akt and FoxOs
| Brain Regions | LY294002+d-fenfluramine / d-fenfluramine (mean ± SEM) | |||||
|---|---|---|---|---|---|---|
| Sample size n=6 | Phopho-Thr308-Akt | p-value | Phopho-Ser253-FoxO3a | p-value | Phopho-Ser256-FoxO1 | p-value |
| Cerebral Cortex | 0.55 ± 0.19 | 0.06 | 0.76 ± 0.07 | 0.02 | 0.66 ± 0.12 | 0.03 |
| Hippocampus | 0.72 ± 0.10 | 0.06 | 0.78 ± 0.10 | 0.07 | 0.70 ± 0.14 | 0.08 |
The above findings, taken in conjunction with reports that FoxOs are regulated by the mood regulating BDNF (48, 55) and the mood stabilizer lithium (58), prompted us to hypothesize that FoxOs may be a transcriptional target of mood disorder treatment. We therefore examined if the monoamine-regulating antidepressant imipramine regulates brain FoxOs. An acute imipramine treatment (30 mg/kg, i.p. for 1 hr) did not cause a significant change in either phosphorylated or total FoxO1 and FoxO3a in the tested brain regions, except a trend toward an increase in phospho-Ser256-FoxO1 was noted in the hippocampus (Table 2).
Table 2.
Acute Imipramine Treatment
| Brain Regions | Phopho-Ser256-FoxO1 | Phopho-Ser253-FoxO3a | ||||
|---|---|---|---|---|---|---|
| Mean ± SEM (% saline) | p-value | Mean ± SEM (% saline) | p-value | |||
| Saline | Imipramine | (n=9) | Saline | Imipramine | (n=9) | |
| Cerebral Cortex | 100.00 ± 10.92 | 98.68 ± 10.92 | 0.96 | 100.00 ± 9.42 | 120.48 ± 18.39 | 0.38 |
| Hippocampus | 100.00 ± 17.32 | 148.13 ± 39.04 | 0.21 | 100.00 ± 9.24 | 111.96 ± 17.95 | 0.60 |
| Striatum | 100.00 ± 10.89 | 113.17 ± 32.67 | 0.37 | 100.00 ± 16.65 | 118.36 ± 16.72 | 0.46 |
Since the therapeutic effects of antidepressants usually occur after prolonged treatment, we then tested if chronic imipramine treatment (20 mg/kg, i.p.) may regulate brain FoxOs. Before testing brain FoxOs, mice were subjected to the FST after receiving chronic imipramine treatment (23 days) to confirm an antidepressant-like effect of this treatment. The result demonstrated that the chronic imipramine treatment used in our experiment reduced the immobility time when compared to saline treatment (Figure 3a). Interestingly, chronic imipramine treatment (28 days) caused significant increases in phospho-Ser256-FoxO1 and phospho-Ser253-FoxO3a in the cerebral cortex, hippocampus, and striatum, whereas the levels of total FoxO1 and FoxO3a were not affected by chronic imipramine treatment (Figure 3b and 3c). This effect of imipramine was accompanied by a moderate increase in phospho-Thr308-Akt but no change was observed in the level of total Akt. Thus, a therapeutically relevant imipramine treatment can inhibit FoxO1 and FoxO3a in mouse brain.
Figure 3.

Regulation of FoxOs and Akt by imipramine. C57BL/6 mice were treated with imipramine (20 mg/kg/d, i.p.) for 23 days before the FST, followed by immunoblotting brain FoxOs and Akt at the end of imipramine treatment (28 days). (a) Mice were subjected to FST 30 min after the 23rd day of saline or imipramine treatment. The immobility (resting time) was recorded during the last 4 min of a 6-min test. Data are mean ± SEM. *p<0.04 (t=2.194, n=10 per group) when values are compared to saline treatment in Student's t-test. (b) Immunoblots of brain FoxO1, FoxO3a, and Akt after saline (-) or imipramine treatment. (c) Quantification of phospho-Ser256-FoxO1 (t-test; CTX: t=-2.628, p=0.020; HIP: t=-2.226, p=0.041; STR: t=-2.229, p=0.041), phospho-Ser253-FoxO3a (t-test; CTX: t=-2.209, p=0.044; HIP: t=-2.456, p=0.027; STR: t=-3.674, p=0.002), and phospho-T308-Akt (t-test; CTX: t=-2.617, p=0.019; HIP: t=-2.425, p=0.027; STR: t=-2.746, p=0.015). Protein levels in imipramine treated samples are calculated as % saline treatment. Data are mean ± SEM. *p<0.05 (n=7-9 per group) when values are compared to saline treatment in Student's t-test.
Serotonin and antidepressants are key regulators of anxiety and mood. Since both serotonin and imipramine increased the inhibitory phosphorylation of FoxO1 and FoxO3a in mouse brain, we hypothesized that mice lacking brain FoxOs may exhibit an anxiolytic or antidepressive behavioral phenotype. To test this hypothesis, we examined behavioral phenotypes of brain FoxO1-knockout and systemic FoxO3a-deficient mice.
Although systemic FoxO1 knockout mice die on embryo day-10 from impaired vasculogenesis (45), the survival of brain-targeted FoxO1 knockout mice has not yet been reported. In order to measure FoxO1-mediated behavioral phenotypes, we generated brain-targeted FoxO1 knockout mice (Figure S1). These mice survived and remained viable through adulthood and bred normally.
To measure anxiety-like behavior, the FoxO1 knockout mice were subjected to the EPMT. In male mice, the time spent in open arms was significantly different among wild-type controls, heterozygous, and homozygous FoxO1 knockout mice. Both heterozygous and homozygous FoxO1 knockout mice stayed a significantly longer time in open arms of the maze during the test period when compared to wild-type controls, whereas there was no difference between the heterozygous and homozygous FoxO1 knockout mice (Figure 4a). Although time in closed arms was only mildly shorter in FoxO1 knockout mice, it was noted that the frequency to entering closed arms was significantly lower in FoxO1 knockout mice than in wild-type controls. Results shown in Figure 4a were a summary of three separate experiments and similar results were observed in all three experiments. These results demonstrate that FoxO1 plays a role in mediating anxiety-like behavior in mice.
Figure 4.



Behavior tests in FoxO1 knockout mice. Homozygous (-/-) and heterozygous (+/-) FoxO1 knockout and matching wild-type (+/+) mice were subjected to the EPMT in (a) male mice (ANOVA; time in open arm: Fgenotype(2,48)=4.119, p=0.022; entries to closed arms: Fgenotype(2,47)=3.411, p=0.041); (b) female mice (ANOVA; entries to closed arms: Fgenotype(2,39)=11.616, p<0.001); (c) the FST in male (ANOVA; Fgenotype(2,46)=4.350, p=0.019) and female (ANOVA; Fgenotype(2,30)=2.608, p=0.045) mice; and (d) the TST in male mice (t-test; t=-3.115, P=0.005). (e) The OFT was performed in male mice at baseline (no drug treatment) and after d-amphetamine treatment (4 mg/kg, i.p., 30 min). Values after d-amphetamine treatment were calculated as % baseline. Data are expressed as mean ± SEM (male: n=18; female: n=16 for each test). Statistical analysis is conducted using one-way ANOVA with pair-wise comparisons of sample means via the Tukey HSD test or Student's t-test (d). *p<0.05 when the testing group is compared to the wild-type mice.
To further test if the anxiolytic phenotype in FoxO1 knockout mice is gender-specific, three groups of female mice were subjected to the EPMT. Although time spent in open arms and in closed arms were not different among female wild-type controls, heterozygous, and homozygous FoxO1 knockout mice, a statistically significant difference was observed in the frequency of entering closed arms, in which female heterozygous and homozygous FoxO1 knockout mice had significantly lower frequency than wild-type controls (Figure 4b). Thus, it appears that both male and female FoxO1 knockout mice carry an anxiolytic behavioral phenotype, but the phenotype is more pronounced in males than in females.
When FoxO1 knockout mice were subjected to the FST, somewhat unexpectedly, both heterozygous and homozygous FoxO1 knockout male mice displayed prolonged immobility in the FST when compared to wild-type controls (Figure 4c), suggesting an increase in depressive behavior in mice lacking brain FoxO1. Similarly, a prolonged immobility was also observed in female homozygous FoxO1 knockout mice. To confirm the depressive behavior in FoxO1 knockout mice, another group of male wild-type and homozygous FoxO1 knockout mice were tested in the TST. Homozygous FoxO1 knockout mice again displayed prolonged immobility (Figure 4d), confirming the depressive phenotype of these mice. The increased immobility time in brain FoxO1 knockout mice was unlikely a result of impaired locomotor activity since FoxO1 knockout mice and wild-type controls had similar travel distance and velocity in the baseline Open Field Test (OFT) (Figure 4e). When the locomotor response of mice to d-amphetamine treatment (4 mg/kg, i.p., 30 min) was tested, the travel distance and velocity in wild-type and FoxO1 knockout mice were increased by 348.23±47.21% and 275.04±43.19%, respectively. Although FoxO1 knockout mice showed a trend towards slightly lower response to d-amphetamine, the difference was not significant.
To test if FoxO1 and FoxO3a have similar effects on anxiety- and mood-relevant behaviors, these behavior tests were applied to a line of mice systemically lacking FoxO3a (FoxO3a-deficient) (61). During the EPMT, both male and female FoxO3a-deficient mice tended to stay longer times in the open arms, but no significant difference was detected among wild-type, heterozygous, and homozygous FoxO3a-deficient mice (Figure 5a). This result suggests that FoxO3a has less impact on anxiety-like behavior than FoxO1. When tested in the OFT, the total travel distance and velocity were also similar among wild-type, heterozygous, and homozygous FoxO3a-deficient mice (Figure 5b), indicating normal locomotor activity in FoxO3a-deficient mice.
Figure 5.

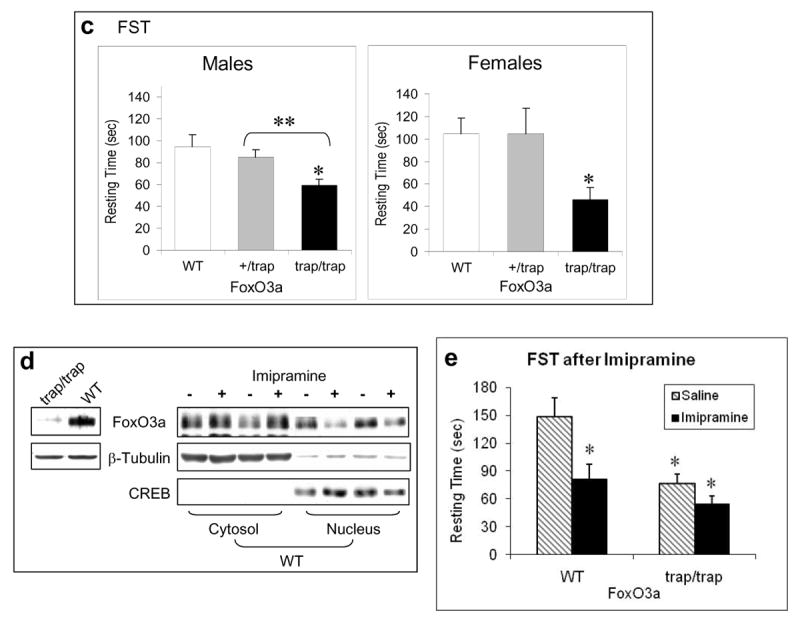
Behavior tests in FoxO3a-deficient mice. Homozygous (trap/trap) and heterozygous (+/trap) FoxO3a-deficient and matching wild-type (WT) mice were subjected to behavioral tests (male: n=10; female: n=9 for each test). (a) The EPMT in male and female mice. (b) Baseline OFT in male mice. (c) The FST in male and female mice. Data are expressed as mean ± SEM. Statistical analysis is conducted using one-way ANOVA (male: Fgenotype(2,30)=4.647, p=0.017; female: Fgenotype(2,15)=5.959, p=0.012) with pair-wise comparisons of sample means via the Tukey HSD test. *p<0.05 when the testing group was compared to the wild-type mice, and **p<0.05 when heterozygous were compared to homozygous FoxO3a-deficient mice. (d) The level of total FoxO3a in the cerebral cortex of FoxO3a-deficient (trap/trap) and wild-type (WT) mice (left panel), and in the cytosolic and nuclear fractions of the cerebral cortex of wild-type mice treated with saline or imipramine for 28 days (right panel). (e) Male FoxO3a-deficient (trap/trap) and littermate wild-type (WT) mice were treated with either saline or imipramine (20 mg/kg/d, i.p.) for 23 days before the FST. Data are mean ± SEM. Statistical analysis is conducted using one-way ANOVA (Fgenotypextreatment (3,25)=8.543, p<0.001) with post hoc multiple group comparisons (n=6-8). Since this analysis involved 6 comparisons, all p-values less than 0.008 were considered significant. *p<0.005 when data from each testing group was compared to the saline-treated wild type group.
Interestingly, male homozygous FoxO3a-deficient mice, in strong contrast to the brain FoxO1 knockout mice, had a significantly lower immobility in the FST when compared to both heterozygous FoxO3a-deficient and wild-type mice (Figure 5c). This antidepressant-like behavioral phenotype required complete deletion of FoxO3a since heterozygous FoxO3a-deficient mice did not display a significant decrease in immobility. Similarly to male mice, a significantly lower immobility was also observed in female homozygous FoxO3a-deficient mice when compared to wild-type controls.
To test if the low expression of FoxO3a in FoxO3a-deficient mouse brain contributes to the antidepressant phenotype, we measured the levels of cytosolic and nuclear FoxO3a after wild type mice were treated with the antidepressant imipramine (20 mg/kg/day, i.p.) for 28 days. Comparing to saline-treated wild-type mice, an increase in the level of cytosolic FoxO3a and a corresponding decrease in the level of nuclear FoxO3a was noticeable in the cerebral cortex of mice treated with imipramine (Figure 5d), therefore suggesting that the lowered level of FoxO3a may contribute to the antidepressant behavior.
Furthermore, to compare the response of FoxO3a-deficient mice versus comparable wild type mice to antidepressant, a group of homozygous FoxO3a-deficient and littermate wild-type mice received daily injection of saline or imipramine (20 mg/kg/day, i.p.) for 23 days. When tested in the FST, there was a significant difference in immobility among the four groups of mice with different genotypes and treatments (Figure 5e). When multiple comparison with Bonferroni correction was conducted using the wild type saline-treated mice as control, imipramine significantly reduced immobility in wild type mice (p<0.0018), and FoxO3a-defidient mice, either without or with imipramine treatment, also exhibited reduced immobility (p<0.0019 and p<0.0001, respectively). On the other hand, when using the FoxO3a-deficient mice with saline treatment as control, the immobility in imipramine-treated mice, either wild type or FoxO3a-deficient, was not different from control.
Discussion
In this study, we tested the regulation of brain FoxO1 and FoxO3a by serotonin and imipramine, and the behavioral manifestations of genetic insufficiency of each. We focused on FoxO1 and FoxO3a because they are abundant in the adult brain (62-64).
The findings that enhancing brain serotonergic activity by d-fenfluramine treatment increases the inhibitory phosphorylation of FoxO1 and FoxO3a in several regions of mouse brain conform with findings in C. elegans by Liang et al. (57), and we confirm that serotonin can regulate FoxO transcription factors in mammalian brain. Although serotonin often is not considered as a primary activator of the PI3K/Akt signaling pathway, several studies in cultured cells and in invertebrates have shown that stimulation of serotonin receptors can result in phosphorylation and activation of Akt (57, 65, 66). The results shown in this study support the conclusion that enhancing serotonin activity exhibits a regulatory effect in Akt-regulated FoxO1 and FoxO3a in brain. Although the specific serotonin receptors mediating this effect remain to be identified, our observations imply that brain FoxOs are important transcription factors on which signals from serotonergic neurotransmission and neurotrophic activity converge. In our experiments, the PI3K inhibitor LY294002 partially reduced the effect of d-fenfluramine. It is possible that the low dose LY294002 is not sufficient to penetrate into all brain areas, but we do not rule out that other signaling pathways other than the PI3K/Akt signaling pathway involved also contribute to the effect of d-fenfluramine on FoxOs.
Unlike d-fenfluramine, which has a selective dual action on serotonin by blocking reuptake and stimulating release, the antidepressant imipramine inhibits monoamine reuptake to enhance serotonergic and noradrenergic activity in the brain. Interestingly, chronic, but not acute imipramine treatment significantly increased phosphorylation of FoxO1 and FoxO3a, as well as moderately increased phosphorylation of Akt. Additional to modulating monoamine neurotransmission, chronic antidepressant treatment, including imipramine, is known to increase brain BDNF (67, 68), a neurotrophin that also phosphorylates and inactivates FoxOs (48, 56). Thus, imipramine may have increased FoxO phosphorylation by enhancing serotonin action and increasing brain BDNF. Since the therapeutic effect of an antidepressant typically requires prolonged treatment, the observation from this study links FoxO1 and FoxO3a to a clinically relevant treatment for anxiety and depression. We examined this role of FoxOs by taking advantage of mice with genetically modified FoxO1 and FoxO3a.
Unlike the systemic FoxO1 knockout mice, mice lacking brain FoxO1 survive through adulthood, thus nestin-directed brain FoxO1 deletion provides a useful model to identify behaviors that involve complex neuronal circuits. Mice lacking brain FoxO1 consistently displayed an anxiolytic behavioral phenotype, which in males only required a partial depletion of FoxO1, indicating that FoxO1 has a prominent role in regulating anxiety-like behaviors. This interesting finding is in accordance with the well-known effect of serotonin on anxiety and the anxiolytic effect of most monoamine reuptake inhibitor antidepressants (69). A somewhat surprising observation from this study is that mice lacking brain FoxO1 appear to have a depression-like behavioral phenotype. A significant difference from the matching wild-type mice suggests that the results represent a FoxO1-specific behavioral alteration. Since mood is a complex behavior regulated diversely in brain by monoamines and neurotrophins in a temporal and spatial-dependent manner (70, 71), we speculate that several factors may contribute to this differential mood/anxiety-related effect of FoxO1. Noticeably, unlike FoxO3a, the distribution of FoxO1 in the brain is mostly confined to a few brain areas, including dentate gyrus and ventral CA3 area of the hippocampus, the amygdalohippocampal region, the piriform cortex, the striatum, the caudate putamen, and the nucleus accumbens (64). Serotonin regulates mood and anxiety by activating, as well as desensitizing many types of serotonin receptors located in different brain regions (72, 73). BDNF, on the other hand, differentially regulates mood and anxiety in selective brain regions (71, 74, 75). Serotonin and BDNF also interact with each other to achieve enhanced regulation of mood and anxiety (76, 77). The localization of FoxO1 may determine its selective regulation by either BDNF or serotonin in a brain region-specific manner. However, a conclusion cannot be reached until the regulation of brain FoxO1 by neuromodulators is further investigated in detail. On the other hand, the findings in this study may also suggest that maintaining FoxO1 transcriptional activity in brain is necessary to stabilize mood and prevent depression developed under environmental changes, such as stress. A limitation of this study was that the brain FoxO1 knockout mice were generated originally from a mouse line containing floxed alleles of FoxO1, 3a, and 4. During selection for behavior tests, no homozygous FoxO3a knockout mouse was included in the tests. Since FoxO4 expression was limited in brain (62, 63), no FoxO4 genotype was excluded during selection, and there was no significant behavioral difference noticed between Nestin-Cre:FoxO4-floxed and wild type mice (data not shown). To avoid this potential limitation, we tested FoxO3a behavior phenotypes in another line of mice that were only lack of FoxO3a.
In contrast to FoxO1 knockout mice, reducing anxiety was not a prominent feature in mice systemically deficient of FoxO3a. Perhaps more interestingly, FoxO3a-deficient mice had significantly less immobility in the FST. The non-responsiveness to chronic imipramine treatment in FoxO3a-deficient mice provides evidence that FoxO3a may be a transcriptional mediator of the antidepressant action of imipramine. The increased phosphorylation of Akt and FoxO3a and the decreased nuclear FoxO3a after chronic imipramine treatment also suggest that the Akt signaling pathway may contribute to the delayed antidepressant effect of imipramine in which transcriptional regulation of gene expression is critical for sustained therapeutic effect (78). Furthermore, our group previously reported that glycogen synthase kinase 3 (GSK3), another major phosphorylation substrate of Akt, can be inhibited by serotonin and antidepressants that increase its inhibitory serine phosphorylation (60), and Beaulieu et. al. (79) reported that the depressive and anxiogenic behaviors of serotonin-deficient mice were blunted when GSK3 was semi-deleted. Those studies, in agreement with our findings in this study, indicate that Akt signaling pathway have an important impact in 5HT-regulated behaviors, and a coordinated regulation of substrates of the Akt-dependent signaling pathway by antidepressants may contribute to their therapeutic effects.
As has been observed in studies in other systems, FoxO1 and FoxO3a have divergent physiological effects despite their physical and functional relatedness (45-47). Findings in this study provides new evidence that the two major FoxO subtypes also exhibit distinct physiological effects in brain, where they preferentially regulate behaviors related to anxiety and mood. Detailed studies in the context of FoxO brain distribution, preferential regulation by serotonin and neurotrophins, and selective molecular targets of each FoxO may be useful in addressing their differential roles in brain. On the other hand, the common regulation of FoxOs by serotonin and antidepressant may add a level of complexity to the physiological consequence when all subtypes of FoxOs are regulated simultaneously by a neuromodulator. Using genetic mouse model that carries more than one FoxO mutant may help further identify the common as well as the divergent functions of FoxOs in brain.
Supplementary Material
Acknowledgments
Disclosure: This study is supported by NIH grants: MH64555 and MH73723 (Li), MH38752 (Jope), CA84628 (DePinho), AI57571 (Peng), and in part by NS47466 (the UAB Behavioral Assessment Core, PI, John Hablitz) and NS57098 (the Alabama Neuroscience Blueprint Core Center, PI, Kevin A Roth). X. Li is also supported by the NARSAD independent investigator award. R.A. DePinho is an American Cancer Society Research Professor and is supported by the Robert A. and Renee E. Belfer Foundation Institute for Innovative Cancer Science. J-H. Paik is a Damon Runyon Fellows supported by the Damon Runyon Cancer Research Foundation.
The authors thank Ms. L. Liu, Ms. L. Wright, and Dr. Z. Mao for technical assistance, S. Weiler and M. Neptune for generation of the FoxO1/3/4-flox/flox mice, UAB Behavior Core for providing the testing facility and instructions, Dr. J. M. Wyss for scientific advice on behavioral tests, Dr. Buffie Clodfelder-Miller for technical advice and assistance on immuno-fluorescence imaging, and Drs. S. Batra and A. A. Bartolucci for assistance in statistical analysis.
Footnotes
Conflict of Interest: The authors reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weigel D, et al. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57:645–58. doi: 10.1016/0092-8674(89)90133-5. [DOI] [PubMed] [Google Scholar]
- 2.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–6. [PubMed] [Google Scholar]
- 3.Katoh M. Human FOX gene family. Int J Oncol. 2004;25:1495–500. [PubMed] [Google Scholar]
- 4.Galili N, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5:230–5. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 5.Davis RJ, et al. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:2869–72. [PubMed] [Google Scholar]
- 6.Parry P, Wei Y, Evans G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer. 1994;11:79–84. doi: 10.1002/gcc.2870110203. [DOI] [PubMed] [Google Scholar]
- 7.Anderson MJ, et al. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics. 1998;47:187–99. doi: 10.1006/geno.1997.5122. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs FM, et al. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278:35959–67. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 9.Kenyon C, et al. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–4. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 10.Hwangbo DS, et al. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–6. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 11.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 12.del Peso L, et al. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene. 1999;18:7328–33. doi: 10.1038/sj.onc.1203159. [DOI] [PubMed] [Google Scholar]
- 13.Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274:15982–5. doi: 10.1074/jbc.274.23.15982. [DOI] [PubMed] [Google Scholar]
- 14.Rena G, et al. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 15.Takaishi H, et al. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc Natl Acad Sci U S A. 1999;96:11836–41. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–6. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 17.Biggs WH, 3rd, et al. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo S, et al. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–92. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 19.Matsuzaki H, et al. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–90. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361–6. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 21.Hu MC, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–37. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 22.Huang H, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102:1649–54. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogt PK, Jiang H, Aoki M. Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle. 2005;4:908–13. doi: 10.4161/cc.4.7.1796. [DOI] [PubMed] [Google Scholar]
- 24.Dijkers PF, et al. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–4. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 25.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277:47928–37. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 26.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bois PR, Grosveld GC. FKHR (FOXO1a) is required for myotube fusion of primary mouse myoblasts. Embo J. 2003;22:1147–57. doi: 10.1093/emboj/cdg116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puigserver P, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–5. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 29.Liu ZP, Wang Z, Yanagisawa H, Olson EN. Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell. 2005;9:261–70. doi: 10.1016/j.devcel.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 30.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura N, et al. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–82. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramaswamy S, et al. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt M, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22:7842–52. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furukawa-Hibi Y, et al. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277:26729–32. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- 35.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 36.Tran H, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 37.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 38.Paik JH, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–23. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 40.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–8. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 41.Seoane J, et al. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–23. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 42.Imae M, et al. Nutritional and hormonal factors control the gene expression of FoxOs, the mammalian homologues of DAF-16. J Mol Endocrinol. 2003;30:253–62. doi: 10.1677/jme.0.0300253. [DOI] [PubMed] [Google Scholar]
- 43.Aoyama H, Daitoku H, Fukamizu A. Nutrient control of phosphorylation and translocation of Foxo1 in C57BL/6 and db/db mice. Int J Mol Med. 2006;18:433–9. [PubMed] [Google Scholar]
- 44.Sandri M, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103:16260–5. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furuyama T, et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–9. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 46.Hosaka T, et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–80. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castrillon DH, et al. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–8. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 48.Zhu W, Bijur GN, Styles NA, Li X. Regulation of FOXO3a by brain-derived neurotrophic factor in differentiated human SH-SY5Y neuroblastoma cells. Brain Res Mol Brain Res. 2004;126:45–56. doi: 10.1016/j.molbrainres.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 49.Fukunaga K, Ishigami T, Kawano T. Transcriptional regulation of neuronal genes and its effect on neural functions: expression and function of forkhead transcription factors in neurons. J Pharmacol Sci. 2005;98:205–11. doi: 10.1254/jphs.fmj05001x3. [DOI] [PubMed] [Google Scholar]
- 50.Gilley J, Ham J. Evidence for increased complexity in the regulation of bim expression in sympathetic neurons: involvement of novel transcriptional and translational mechanisms. DNA Cell Biol. 2005;24:563–73. doi: 10.1089/dna.2005.24.563. [DOI] [PubMed] [Google Scholar]
- 51.Chong ZZ, Li F, Maiese K. Group I metabotropic receptor neuroprotection requires Akt and its substrates that govern FOXO3a, Bim, and beta-catenin during oxidative stress. Curr Neurovasc Res. 2006;3:107–17. doi: 10.2174/156720206776875830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obexer P, et al. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007;14:534–47. doi: 10.1038/sj.cdd.4402017. [DOI] [PubMed] [Google Scholar]
- 53.Hillion JA, et al. Involvement of Akt in preconditioning-induced tolerance to ischemia in PC12 cells. J Cereb Blood Flow Metab. 2006;26:1323–31. doi: 10.1038/sj.jcbfm.9600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Won CK, Ji HH, Koh PO. Estradiol prevents the focal cerebral ischemic injury-induced decrease of forkhead transcription factors phosphorylation. Neurosci Lett. 2006;398:39–43. doi: 10.1016/j.neulet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 55.Zheng WH, Kar S, Quirion R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J Neurochem. 2002;80:1049–61. doi: 10.1046/j.0022-3042.2002.00783.x. [DOI] [PubMed] [Google Scholar]
- 56.Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- 57.Liang B, et al. Serotonin targets the DAF-16/FOXO signaling pathway to modulate stress responses. Cell Metab. 2006;4:429–40. doi: 10.1016/j.cmet.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Mao Z, Liu L, Zhang R, Li X. Lithium reduces FoxO3a transcriptional activity by decreasing its intracellular content. Biol Psychiatry. 2007;62:1423–30. doi: 10.1016/j.biopsych.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Li X, et al. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- 60.Li X, et al. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin L, Hron JD, Peng SL. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21:203–13. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 62.Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349:629–34. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biggs WH, 3rd, Cavenee WK, Arden KC. Identification and characterization of members of the FKHR (FOX O) subclass of winged-helix transcription factors in the mouse. Mamm Genome. 2001;12:416–25. doi: 10.1007/s003350020002. [DOI] [PubMed] [Google Scholar]
- 64.Hoekman MF, Jacobs FM, Smidt MP, Burbach JP. Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expr Patterns. 2006;6:134–40. doi: 10.1016/j.modgep.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Cowen DS, Johnson-Farley NN, Travkina T. 5-HT receptors couple to activation of Akt, but not extracellular-regulated kinase (ERK), in cultured hippocampal neurons. J Neurochem. 2005;93:910–7. doi: 10.1111/j.1471-4159.2005.03107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cowen DS. Serotonin and neuronal growth factors - a convergence of signaling pathways. J Neurochem. 2007;101:1161–71. doi: 10.1111/j.1471-4159.2006.04420.x. [DOI] [PubMed] [Google Scholar]
- 67.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–47. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–72. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feighner JP. Overview of antidepressants currently used to treat anxiety disorders. J Clin Psychiatry. 1999;60 22:18–22. [PubMed] [Google Scholar]
- 70.Duman RS. Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry. 1998;44:324–35. doi: 10.1016/s0006-3223(98)00031-6. [DOI] [PubMed] [Google Scholar]
- 71.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 72.Weisstaub NV, et al. Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science. 2006;313:536–40. doi: 10.1126/science.1123432. [DOI] [PubMed] [Google Scholar]
- 73.Gingrich JA. Mutational analysis of the serotonergic system: recent findings using knockout mice. Curr Drug Targets CNS Neurol Disord. 2002;1:449–65. doi: 10.2174/1568007023339003. [DOI] [PubMed] [Google Scholar]
- 74.Kalueff AV, Avgustinovich DF, Kudryavtseva NN, Murphy DL. BDNF in anxiety and depression. Science. 2006;312:1598–9. doi: 10.1126/science.312.5780.1598. author reply 1598-9. [DOI] [PubMed] [Google Scholar]
- 75.Rasmusson AM, Shi L, Duman R. Downregulation of BDNF mRNA in the hippocampal dentate gyrus after re-exposure to cues previously associated with footshock. Neuropsychopharmacology. 2002;27:133–42. doi: 10.1016/S0893-133X(02)00286-5. [DOI] [PubMed] [Google Scholar]
- 76.Ren-Patterson RF, et al. Loss of brain-derived neurotrophic factor gene allele exacerbates brain monoamine deficiencies and increases stress abnormalities of serotonin transporter knockout mice. J Neurosci Res. 2005;79:756–71. doi: 10.1002/jnr.20410. [DOI] [PubMed] [Google Scholar]
- 77.Ren-Patterson RF, et al. Gender-dependent modulation of brain monoamines and anxiety-like behaviors in mice with genetic serotonin transporter and BDNF deficiencies. Cell Mol Neurobiol. 2006;26:755–80. doi: 10.1007/s10571-006-9048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duman RS. Pathophysiology of depression: the concept of synaptic plasticity. Eur Psychiatry. 2002;17 3:306–10. doi: 10.1016/s0924-9338(02)00654-5. [DOI] [PubMed] [Google Scholar]
- 79.Beaulieu JM, et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105:1333–8. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.