

SUMMARY
FK506 binding protein 12 (FKBP12) binds the immunosuppressant drugs FK506 and rapamycin and regulates several signaling pathways, including mammalian target of rapamycin (mTOR) signaling. We determined whether the brain-specific disruption of the FKBP12 gene altered mTOR signaling, synaptic plasticity, and memory. Biochemically, the FKBP12-deficient mice displayed increases in basal mTOR phosphorylation, mTOR-Raptor interactions, and p70 S6 kinase (S6K) phosphorylation. Electrophysiological experiments revealed that FKBP12 deficiency was associated with an enhancement in long-lasting hippocampal long-term potentiation (LTP). The LTP enhancement was resistant to rapamycin, but not anisomycin, suggesting that altered translation control is involved in the enhanced synaptic plasticity. Behaviorally, FKBP12 conditional knockout (cKO) mice displayed enhanced contextual fear memory, and autistic/obsessive-compulsive-like perseveration in several assays including the water maze, Y-maze reversal task, and the novel object recognition task. Our results indicate that FKBP12 plays a critical role in the regulation of mTOR-Raptor interactions, LTP, memory, and perseverative behaviors.
Introduction
FK506-binding proteins (FKBPs) originally were described as ubiquitously expressed immunophillins that mediate the pharmacological activities of naturally occurring macrolide immunosuppressants such as FK506 and rapamycin. It is now known that FKBPs have a broader range of functions. FKBPs are peptidyl-prolyl cis-trans isomerases (PPiases) involved in the conversion of cis-proline residues to a less sterically restricted trans conformation. FKBPs also are involved in the regulation of intracellular calcium release, gene transcription, protein translation, and cellular trafficking (Harrar et al., 2001). FKBP12, the smallest of member of the FKBP family, has a basic domain, a PPiase catalytic domain, and a drug-binding pocket, all of which are characteristic of the FKBP family.
FKBP12 modulates the activity of several receptor complexes, including ryanodine receptors (RyRs), transforming growth factor-β receptor complex, and inositol-P3-receptors (Harrar et al., 2001), but also influences protein kinase signaling. FKBP12 bound to rapamycin regulates the kinase, mammalian target of rapamycin (mTOR, also known as FRAP or RAFT) (Jacinto and Hall, 2003). The regulation of mTOR and its downstream effectors are highly conserved from yeast to humans (Hay and Sonenberg, 2004). The most studied function of mTOR is its role in regulating protein translation, primarily cap-dependent translation initiation. mTOR phosphorylates 4E-BP, an inhibitory protein that acts to sequester the translation initiation factor eIF4E (Klann and Dever, 2004). Another important target of mTOR is p70 S6 kinase (S6K), which has been shown to phosphorylate and activate eIF4B, thereby stimulating the DEAD box helicase eIF4A to unwind the inhibitory secondary structure in the 5’ untranslated region of eukaryotic mRNAs (Raught et al., 2004). Thus, activation of mTOR results in the stimulation of translation initiation via the regulation of both 4E-BP and S6K.
FKBP12 is the intracellular receptor for rapamycin, a commonly utilized inhibitor of mTOR signaling. Rapamycin-bound FKBP12 binds and inhibits mTOR (Brown et al., 1994). mTOR also is regulated by other associated proteins (Kim et al., 2002; Loewith et al., 2002; Sarbassov et al., 2004), including the regulatory associated protein of mTOR (Raptor); mTOR bound to Raptor is commonly referred to as mTOR complex 1 (mTORC1). Raptor enhances the interaction of mTOR with S6K, promoting cell growth. mTORC1 formation is inhibited by rapamycin, but the extent of the inhibition is dependent on the nutrient state of the cell, suggesting the existence of variable mTOR-Raptor interaction states (Kim et al., 2002).
New protein synthesis is required for many long-lasting forms of synaptic plasticity and long-term memory (LTM) (Kelleher et al., 2004b; Klann and Dever, 2004). Given the central role of mTOR in translation control, ascertaining its regulation in neurons is vital to elucidating the regulation of protein synthesis during synaptic plasticity and memory. We examined the effects of the genetic deletion of the FKBP12 protein on mTOR signaling, synaptic plasticity and memory. In biochemical experiments we found that deletion of FKBP12 was associated with enhanced mTOR phosphorylation, an increase in the interaction between mTOR and Raptor, and enhanced phosphorylation of the mTOR target S6K. In electrophysiological studies we found that the deletion of FKBP12 resulted in enhanced late phase LTP (L-LTP) that was resistant to rapamycin, but was blocked by inhibitors of protein synthesis. Behavioral experiments revealed that the FKBP12 mutant mice exhibit enhanced contextual fear, repetitive behavior, and perseveration in several assays of memory, phenotypes that have been observed in several cognitive disorders, including autism spectrum disorder (ASD), obsessive-compulsive disorder (OCD), schizophrenia, and Huntington’s disease. Taken together, our findings demonstrate that FKBP12 normally acts to constrain the mTOR signaling during long-term synaptic plasticity, memory, and perseverative behaviors.
RESULTS
FKBP12 is selectively disrupted in the hippocampal area CA1
A general deficiency in FKBP12 produces an embryonic lethal phenotype due to altered cardiac development (Shou et al., 1998). For this reason, we employed a strategy of selective FKBP12 ablation using a “floxed” allele of FKBP12 (Fkbp12fl) crossed with a brain-specific Cre recombinase mouse line (T-29). The maximal onset of Cre recombinase expression in this mouse line is delayed until approximately three weeks after birth allowing for normal development in the presence of FKBP12. Furthermore, Cre expression is primarily restricted to area CA1 of the hippocampus and the forebrain (Tsien et al., 1996). The presence of the Fkbp12fl allele or Cre transgene was determined using PCR specific primers (Figure 1a).
Figure 1. FKBP12 is selectively deleted from the mouse hippocampus and forebrain.

(a) PCR identification of alleles of Fkpb12fl and CAMKIIα driven Cre. (b) Western blot analysis using antibodies to FKBP12 shows elimination of FKBP12 hippocampal area CA1. (c) FKBP12 is primarily localized to cell body layer in area CA1. (A) In wild-type mice, FKBP12 is highly enriched in the cell body layer. (B) 40X magnification of cell body layer in wild-type mice. (C) FKBP12 is largely absent in FKBP12 cKO mice. (D) 40X magnification of the cell body layer in FKBP12 cKO mice. (d) FKBP12 is present in synaptoneurosomes of wild-type (WT) mice and is reduced in FKBP12 cKO (KO) mice. (e) CAMKIIα driven Cre disruption of FKBP12 in other regions of the brain. Expression is quantified as % of control, wild-type expression for the tissue and brain region examined. FKBP12 cKO expression, area CA1: 10, area CA3/DG: 42, cerebellum: 87, midbrain: 64, prefrontal cortex: 22, amygdala: 21, diaphragm: 104, liver: 99, heart: 102.3. 20 μg/ lane. (* p<.05, ** p<.01, *** p<.005, ANOVA) (f) FKBP12 protein levels in KO are expressed at similar levels to those in WT mice in tissues outside the nervous system. Quantification included with Figure 1e, mean ± SEM. Lysates prepared from tissues extracted from WT and KO mice, 75 μg/ lane, β-actin controls not shown.
The absence of FKBP12 in hippocampal area CA1 was confirmed by immunoblotting and immunohistochemical staining (Figures 1b and 1c). To examine the possibility that a closely related FKBP12 homolog, FKBP12.6, was upregulated by deletion of FKBP12, we utilized antibodies that detect both proteins (Figure 1b). Upregulation of FKBP12.6 did not occur in response to the genetic disruption of FKBP12. Given the recent finding that FKBP38 can function as a direct inhibitor of mTOR (Bai et al., 2007), we also examined FKBP38 levels in the brain and found that FKBP38 is expressed at low levels in the cortex and hippocampus (Supplemental Figure 1d). Interestingly, in a survey of several tissues we found that FKBP12 levels were highest in the mouse prefrontal cortex and hippocampus, and were expressed in a pattern inversely correlated with that of FKBP38 (Figure 1e, Supplemental Figure 1d). In wild-type mice, FKBP12 protein expression was detected in the somatic layers in area CA1 of the hippocampus (Figure 1b) and in area CA3 (data not shown). In FKBP12 conditional knockout (cKO) mice the staining for FKBP12 was nearly absent. Higher resolution examination showed dense staining of FKBP12 in the soma, with less dense but punctate staining in the dendritic layer of stratum radiatum (Figure 1c, Panel A, B). This staining was largely absent in FKBP12 mutant mice (Figure 1c, Panel C, D). Fractionation experiments with hippocampal homogenates revealed that FKBP12 is normally present in the synaptoneurosome fraction and the cellular supernatant (Figure 1d). To determine whether the transgenic Cre line we used for this study was expressed in other parts of the brain, we examined FKBP12 levels in other brain regions and tissues. We found nearly normal levels of FKBP12 in the cerebellum, but found reduced levels of FKBP12 in the midbrain (Figure 1e). Strikingly, FKBP12 levels were reduced to ~20% of wild-type levels in the prefrontal cortex and amygdala, with expression reduced to levels similar to those displayed in hippocampal area CA1 of the cKO mice (Figure 1e). Finally, to confirm that the gene was not disrupted in other tissues, we examined FKBP12 levels in muscles and organs of the periphery and observed that FKBP12 expression was similar in mutant and wild-type mice (Figure 1e, f). We also examined basal FKBP12 expression in mice that were homozygous for the floxed FKPB12 allele but did not express Cre recombinase. We found that FKBP12 was expressed at similar levels to mice homozygous for the wild-type FKBP12 allele (Supplemental Figure 1a). In addition, FKBP12 protein was removed from area CA1 as early as P14 (Supplemental Figure 1b). However, the removal of FKBP12 did not occur early enough in development or to a degree sufficient to result in lethality, as expected numbers of cKO pups were generated (data not shown). In summary, forebrain-specific disruption of FKBP12 was achieved bypassing the critical developmental period for which FKBP12 is required.
mTOR phosphorylation, mTOR-Raptor interactions, and S6K phosphorylation are enhanced in FKBP12 cKO mice
FKBP12 serves as an intracellular receptor for rapamycin and mediates the activity of this immunosuppressant. Rapaymycin binds FKBP12 and occupies a conserved binding domain within mTOR, blocking its normal signaling (Kim et al., 2002). Given the unlikely scenario that FKBP12 evolved to bind macrolide toxins, we hypothesized that the loss of FKBP12 might result in enhanced mTOR signaling. To test this hypothesis, we analyzed the phosphorylation of mTOR at residues correlated with its activation in the FKBP12-deficient mice. In FKBP12 cKO mice, we observed an increase in the level of mTOR phosphorylation at serine-2448 in hippocampal area CA1 homogenates. These findings suggest that FKBP12 suppresses mTOR signaling even in the absence of rapamycin.
We also examined the phosphorylation of several downstream targets of mTOR. Although we detected a trend toward increased levels of phosphorylated 4E-BP in the FKBP12 cKO mice, the differences were not statistically significant (Figure 2b). Another target of mTOR is S6K. It is phosphorylated at serine-389 by mTOR and threonine-421 by extracellular signal regulated kinase (ERK) (Zhang et al., 2001). We observed increased phosphorylation of S6K at threonine-389, but no difference in phosphorylation at threonine-421/424 (Figure 2b). These results indicate that although mTOR signaling may be enhanced homeostatically in the FKBP12 cKO mice, its downstream targets are differentially regulated.
Figure 2. mTOR phosphorylation, S6K phosphorylation, and Raptor/mTOR interactions are increased in FKBP12 cKO mice.

(a) mTOR phosphorylation is increased in FKBP12 cKO mice. Wild-type (WT) n=5, FKBP12 cKO (KO) n=6, **p<0.01. (b) Western blot analyses of downstream mTOR targets. Proteins examined: p-4EBP phospho-4EBP1 (Thr 37/46); 4E-BP; p-S6K1(421), phospho-S6K1 (Thr 421/424); p-S6K(389), phospho-S6K1 (Ser 389); β-Tub, beta-tubulin; FKBP12. n=4, each genotype. WT n=5, KO n=6, *p<.05. (c) Raptor binding to mTOR is increased in FKBP12 cKO mice. Immunoprecipitation of protein isolated from hippocampal lysates with antibodies to mTOR (IP:mTOR) and Raptor (IP:Raptor). IP:mTOR, n=5; IP:Raptor, n=3 (*p<.05, Student’s t-test).
The complex formed by FKBP12-rapamycin occupies the same binding site within mTOR that is required for interaction with Raptor, a protein that facilitates mTOR signaling. Because the FKBP12-rapamycin complex displaces Raptor, we hypothesized that a deficiency in FKBP12 would enhance the interaction between mTOR and Raptor (Kim et al., 2002). To test this possibility, we isolated tissue from area CA1 and immunoprecipitated mTOR protein complexes using an mTOR antibody. We found increased levels of mTORC1 in FKBP12 cKO mice when compared to wild-type mice (Figure 2c). These differences were not due to alterations in total levels of either Raptor or mTOR. Similar results were obtained when we used a Raptor antibody for the immunoprecipitation (IP) (Figure 2c). Furthermore, when Raptor was examined in extracts obtained from the unbound IP supernatant, reduced Raptor levels were present in fractions from the cKO mice (Supplemental Figure 1c). These increases were quantified and correlated with the levels of increased p-mTOR in the FKBP12 cKO mice. Moreover, because Raptor promotes mTOR binding to S6K, one would predict that the IPs prepared from FKBP12 cKO mice would yield increased S6K. Indeed, we observed an increase in S6K levels in the IPs from the FKBP12 mutant mice (Supplemental Figure 1c). Together, these data support the idea that FKBP12 is normally involved in the suppression of mTOR activity and signaling.
FKBP12 cKO mice display normal basal synaptic transmission and E-LTP
We next examined synaptic plasticity at the Schaffer collateral-CA1 synapse in hippocampal slices from the FKBP12 mutant mice. Because the absence of FKBP12 can increase calcium leak from the ryanodine receptor, an intracellular calcium release channel, it was possible that the loss of FKBP12 would affect basal synaptic function (Berridge, 1998). We found that basal synaptic transmission, measured as synaptic output in response to a stimulatory input, was normal in FKBP12 cKO mice (Figure 3a). We next examined whether FKBP12 was required for paired-pulse facilitation (PPF), a form of presynaptic plasticity evoked by two temporally-linked stimuli (Katz and Miledi, 1968). PPF was normal in FKBP12 cKO mice when compared to wild-type mice at several time intervals (Figure 4b). The same measurements of synaptic function also were examined in FKBP12 heterozygote (CamKIIα-Cre+/Fkbp12fl/+) mice and were indistinguishable from both wild-type and FKBP12 homozygote cKO mice (data not shown).
Figure 3. FKBP12 cKO mice have normal basal synaptic transmission and express normal E-LTP.

(a) Input versus output plot indicating that wild-type and FKBP12 cKO mice have comparable fEPSP slopes evoked by increasing stimulation. Wild-type (WT) mice, FKBP12 cKO mice (KO), n= 20 slices, 12 mice per genotype (p>.05, Student’s t-test). (b) FKBP12 cKO mice exhibit normal PPF compared to their wild-type littermates. The percent facilitation, determined by the ratio of the second fEPSP to the first fEPSP, is shown at interpulse intervals from 10 to 300 ms. WT, n=25 slices, KO, n=26 slices, 14 mice per genotype. (c) A single train of HFS evoked similar levels of E-LTP in wild type and FKBP12 cKO mice that decayed to baseline after 80 minutes. WT, n=12 slices; KO, n=13 slices; 6-9 mice per genotype (p>.05, ANOVA). (d) A single pattern of TBS evoked similar levels of E-LTP in WT and KO mice that decayed to baseline after 80 minutes. WT, n=14 slices; KO, n=15 slices; 7-10 mice per genotype (p>.05, ANOVA).
Figure 4. FKBP12 cKO mice express enhanced L-LTP and L-LTP induction stimulates mTOR-Raptor interactions.

(a) Representative traces of fEPSPs in slices from wild-type (WT) and FKBP12 cKO (KO) mice that received four trains of HFS. (b) Four trains of HFS elicited enhanced L-LTP in FKBP 12 KO mice compared to that evoked in WT mice. WT, n=12 slices; KO, n=14 slices; 5-8 mice per genotype (p<.05, ANOVA). (c) Representative traces of fEPSPs from WT and KO slices that received three patterns of TBS. (d) Three patterns of TBS elicited L-LTP in FKBP12 cKO mice that were greater that that evoked in wild-type mice 90 minutes post-stimulation. WT, n=8 slices; KO, n=9 slices; 4-6 mice per genotype (p<0.05, ANOVA). For both 4X HFS and 3X TBS: (WT) a, baseline, c post stimulation. (KO) b, baseline, d, post stimulation. (e) Raptor binding to mTOR increases following L-LTP-inducing stimulation. In FKBP12 cKO mice, Raptor binding to mTOR is near saturation. (f) Following stimulation of the Schaffer collaterals to area CA1 increased Raptor binding is observed in area CA1, but not area CA3. (g) mTORC1 formation increases 10 minutes after HFS stimulation in wild-type slices but not KO slices. mTORC1 levels return to baseline 60 minutes post-HFS in wild type slices but remain significantly elevated in KO slices compared to 10 minutes post HFS but not basal (unstimulated) slice. Steady-state raptor association with mTOR is higher initially in KO slices (No HFS: WT, n=6; KO, n=6) (HFS+ 10’; WT, n=6; KO, n=6) (HFS +60’WT, n=5; KO, n=5) * p<0.05, ** p< 0.01, ANOVA.
We next examined early phase LTP (E-LTP) in hippocampal slices prepared from the FKBP12 cKO mice. E-LTP was elicited by two different patterns of stimulation, either with one train of high-frequency stimulation (HFS) or a single pattern of theta-burst stimulation (TBS). A single train of either HFS (Figure 3c) or TBS (Figure 3d) elicited E-LTP in slices from the FKBP12 cKO mice that were indistinguishable from wild-type littermates. These results suggest that the loss of FKBP12 has no apparent effect on the underlying molecular mechanisms involved in the establishment of transient (protein-synthesis independent) forms of hippocampal LTP.
FKBP12 cKO mice have enhanced L-LTP and L-LTP induction increases mTORC1 interaction
To determine whether the removal of FKBP12 altered late-phase LTP (L-LTP), we stimulated hippocampal slices with four spaced trains of HFS. Following this induction protocol, L-LTP in the FKBP12-deficient mice was significantly enhanced when compared to their wild-type littermates (representative traces are shown in Figure 4a). Cumulative data from several experiments indicated that L-LTP in FKBP12 cKO mice was increased for the duration of the three-hour experiment compared to L-LTP in control mice (Figure 4b). Similar results were obtained when L-LTP was elicited by multiple patterns of TBS (Figures 4c and 4d). These results indicate that the loss of FKBP12 enhances long-lasting forms of LTP in area CA1 that normally require protein synthesis.
We also examined whether the relative levels of mTORC1 were dynamically regulated. To accomplish this, we elicited L-LTP in hippocampal slices using four trains of HFS, and immunoprecipitated mTOR protein complexes. Analysis of IP complexes isolated 10 minutes after the final train of HFS revealed a robust increase in the overall level of mTOR-Raptor binding (Figure 4e). The observed increases in mTORC1 formation also was specific to the L-LTP stimulation as the observed increase in mTOR-Raptor association was only observed in area CA1 and not other regions of the hippocampus that were not stimulated (Figure 4f). Interestingly, we found no further increase in mTORC1 formation in FKBP12 mutant mice 10 minutes after HFS, suggesting that mTORC1 levels were near saturation (Figures 2c and 4e). However, when we examined mTORC1 levels 60 min post-HFS stimulation in wild-type slices, we found mTORC1 levels had returned to pre-stimulation levels, whereas mTORC1 levels in FKBP12 mutant slices remained elevated. Although not significantly different from unstimulated (no HFS) slices, the overall levels of mTORC1 in FKBP12 cKO slices at 60 min post-HFS were significantly higher than those 10 minutes post-HFS (Figure 4g), suggesting that newly formed mTORC1 in FKBP12-deficient mice is resistant to the molecular processes down-regulating this interaction in wild-type mice. These data indicate that the interaction between Raptor and mTOR is dynamically regulated and that the modulation of this interaction is a circuit specific.
Enhanced L-LTP in FKBP12 cKO mice is insensitive to rapamycin but sensitive to inhibitors of protein synthesis
Rapamycin inhibits L-LTP without affecting the early phase of synaptic enhancement (Tang et al., 2002), consistent with the notion that mTOR-dependent regulation of translation initiation is required for more durable, long-lasting forms of LTP. Because FKBP12 is the intracellular receptor that binds rapamycin, one would predict that L-LTP in the FKBP12-deficient slices would be resistant to rapamycin. Consistent with this prediction, rapamycin blocked L-LTP evoked by multiple trains of HFS in wild-type slices, but did not significantly affect L-LTP in slices from the FKBP12 cKO mice (Figures 6a and 6b). This result supports a role for FKBP12 as a mediator for rapamycin-dependent blockade of L-LTP and provides evidence that homologs of FKBP12, like FKBP12.6, do not play redundant molecular roles for FKBP12 during L-LTP.
Figure 6. FKBP12 cKO mice display enhanced contextual associative fear memory, altered novelty interaction, and increased perseverative/repetitive behavior.

(a) Mean freezing behavior of wild type (WT) and FKBP12 cKO (KO) mice. Both genotypes performed similarly during training and had similar freezing behavior in both STM contextual and cued memory tests. FKBP12 KO mice display enhanced freezing during LTM contextual fear memory testing but no difference in cued associative fear memory (* p>.05, One way ANOVA). (b) FKBP12 cKO mice display enhanced contextual LTM in the absence of STM testing (* p=<.05, One way ANOVA). (c) mTORC1 levels increase in wild-type mice following fear conditioning. mTOR immunoprecipate complexes isolated from untreated mice (naïve), training environment exposure only (con), and fear conditioned (context+shock, FC) and allowed to recover 15 or 60 min (FC 15’ or FC 60’). mTORC1 levels compared to naïve wild-type control. Naïve levels: KO, 179 ± 21%. Context: WT, 156 ± 28%; KO, 208 ± 30%. FC 15’: WT, 172 ± 23%; KO: 214 ± 31%. FC 60’: WT, 104 ± 9%; KO: 176 ± 18% (* p<.05, ** p<.01, ANOVA) WT n=5, KO n=4 for each time point. (d) FKBP12 cKO mice display greater repetitive behavior as determined by marble burying during 30-min test period. Marbles buried, WT: 10.9 ± 1.9; KO: 15.3 ± 0.9 (* p<.05, ANOVA) (e) FKBP12 cKO mice display preference for the familiar object during object recognition analysis. Dashed line represents PI=0.5, or neutral preference. WT mice, n=11; KO, n=10 (*, + p<0.05, ++ p<.01, ANOVA).
Under normal conditions, L-LTP is critically dependent on the de novo synthesis of mRNAs and proteins (Frey and Morris, 1997). Overlaying these processes are complex signal transduction cascades that regulate the synthesis of these new gene products. Because the loss of FKBP12 has the potential to not only influence protein synthesis by modulating mTOR signaling, but also affect calcium signaling via RyRs or phosphatase signaling via calcineurin, we next determined whether the enhanced L-LTP in FKBP12-deficient mice required protein synthesis. Incubation of slices with the protein synthesis inhibitor anisomycin significantly attenuated L-LTP in both FKBP12 cKO and wild-type mice (Figures 5c and 5d). This was especially evident at later time points (t>160 minutes), when L-LTP in slices from the FKBP12 mutant mice was indistinguishable from that in slices from wild-type mice (Figure 5d). There was a trend for enhanced potentiation during intermediate time points (t>60 minutes and t<160 minutes) indicating the possibility that the removal of FKBP12, in addition to altering mTOR signaling, might influence other molecules that contribute to the enhanced L-LTP observed in these mice. Although anisomycin treatment prevented the L-LTP enhancement in FKBP12 cKO slices following four trains of HFS, a significant enhancement was still observed in the early phase of potentiation following the induction of TBS-LTP (Figure 4d). This phase of LTP is resistant to inhibitors of protein synthesis; thus, we cannot rule out the possibility that in addition to enhancing protein synthesis-dependent LTP, FKBP12 removal alters the kinetics of calcium signaling, possibly through altered RyR/IP3 receptor activity, in the early phase of potentiation following some patterns of stimulation.
Figure 5. Enhanced L-LTP in FKBP12 cKO mice is resistant to rapamycin, but sensitive to anisomycin.

(a) Representative fEPSP traces in slice from wild type (WT) and FKBP12 cKO (KO) mice that received four trains of HFS in the presence of rapamycin or vehicle. (b) Four trains of HFS in the presence of rapamycin elicited enhanced L-LTP in FKBP12 cKO mice but blocked L-LTP wild-type mice. WT+vehicle, n=8 slices; KO+vehicle, n=8 slices; WT+rapamycin, n=12 slices; KO+rapamycin, n=19 slices; 5-8 mice per genotype and treatment (p<.05, ANOVA). (c) Representative fEPSP traces from WT and KO slices that received four trains of HFS in the presence of either anisomycin or vehicle. (d) Four trains of HFS in the presence of anisomycin blocked L-LTP in both FKBP12 KO mice and their WT mice. WT+vehicle, n=9 slices; KO+vehicle, n=14 slices; WT+anisomycin, n= 11 slices; KO+anisomycin, n=11 slices; 6-10 mice per genotype (p<0.05, ANOVA). For both rapamycin and anisomycin treatments: (WT) a, baseline, c post stimulation+drug. (KO) b, baseline, d, post stimulation+drug.
FKBP12 cKO mice have enhanced associative contextual memory and altered object recognition memory
To determine whether the lack of FKBP12 alters associative learning and memory, we tested the FKBP12 cKO mice with a conditioned fear paradigm. In this paradigm, mice learn to fear an environmental context or an emotionally neutral conditioned stimulus (CS, white noise) resulting from its temporal association with an aversive unconditioned stimulus (US, foot shock). When exposed to the same environmental context (contextual conditioning) or CS (cued conditioning) at a later time, conditioned mice demonstrate a stereotypical fear response, termed freezing (Fanselow, 1984; Phillips and LeDoux, 1992).
For contextual and cued conditioning, FKBP12 cKO mice and their wild-type littermates were trained with two CS/US pairings. Both groups of mice displayed similar levels of freezing prior to training (data not shown) and exhibited comparable short-term memory (STM) performance following both contextual (one hour after training) and cued training (two hours after training) (Figure 6a). Combined these results suggest that FKBP12-deficient mice can learn and remember to associate neutral stimuli with an aversive event when tested following a short delay interval. Importantly, these data also demonstrate that the FKBP12-deficient mice display similar levels of stereotypic freezing behavior relative to wild-type mice. When long-term memory (LTM) was examined 24 hours after training, the FKBP12 cKO mice demonstrated a significant increase in freezing to the contextual cues associated with the footshock. In contrast, both genotypes showed similar levels of freezing to the auditory cue. These findings suggest that FKBP12-deficient mice display enhanced contextual memory but not cued memory following a 24-hr delay (Figure 6a). FKBP12 mutant and wild-type mice trained using a single CS-US pairing displayed low levels of freezing that were indistinguishable from one another (data not shown).
The initial associative fear memory experiments tested the same mice for both STM and LTM memory 24-25 hours after training. One possible explanation for the observed fear memory enhancement in FKBP12 cKO mice is that they have impaired extinction where normally, prior contextual exposure diminishes freezing after 24 hours (Lattal and Abel, 2001). To test this possibility, mice were tested for contextual fear memory 24 hours, but not one hour, after training. Similar to the previous findings, FKBP12-deficient mice displayed an increase in freezing behavior compared to wild-type mice following the 24-hour delay interval. Thus, the memory augmentation observed in FKBP12 mutant mice is likely due to enhanced memory formation rather than an impairment in extinction. These results indicate that FKBP12 normally plays a role regulating the formation of long-term associative contextual fear memory.
We next examined whether contextual conditioning in wild-type mice increased mTORC1 levels in a manner comparable to that following L-LTP induction. Mice were trained using the conditioned fear paradigm and extracts were prepared from hippocampal area CA1. Experimental groups included mice that had been directly removed from their home cage (naïve), mice that were exposed to the training environment but were not exposed to the shock (US) or tone (CS) (context), or mice that received fear conditioning (US/CS) and were allowed to recover for either 15 or 60 minutes after training (context + shock 15’, context + shock 60’). Compared to untrained mice, we detected a significant increase in mTORC1 formation in mice exposed to the fear conditioning paradigm 15 minutes post-training (Figure 6c). Although there was a similar trend for context exposure alone, these results were not significant. FKBP12-deficient mice exhibited an analogous trend for increased mTORC1 formation in the context alone situation and 15 minutes post fear conditioning, but these increases were not statistically significant. Similar results were detected in area CA3 following fear conditioning (Supplemental Figure 3d). The increases in mTORC1 formation following contextual fear conditioning suggest that mTORC1 signaling is altered by learning that takes place during exposure of the mice to the US/CS in the novel training environment.
The enhanced fear memory displayed by the FKBP12 cKO mice could be due to subtle differences in other expression of other behaviors such as decreased activity or increased ‘anxiety’. Although it is unlikely that the mutant mice have decreased activity or increased anxiety because the FKBP12-deficient and wild-type mice displayed similar levels of freezing following the short-test interval, additional behavioral measures were employed to more directly assess activity and ‘anxiety-like’ traits in an unconditioned paradigm (Bouwknecht and Paylor, 2008). To determine whether FKBP12-deficient mice displayed a generalized decrease in activity and/or increase in anxiety in other settings, we performed open field analysis (OFA). OFA testing revealed no significant differences between FKBP12 cKO and wild-type mice in overall time spent moving, resting, or rising vertically. Similarly, time spent in the center of the open field or performing stereotypical movement also was indistinguishable between genotypes (Supplemental Figure 2a). Another behavioral task employed to examine repetitive and stereotypic behavior is the marble burying task. The marble burying assays the tendency of mice to dig and bury objects, and alterations in marble burying reflects changes in this normal perseverative/repetitive behavior. The results from the marble-burying test revealed that the FKBP12 cKO mice bury significantly more marbles compared to their wild-type littermates (Figure 6d), indicating that the FKBP12 cKO mice display increased repetitive stereotypic/perseverative behavior in this setting.
We next examined the FKBP12 cKO mice for novel object recognition memory. The novel object test can be used to examine STM and LTM, as well as anxiety in mice (Ennaceur and Delacour, 1988). In this test, the mice were exposed and habituated to two different, but neutrally attractive objects. On the test day, the mice were exposed to one of the previously presented objects (the familiar object) and a new object (the novel object). Time spent exploring both objects was recorded and an index generated to measure preference for the novel object. As expected, wild-type mice preferred the novel object during the testing period. In contrast, the FKBP12 cKO mice preferred the familiar object during novel object testing (Figure 6e). The difference between wild-type and FKBP12-defcient mice could not be accounted for by exploration time or inherent preference value of the novel object (Figure 6e). There are several explanations for these findings. First, the data could indicate that the FKBP12 mutant mice display a memory deficit during the execution of this particular task. However, this is unlikely because if the FKBP12-deficient mice did not remember the objects then they would have displayed an equal preference for both the familiar and novel object, yet they showed a clear preference for the familiar object. Second, the FKBP12-deficient mice could have an increase in ‘anxiety’ toward the novel object, which we also believe is unlikely given that they do not display increased anxiety in other settings. The final possible explanation is that the FKBP12 mutant mice exhibit a preference for the familiar object, reflecting either perseveration for the familiar object or an inability to inhibit responding to the familiar object.
FKBP12 cKO mice display enhanced perseveration following RAP MWM training and Y-maze reversal task
To test the hypothesis that FKBP12-deficient mice display increased perseveration and/or impaired inhibition, we examined the performance of FKBP12-deficient mice on two water-escape tests. In the first test, we used the Morris water maze (MWM) (Morris, 1984). During ‘standard’ hidden-platform training the FKBP12 cKO mice and their wild-type displayed similar escape latencies to locate the hidden platform (Supplemental Figure 3a). During a probe test on day eight, the two groups of mice both displayed spatially-biased search behavior in the training quadrant relative to the other quadrants (Supplemental Figure 3b, 3c), indicating that the wild-type and FKBP12-deficient mice located the hidden platform using similar spatial search strategies. The two groups of mice also performed similarly on an earlier probe test, conducted on day four of the eight-day training (Supplemental Figure 3d). Using a four-day protocol, performance of the wild-type and FKBP12 cKO mice also was indistinguishable (data not shown).
To more specifically determine whether the FKBP12-deficient mice displayed perseverative behavior in the MWM, we challenged their ability to learn the location of the hidden platform when moved to different locations using a modified version of the delayed match to place MWM paradigm, termed the repeated acquisition paradigm (RAP, see Experimental Procedures for more detail). This MWM paradigm tests the ability of the mice to use information gathered during a brief period of time and forces them to employ more transient forms of memory. FKBP12 cKO mice performed similarly to their wild-type littermates during the reference memory phase of the training (Figure 7b). The FKBP12 cKO mice displayed notable perseveration for the original reference training platform location (Supplemental Figure 3e), but their escape latency times were not different compared to wild-type littermates. However, on the second day of training, when the hidden platform had been moved to the opposite quadrant of reference memory training, FKBP12 cKO mice showed reduced latency time compared to wild-type mice (Supplemental Figure 3e). This difference was not due to altered swim speed (data not shown), visual deficiency, or quadrant preference as escape latency times using a visible cue (a flag placed on top of the submerged platform) were identical for both groups (Figure 7a). The enhancement, although small, became more apparent during later phases of the RAP paradigm.
Figure 7. FKBP12 cKO mice have normal spatial learning, reference and working memory following RAP MWM and Y-maze reversal training mice but enhanced perseveration for former escape location and initial Y-maze escape arm.

(a) Escape latency times during initial nine days of RAP training. (b) Previous testing day platform crossings (* p<.05). For RAP MWM, WT, n=11; KO, n=9 (* p<0.05, ANOVA). The mean of time spent in each maze quadrant during the probe test. (c) Percent correct arm choice by trial block number during training, testing and reversal phase of Y-maze experiment. FKBP12 KO mice perseverate more for the original training arm then their wild-type littermates (* p<0.05, ** p<0.01, ANOVA). (d) The number of correct arm choices during training phase (20 trials) of Y-maze reversal task, WT=17.3 ± 0.5 KO=17.6 ± 0.5. The number of trials needed to reach criterion (9/10 correct) during reversal phase of Y-maze reversal task, WT=18.5 ± 1 KO= 23.3 ± 1.4. For Y-Maze, WT, n=13; KO, n=12 (* p<0.05, ANOVA).
Following the reference, reversal and visible portions of the RAP paradigm, the submerged platform was moved to random locations within the water maze. This location was used for only one day and was not repeated. The mice then were challenged to find the new location in four trials, one after the other. Although the FKBP12-deficient mice appeared to display slighter faster acquisition of the new platform location on trial to trial comparison, they displayed indistinguishable escape latencies over the course of the four-trial block (each trial block is contained by a single novel platform location) compared to wild-type mice (Supplemental Figure 3f). During the course of the test, both wild-type and FKBP12 cKO mice displayed a tendency to search “old” platform locations while trying to escape the water maze. An examination of these “previous platform” crossings (PPCs) revealed that FKBP12 cKO mice showed a significantly increased number of PPCs when compared to their wild-type littermates (Figure 7b). Together these results suggest that FKBP12 is involved in inhibiting the tendency to perseverate and inhibit responding to previously reinforced locations.
To further challenge this ‘increased perseveration/decreased inhibition’ hypothesis in FKBP12 cKO mice, we used a water-based Y-maze assay. In brief, mice were trained to locate an escape platform located in one arm of a Y-shaped maze. Upon successful acquisition of the task, mice were given a reversal test where the position of the escape platform was reversed to the alternate arm and ability to learn the new location was measured. The number of trials to criterion (9/10 correct) on the reversal test was determined. During training, FKBP12-deficient mice and their wild-type littermates acquired the initial escape arm with equal efficacy (Figure 7c), indicating that FKBP12 is not involved in this form of simple position learning. However, when the escape arm was reversed, the FKBP12 cKO mice required ~25% more trials to achieve the same escape success criteria compared to wild-type littermates (Figure 7d). These findings further demonstrate that FKBP12-deficient mice have impaired performance in learning and memory assays that require an inhibition of previously reinforced responses. Performance in this assay is governed predominantly by cortical-striatal function. Although not reduced to the same levels as those found in hippocampal area CA1, FKBP12 levels were significantly decreased in the midbrain of the FKBP12 cKO mice (Figure 1e), which may be sufficient to explain these results. These results combined with the RAP MWM results and novel object recognition data suggest that FKBP12 mice display increased perseverative behaviors.
Discussion
FKBP12 is a modulator of signaling pathways known to be involved in synaptic plasticity, behavioral plasticity, and memory formation. Thus, it is important to define the mechanism for the FKBP12-dependent modulation of these processes. Previous studies had shown that the complete depletion of FKBP12 resulted in near total lethality, precluding studies aimed at the examination of its role in the cellular and molecular processes underlying learning and memory. We have taken advantage the Cre-lox expression system to achieve both spatially- and temporally-restricted deletion of the FKBP12 gene to circumvent this problem. We found that deletion of the FKBP12 gene resulted in biochemical alterations in the mTOR signaling pathway, enhanced L-LTP, increased memory following associative fear conditioning, and enhanced memory perseveration in the novel object recognition task, RAP version of the MWM and Y-maze reversal task. In addition, FKBP12-deficient mice displayed an increase in repetitive/perseverative behavior in the marble burying assay. Our studies are the first to demonstrate a critical role for an immunophilin in learning, memory, and perseverative behaviors, and to define a role for FKBP12 in the regulation of mTOR in the absence of macrolide toxins. Moreover, these studies may offer insight into the molecular underpinnings of repetitive behaviors and perseveration phenotypes presented in ASD, OCD, schizophrenia, and neurodegenerative disorders.
FKBP12 regulates mTOR/Raptor interactions
It is unlikely that FKBP12 evolved to normally interact with macrolide immunosuppressants. Clues to its normal cellular function can however be gleaned from the observation that rapamycin blocks mTOR signaling. Moreover, this interaction disrupts the interaction between mTOR and Raptor at the FKBP12-binding domain (FRB) on mTOR (Cafferkey et al., 1993). Not only does Raptor play a positive role in mTOR signaling but it also interacts with mTOR at the FRB site. Thus, it is possible that FKBP12 normally functions to repress mTOR activity by acting as the intracellular adaptor/receptor for an as yet unidentified endogenous inhibitor of neuronal mTOR activity, perhaps via modulation of mTOR/Raptor binding. This idea is further supported by recent findings establishing direct inhibition of mTOR in the absence of rapamycin. FKBP38, an FKBP family member formed from an FKBP-C domain (essentially a minimal consensus FKBP12 sequence) and a pair of protein-protein interaction domains, was shown to bind Rheb, a potent activator of mTOR (Bai et al., 2007). This same study showed that HEK-293 cells over expressing FKBP38 displayed decreased mTOR activity and conversely, cells in which the levels of FKBP38 had been decreased with siRNA showed enhanced mTOR signaling. Deletion analysis of FKBP38 showed that its FKBP-C domain, a region highly homologous to FKBP12, was sufficient for mTOR binding (Bai et al., 2007). However, FKBP38 is expressed at very low levels in the brain and is not upregulated in response to FKBP12 removal (Supplemental Figure 1d), diminishing the likelihood that FKBP38 is the major FKBP regulating neuronal mTOR activity. Instead, we hypothesize that FKBP12 serves as the critical FKBP suppressor of neuronal mTOR signaling activity.
The idea that FKBP12 suppresses mTOR signaling is supported by several lines of experimental evidence presented herein. First, enhanced phosphorylation of serine-2448 on mTOR was observed in FKBP12-deficient mice. Although not a direct assay of mTOR activity, phosphorylation at this site previously was shown to increase following stimulation of mTOR signaling with insulin, interleukin-3, or Akt activation (Reynolds et al., 2002; Scott et al., 1998). Second, in FKBP12 cKO mice, we observed an increase in the amount of Raptor associated with mTOR (Figure 2c). This finding is more important when examined in the context of hippocampal synaptic plasticity and memory, where L-LTP-inducing stimuli and fear conditioning in wild-type mice produce a similar increase in mTORC1 formation (Figures 4e, 4g and 6c). Finally, we observed enhanced phosphorylation of S6K, a translational regulatory kinase and downstream target of mTORC1. Taken together, these data strongly suggest that FKBP12, even in the absence of rapamycin, normally acts to suppress mTORC1 signaling. Interestingly, not all downstream targets of mTORC1 were affected by removal of FKBP12. Notably, basal 4E-BP phosphorylation was unaffected. This result is consistent with the idea that when FKBP12 is ablated, mTORC1 signaling is sensitized and/or partially activated, but remains under the control of other regulatory elements. Multiple signaling pathways converge on mTOR to promote its full activation in response to a stimulus (Kelleher et al., 2004a). In this way, mTOR signaling required for long-lasting synaptic change may be a node at which signal coincidence detection takes place, similar to activation of the NMDA receptor and ERK (Sweatt, 2001; Wittenberg and Tsien, 2002).
FKBP12 could regulate mTOR function in at least two ways. One obvious mechanism is via its proyl-isomerase activity. FKBP12 may simply modulate protein folding associated with the interaction states that regulate mTOR activity. In addition to Raptor, mTOR is found to complex with at least four different proteins (Bai et al., 2007; Hay and Sonenberg, 2004; Vander Haar et al., 2007). One of these proteins may act as a state conditional repressor of mTOR activity. Intriguingly, although Raptor has been shown to positively affect mTOR signaling in genetic and RNAi studies, it also has been shown to negatively affect mTOR activity in vitro (Kim et al., 2002). It may be that FKBP12 is involved in transitional modification of Raptor between activation (stimulated) and inhibitory (basal) states (Figure 8a). A second possibility is that FKBP12 might act as an intracellular mediator for an unidentified endogenous inhibitor(s) of mTOR or may be a direct inhibitor of mTOR activity by precluding Raptor binding (Figure 8b). Signaling involved in triggering protein synthesis could displace FKBP12 through interaction with an unknown adaptor; recent studies suggest Rheb as attractive candidate for this function (Bai et al., 2007) (Figure 8b). A possible explanation for the finding that some but not all mTOR substrates are upregulated in FKBP12 mutant mice is that FKBP12 regulates only Raptor-mediated mTOR signaling. Other substrates such as 4E-BP may be under the regulation of another mTORC1 scaffolding protein such as proline-rich Akt/PKB substrate-40 (PRAS-40) (Vander Haar et al., 2007). Interactions between mTOR, FKBP12, and endogenous inhibitors may be either intrinsically weak or dependent on multiple proteins in the complex, which may explain why traditional immunoprecipitation experiments have failed to identify them. Indeed, two mTOR interactors were identified only in experiments that pulled down mTOR-binding proteins under crosslinking or less disruptive detergent conditions (Kim et al., 2002; Sarbassov et al., 2004). In either scenario, immunosuppressants would act as a conformational mimetics of a normal cellular inhibitor(s) that stabilizes FKBP12 interaction with mTOR, blocking Raptor access to the FRB domain.
Figure 8. Models for FKBP12 modulation of Raptor/mTOR interactions to promote mTOR signaling.
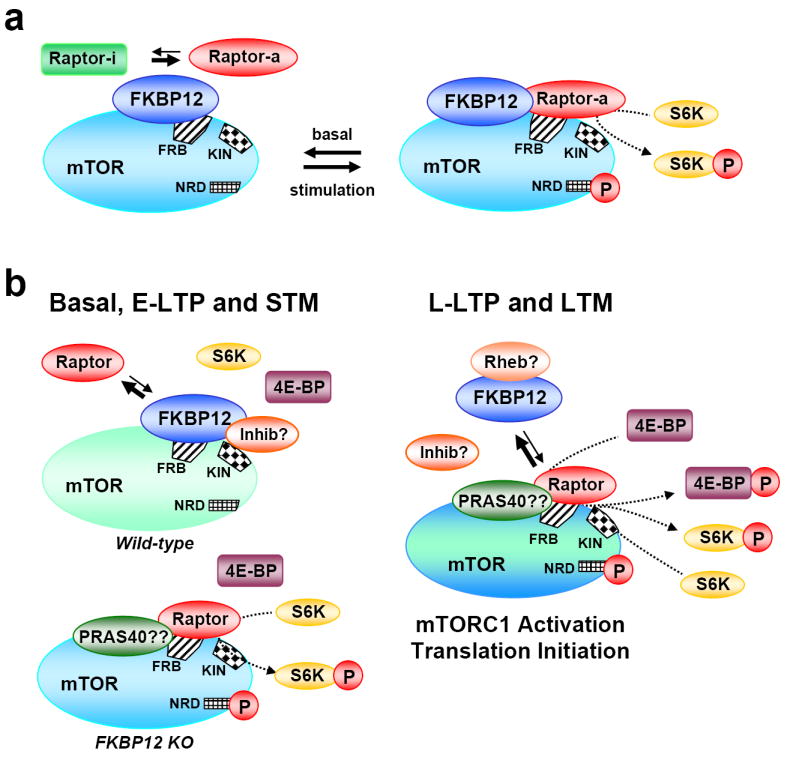
(a) One of the known mTOR-binding proteins (Raptor is used for this example) exists in two states mediated by FKBP12 binding and/or FKBP12 isomerase activity. One state, Raptor-i represses mTOR signaling, the other state, Raptor-a, promotes mTOR signaling. Following upstream activation, Raptor-i is shifted to Raptor-a, which has greater affinity for mTOR, and mTOR signaling is promoted. (b) FKBP12 serves as the intracellular scaffold for an mTOR inhibitory factor(s) or directly competes with Raptor for the mTOR FRB site. In either scenario, FKBP12 represses mTOR activity by blocking Raptor interaction with mTOR. In the FKBP12 cKO mouse, Raptor interaction with mTOR is increased. Increased mTORC1 levels results in enhanced S6K but not 4E-BP phosphorylation preventing translational initiation. Protein synthesis inducing stimulation (i.e. L-LTP), signals either: 1) the displacement of inhibitory factor(s) interacting with FKBP12 or 2) sequesters FKBP12 from mTOR allowing Raptor access to the FRB. Protein synthesis inducing signaling in either wild-type or FKBP12 mutant neurons promotes mTOR access to 4E-BP, possibly through activation of additional mTORC1 associated scaffolds (i.e. PRAS40), allowing translational initiation. Ras-homolog enriched in the brain (Rheb), proline-rich Akt/PKB substrate-40 kd (PRAS-40), FKBP12-binding domain (FRB), KIN (mTOR kinase catalytic domain), NRD (domain, site of serine 2448 phosphorylation).
FKBP12 represses protein synthesis dependent LTP
Paralleling the enhanced memory in the FKBP12 cKO mice is the finding of enhanced L-LTP. The L-LTP enhancement in the FKBP12 cKO mice was insensitive to rapamycin, but sensitive to a general protein synthesis inhibitor (Figure 5d). Although FKBP12 could also affect calcineurin and RyR signaling, the existing studies argue against these pathways as being responsible for the observed enhancement of LTP. Enhanced calcineurin activity would be predicted by the removal of FKBP12; however enhanced calcineurin should result in reduced L-LTP and decreased learning and memory performance (Hoeffer et al., 2007; Malleret et al., 2001; Winder et al., 1998). Similarly, enhanced RyR function resulting from FKBP12 removal is unlikely source for the phenotypes we observe (Van Acker et al., 2004). Deletion of RyR3, the predominant ryanodine receptor expressed in the hippocampus, results in enhanced performance in the MWM and enhanced E-LTP (Futatsugi et al., 1999). Several studies have reported genetic deletions that resulted in the transformation of E-LTP to L-LTP (Banko et al., 2005; Costa-Mattioli et al., 2005; Malleret et al., 2001; Thomas et al., 1996) but only rarely have enhancements of L-LTP been observed (Barco et al., 2003; Wang et al., 2004).
Several features of the observed L-LTP enhancement are particularly intriguing. First, basal synaptic transmission is expressed normally in FKBP12-deficient mice. This is important because FKBP12 has the potential to affect RyR and CaN activity, both of which influence synaptic function (Futatsugi et al., 1999; Nishiyama et al., 2000; Winder et al., 1998). Unlike previous studies reporting shifts to L-LTP following stimulation that normally produces E-LTP (a single train of HFS), E-LTP in the FKBP12 cKO mice was normal. This suggests that FKBP12 does not strongly influence the molecular mechanisms regulating transient change in synaptic strength but instead plays a specific role in processes that control persistent changes in synaptic function. This idea is further supported by the observation that the L-LTP enhancement in the FKBP12 mutant mice is sensitive to protein synthesis blockade (Figure 5d). This is important for two reasons: it implicates mTOR signaling, which is critical for translational initiation, and it eliminates the possibility that the enhanced L-LTP is solely a product of prolonged short-term signaling (i.e. enhanced calcium release from intracellular stores).
A parallel can be drawn between the L-LTP and behavioral phenotypes in the FKBP12 cKO mice. In the both short-term plasticity and STM, i.e. those more involving transient molecular processes, enhancements were not observed in the FKBP12 cKO mice. Enhancements were observed in the FKBP12 cKO mice only in situations where signaling that regulates long-lasting neuronal changes took place. Although a causal relationship between enhanced L-LTP and enhanced memory perseveration has not been shown previously, we propose that this is a potential cellular mechanism for the LTM enhancements in FKBP12 mutant mice.
The loss of FKBP12 enhances stereotypic/repetitive and perseverative behaviors
Our behavioral studies revealed augmented memory and perseveration in the FKBP12 mutant mice. First, FKBP12 cKO mice demonstrated enhanced associative contextual fear memory (Figure 6a). Second, FKBP12-deficient mice displayed significantly altered performance in the novel object recognition test, interacting more with the familiar object than the novel object (Figure 6e). Third, FKBP12 mutant mice demonstrated increased perseveration for previous platform locations during the RAP MWM and Y-maze assays (Figure 7b). Finally, the FKBP12 cKO mice displayed increased repetitive behavior in the marble burying task (Figure 6d). These behavioral phenotypes suggest that FKBP12 regulates neuronal signaling that controls the manifestation of traits that are reminiscent of several neurological disorders including ASD, OCD, and schizophrenia.
The performance of the mutant mice in the novel object recognition test is interesting because FKBP12 cKO mice displayed a preference for the familiar object compared to the novel object (Figure 6e). Although there are several explanations for these results (see Results section), we believe the most parsimonious explanation and one that is consistent with our other findings, is that FKBP12 is important for behavioral inhibition and in its absence mice have increased perseveration. In fact, results from the MWM and Y-maze tests clearly indicate that FKBP12 cKO mice have more difficulty inhibiting previously reinforced responses relative to wild-type littermates. The object recognition, MWM, and Y-maze tests are all learning and memory-based assays, suggesting that FKBP12 may play a significant role in perseveration/inhibition related to cognition and memory. The enhanced contextual fear conditioning is also interesting to consider in this context. Unlike wild-type mice, contextual LTM was equally enhanced in FKBP12 cKO mice regardless of the previous exposure during STM testing. This result, combined with the finding that STM was normally expressed in FKBP12-deficient mice, indicates that FKBP12 may play a role in signaling that supports long-term, but not transient, information storage. The memory enhancements observed in FKBP12 cKO mice may be due to a shift in the molecular signaling mechanisms regulating balance of memory formation and removal, shifting the overall process to favor a type of ‘memory perseveration’. However, the marble burying data suggest that the increase in perseveration may also generalize to domains of CNS function beyond cognitive domains. Collectively, these results suggest that FKBP12 regulates the ability of the mouse to inhibit particular responses, thereby leading to increased perseveration.
Perseveration is a feature of a number of neurodegenerative illnesses, such as AD and Parkinson’s disease (PD) (Griffin et al., 2005; Joseph, 1999) and is symptom of conditions derived from ‘organic’ sources of neurological disorder, such as ASD and OCD. Our data highlight the increasingly important role of mTOR signaling in the expression of several neurological diseases. The dysregulation of mTOR signaling has been directly implicated in the in the expression of a cellular marker of neuronal loss in a mouse model of PD (Malagelada et al., 2006). Studies of mice with a restricted disruption of phosphatase and tensin homolog (PTEN), a tumor suppressor gene involved in upstream regulation of mTOR signaling, displayed behaviors consistent with ASD (Kwon et al., 2006). Protein synthesis is upregulated in mouse and Drosophila models of fragile X syndrome (FXS) (Bolduc et al., 2008; Dolen et al., 2007; Hou et al., 2006). These findings suggest that mTOR signaling may also be enhanced in FXS, a mental retardation syndrome whose symptoms include severe perseverative behavior and is the best characterized genetic cause of autism (DiCicco-Bloom et al., 2006). Our findings suggest that dysfunction in the regulation of mTORC1 by FKBP12 results in increased repetitive/perseverative behaviors such as those observed in ASD. To further explore the role for FKBP12 in ASD-like traits, it will be important to evaluate whether the FKBP12-deficient mice have altered social behavior (e.g. social interactions, sociability, and recognition) and/or altered communication (e.g. ultrasonic vocalization), two additional hallmarks of ASD.
Defining the role of FKBP12 in the brain represents an opportunity to improve our overall understanding of mTOR signaling and its relationship to long-term plasticity, behavioral perseveration, and memory. The mTOR signaling cascade represents a critical intersection for the convergence of multiple upstream neuronal signaling pathways and an equally divergent number of downstream effectors that are involved in learning and memory. Given that FKBP12 is a modulator, but not a required component, of mTOR signaling, it may be an ideal target for therapeutic drug development aimed at ameliorating some of the mTOR-related pathologies of neurological disease. Perseverative behaviors associated with ASD, OCD, and other related neurological disorders are widely believed to be developmentally established, fixed in utero by genetic, hormonal, and environmental factors (Moldin et al., 2006). Because our studies indicate that postnatal dysregulation of mTOR signaling can result in perseverative/repetitive phenotypes, they challenge the idea that some aspects of these conditions are developmentally predetermined (Zoghbi, 2003).
Experimental Procedures
FKBP12 cKO mice
Mice with floxed FKBP12 (Fkbp12fl) alleles were generated as described previously (Tang et al., 2004). Mice expressing the cre recombinase Tg(CamkIIα-cre) (T-29-1), referred to as CamKIIα-Cre were kindly provided by Dr. S. Tonegawa. Mice were genotyped using Cre-specific primers and primers that identify floxed alleles of the FKBP12 locus. The wild-type mice used in this study were CamKII-Cre+/Fkbp12+/+ and cKO mice used in this study were CamKII-Cre+/Fkbp12fl/fl.
Immunoblotting
Soluble protein extracts were prepared by homogenizing the tissue samples in ice-cold buffer. Synaptoneurosome preparations were completed as described previously (Villasana et al., 2006). Proteins were resolved on SDS-polyacrylamide gels using standard techniques. See Supplemental Methods for details of buffer and antibody preparations.
Immunoprecipitation
Transverse hippocampal slices (400 μm) were prepared using conventional techniques. Slices were maintained at 30°C in an interface chamber perfused with oxygenated artificial cerebrospinal fluid (ACSF). See Supplemental Methods for more detail. Efficiency of the immunoprecipitation was determined by examining the supernatant and wash fractions obtained from the procedure (Supplemental Figure 1c).
Electrophysiology
Field EPSPs were recorded from slices prepared in fashion identical to those used for immunoprecipitation. Slices were incubated in an interface chamber as described previously (Banko et al., 2005) and in the Supplemental Methods. When indicated, ASCF was incubated with 20 nM rapamycin (Cell signaling Technology, Beverly, MA) or 40 μM anisomycin (Sigma-Aldrich, St. Louis, MO).
Associative Conditioned Fear
The training sessions for contextual and cued fear conditioning consisted of a 150 sec exploration period followed by two CS-US pairings separated by one min (foot-shock intensity 0.9 mA, two s duration; tone 85 db white noise, 30 s duration). Contextual memory tests were performed in the training chamber after one and 24 hrs. Cued memory tests were performed in an environmentally altered testing chamber either two or 24 hrs following training. Baseline freezing was monitored (three min) prior to presentation of the tone (85 db white noise, three min duration).
Marble Burying
Mice were placed individually in Plexiglas cages containing fresh five cm deep bedding, along with 20 small black marbles arranged in five evenly spaced rows of four marbles. Testing was conducted for 30-min under normal room lighting and white noise conditions. After the test period, mice were removed and the unburied marbles were counted. Marbles were considered buried if they were at least one-half covered with bedding.
Novel Object Recognition
Each genotype was pretested for preference to the objects used during the familiar and novel phases of the experiment. The amount of time spent exploring the novel object was divided by the amount of time exploring both the novel and familiar objects using a video tracking system. The resulting value was divided by total time to generate a preference index (PI). See Supplemental Methods for more detail.
Morris Water Maze and Repeated Acquisition Paradigm
MWM experiments were performed as previously described (Banko et al., 2005). The RAP protocol was adapted from previous studies (Gimenez-Llort et al., 2005). The trajectories of the mice were recorded with a video tracking system (HVS Image Analyzing VP-200). See Supplemental Methods for more detail.
Arm Reversal in Y-Maze
Mice were trained to locate a submerged escape platform in one arm of a Y-shaped maze for 20 trials. Once mice that achieved a 90% success criterion, the escape arm were reversed and the mice were tested to determine the latency to find the new escape location. See Supplemental Methods for more detail.
Supplementary Material
Acknowledgments
We thank Drs. Jonathan Levenson, Faridis Serrano, Lingfei Hou, Caterina Hernandez, Corrine Spencer, Paula Aracena, and J. Lebowski providing reagents and expert advice. We also thank the Baylor College of Medicine MRDDRC Neurobehavioral Core (HD24064) for expert technical assistance. This work was supported by NIH grants NS048037 and NS034007 (E.K.), T32 grant HL07676 (C.A.H.) and AR41802 (SLH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets. 2003;7:101–114. doi: 10.1517/14728222.7.1.101. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila Fragile X mutants impairs long-term memory. Nature neuroscience. 2008 doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwknecht JA, Paylor R. Pitfalls in the interpretation of genetic and pharmacological effects on anxiety-like behaviour in rodents. Behav Pharmacol. 2008;19:385–402. doi: 10.1097/FBP.0b013e32830c3658. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Cafferkey R, Young PR, McLaughlin MM, Bergsma DJ, Koltin Y, Sathe GM, Faucette L, Eng WK, Johnson RK, Livi GP. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol Cell Biol. 1993;13:6012–6023. doi: 10.1128/mcb.13.10.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, Schmitz C, Schultz RT, Crawley J, Young LJ. The developmental neurobiology of autism spectrum disorder. J Neurosci. 2006;26:6897–6906. doi: 10.1523/JNEUROSCI.1712-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Shock-induced analgesia on the formalin test: effects of shock severity, naloxone, hypophysectomy, and associative variables. Behav Neurosci. 1984;98:79–95. doi: 10.1037//0735-7044.98.1.79. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G, Tanaka K, Itohara S, Mikoshiba K. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron. 1999;24:701–713. doi: 10.1016/s0896-6273(00)81123-x. [DOI] [PubMed] [Google Scholar]
- Gimenez-Llort L, Masino SA, Diao L, Fernandez-Teruel A, Tobena A, Halldner L, Fredholm BB. Mice lacking the adenosine A1 receptor have normal spatial learning and plasticity in the CA1 region of the hippocampus, but they habituate more slowly. Synapse (New York, N.Y) 2005;57:8–16. doi: 10.1002/syn.20146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O’Connor R, O’Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. Journal of neurochemistry. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- Harrar Y, Bellini C, Faure JD. FKBPs: at the crossroads of folding and transduction. Trends Plant Sci. 2001;6:426–431. doi: 10.1016/s1360-1385(01)02044-1. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Dey A, Sachan N, Wong H, Patterson RJ, Shelton JM, Richardson JA, Klann E, Rothermel BA. The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J Neurosci. 2007;27:13161–13172. doi: 10.1523/JNEUROSCI.3974-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- Joseph R. Frontal lobe psychopathology: mania, depression, confabulation, catatonia, perseveration, obsessive compulsions, and schizophrenia. Psychiatry. 1999;62:138–172. doi: 10.1080/00332747.1999.11024862. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004a;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004b;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Abel T. Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci. 2001;21:5773–5780. doi: 10.1523/JNEUROSCI.21-15-05773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26:9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Moldin SO, Rubenstein JL, Hyman SE. Can autism speak to neuroscience? J Neurosci. 2006;26:6893–6896. doi: 10.1523/JNEUROSCI.1944-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. Embo J. 2004;23:1761–1769. doi: 10.1038/sj.emboj.7600193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds THt, Bodine SC, Lawrence JC., Jr Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem. 2002;277:17657–17662. doi: 10.1074/jbc.M201142200. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC., Jr Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1998;95:7772–7777. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. Journal of neurochemistry. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Ingalls CP, Durham WJ, Snider J, Reid MB, Wu G, Matzuk MM, Hamilton SL. Altered excitation-contraction coupling with skeletal muscle specific FKBP12 deficiency. Faseb J. 2004;18:1597–1599. doi: 10.1096/fj.04-1587fje. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Van Acker K, Bultynck G, Rossi D, Sorrentino V, Boens N, Missiaen L, De Smedt H, Parys JB, Callewaert G. The 12 kDa FK506-binding protein, FKBP12, modulates the Ca(2+)-flux properties of the type-3 ryanodine receptor. J Cell Sci. 2004;117:1129–1137. doi: 10.1242/jcs.00948. [DOI] [PubMed] [Google Scholar]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nature cell biology. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Villasana LE, Klann E, Tejada-Simon MV. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods. 2006;158:30–36. doi: 10.1016/j.jneumeth.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Wittenberg GM, Tsien JZ. An emerging molecular and cellular framework for memory processing by the hippocampus. Trends Neurosci. 2002;25:501–505. doi: 10.1016/s0166-2236(02)02231-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dong Z, Nomura M, Zhong S, Chen N, Bode AM, Dong Z. Signal transduction pathways involved in phosphorylation and activation of p70S6K following exposure to UVA irradiation. The Journal of biological chemistry. 2001;276:20913–20923. doi: 10.1074/jbc.M009047200. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.