

Abstract
NOD/ShiLtJ (previously NOD/LtJ) inbred mice show polygenic autoimmune disease and are commonly used to model autoimmune-related Type I diabetes, as well as Sjogren’s syndrome. They also show rapidly progressing hearing loss, partly due to the combined effects of Cdh23ahl and Ahl2. Congenic NOD.NON-H2nb1/LtJ mice, which carry corrective alleles within the H2 histocompatibility gene complex, are free from diabetes and other overt signs of autoimmune disease, but still exhibit rapidly progressive hearing loss. Here we show that cochlear pathology in these congenics broadly includes hair cell and neuronal loss, plus endocochlear potential (EP) decline from initially normal values after 2 months of age. The EP reduction follows often dramatic degeneration of capillaries in stria vascularis, with resulting strial degeneration. The cochlear modiolus in the congenic mice also features perivascular inclusions that resemble those in some mouse autoimmune models. We posit that cochlear hair cell/neural and strial pathology in NOD.NON-H2nb1 mice arise independently. While sensory cell loss may be closely tied to Cdh23ahl and Ahl2, the strial microvascular pathology and modiolar anomalies we observe may arise from alleles on the NOD background related to immune function. Age-associated EP decline in NOD.NON-H2nb1 mice may model forms of strial age-related hearing loss caused principally by microvascular disease. The remarkable strial capillary loss in these mice may also be useful for studying the relation between strial vascular insufficiency and strial function.
Keywords: Cochlea, presbycusis, stria vascularis, marginal cells, basal cells, intermediate cells, autoimmunity, capillary
Introduction
Among the multiple forms of age-related hearing loss (ARHL), the form associated with degeneration of cochlear stria vascularis (strial ARHL) has been suggested to demonstrate the clearest genetic influences in humans (Schuknecht et al., 1974; Gates et al., 1999). Given the high degree of genetic standardization of laboratory mice, mouse models should be useful for identifying candidate strial ARHL-promoting genes. However, few mouse strains have been shown to possess the essential feature of this condition, namely delayed decline in the endocochlear potential (EP). Through a detailed comparison of BALB/cJ (BALB) and C57BL/6J (B6) mice, we showed that BALBs exhibit a lifelong EP pattern that is predicted by the density of strial marginal cells (Ohlemiller et al., 2006), while the overall appearance of the stria remains largely normal. Since each strial cell type expresses a unique complement of K+ channels and pumps (Wangemann, 2002; Hibino and Kurachi, 2006), altering the cellular makeup of the stria—even without extensive degeneration—may critically alter the balance of K+-regulating machinery. It is thus interesting that a delayed decrease in EP has been reported in knockout mice that may yield an imbalance of K+ pumps also existing in BALB mice (Diaz et al., 2007). BALB mice, as well as Mongolian gerbils (Schulte and Schmiedt, 1992; Spicer and Schulte, 2005), may model a marginal cell-initiated form of strial ARHL suggested to predominate in humans (Schuknecht et al., 1974; Schuknecht, 1993). Nevertheless, other origins of strial ARHL are likely. Another commonly proposed etiology links strial dysfunction and loss to strial microvascular pathology (Hawkins et al., 1972; Johnsson and Hawkins, 1972; Gratton et al., 1996). Strial vascular insufficiency could easily impair the energetically demanding process of K+ regulation, and might arise as a complication of systemic hypertension (Tachibana 1984; Farkas et al., 2000), diabetes mellitus (McQueen et al., 1999; Frisina et al., 2006; Geesaman, 2006), hyperlipoproteinemia (Spencer, 1973; Pillsbury, 1986; Saito et al., 1986), hyperlipidemia (Sikora et al., 1986; Suzuki et al., 2000), or autoimmune disease (Pallis et al., 1994; Mouadeb and Ruckenstein, 2005). In a cross-strain survey of aging mice, we noted EP decline from initially normal values in NOD.NON-H2nb1/LtJ mice, beginning after 2 mos of age. These mice are congenic to NOD/ShiLtJ (formerly NOD/LtJ [NOD]), which show polygenic autoimmune disease, and are commonly used to model Type I diabetes (Rothe et al., 2001; Ikegami et al., 2003) and Sjogren’s syndrome (van Blokland and Versnel, 2002; Ding et al., 2006). Overt autoimmune pathology in NOD appears to require H2g7 histocompatibility alleles, which have been replaced in the congenics by corrective alleles derived from NON/LtJ mice. The congenics retain some diabetogenic or pro-autoimmune alleles (see Discussion), but are not diabetic, and do not show outward autoimmune disease. The NOD.NON-H2nb1/LtJ congenics were previously shown to have early hearing loss, and found to carry at least two alleles that promote progressive hearing loss: Cdh23ahl, and Ahl2 (Johnson and Zheng, 2002). Because of the potential relation between immune dysfunction, microvascular disease and strial pathology, we examined the cellular correlates of progressive hearing loss and EP decline in the NOD congenic line. Here we show that EP reduction in these mice is associated with strial loss subsequent to often dramatic microvascular degeneration. Although the microvascular pathology may reflect residual autoimmune processes on the NOD background, similarity between the strial pathology of the NOD congenics and other autoimmune models is limited. Other factors, including abnormal lipid accumulation, may play a role. While it is not clear that strial degeneration and EP decline in NOD.NON-H2nb1/LtJ mice make them a mechanistic model of strial ARHL, marked EP reduction in these mice occurs only in some animals, and thus appears more ‘aging-like’ and less deterministic than has been claimed for mouse autoimmune models (Ruckenstein et al., 1999b). Therefore these mice may usefully model age-related strial pathology whose origin lies in microvascular disease.
Methods
Animals
Mice were obtained from NOD.NON-H2nb1/LtJ (NOD) breeders purchased from The Jackson Laboratory (Stock #001626). Subjects included 22 mice of mixed gender ranging from 1.5–26.0 mos in age.
CAP recording
For compound action potential (CAP) recording, animals were anesthetized (60 mg/kg sodium pentobarbital, IP) and positioned ventrally in a custom headholder. Core temperature was maintained at 37.5 ± 1.0 °C using a thermostatically-controlled heating pad in conjunction with a rectal probe (Yellow Springs Instruments Model 73A). An incision was made along the midline of the neck and soft tissues were blunt dissected and displaced laterally to expose the trachea and animal's left bulla. A tracheostomy was then made and the musculature over the bulla was cut posteriorly to expose the bone overlying the round window. Using a hand drill, a small hole was made over the round window. The recording electrode was a silver wire insulated with epoxy except for the tip, inserted into round window antrum by micromanipulator. Additional electrodes inserted into the neck musculature and hind leg served as reference and ground, respectively. Electrodes were led to a Grass P15 differential amplifier (100–3,000 Hz, ×100), then to a custom amplifier providing another x1,000 gain, then digitized at 30 kHz using a Cambridge Electronic Design Micro1401 in conjunction with SIGNAL™ and custom signal averaging software operating on a 120 MHz Pentium PC. Sinewave stimuli generated by a Hewlett Packard 3325A oscillator were shaped by a custom electronic switch to 5 ms total duration, including 1 ms rise/fall times. The stimulus was amplified by a Crown D150A power amplifier and output to a KSN1020A piezo ceramic speaker located 7 cm directly lateral to the left ear. Stimuli were presented freefield and calibrated using a B&K 4135 ¼ inch microphone placed where the external auditory meatus would normally be. Toneburst stimuli at each frequency and level were presented 100 times at 3/sec. The minimum sound pressure level required for visual detection of a response (N1) was determined at 5, 10, 20, 28.3, and 40 kHz, using a 5 dB minimum step size.
Endocochlear potential recording
The EP was measured immediately after CAP recording. Using a fine drill, a hole was made in the left cochlear capsule directly over scala media of the lower basal turn. Glass capillary pipettes (40–80 MΩ) filled with 0.15 M KCl were mounted on a hydraulic microdrive (Frederick Haer) and advanced until a stable positive potential was observed that did not change with increased electrode depth. The signal from the recording electrode was led to an AM Systems Model 1600 intracellular amplifier.
Histological processing
At the end of recording, animals were overdosed and perfused transcardially with cold 2.0% paraformaldehyde/2.0% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). Each cochlea was rapidly isolated, immersed in the same fixative, and the stapes was immediately removed. Complete infiltration of the cochlea by fixative was ensured by making a small hole at the apex of the cochlear capsule, and gently circulating the fixative over the cochlea using a transfer pipet. After decalcification in sodium EDTA for 72 hours, cochleae were post-fixed in buffered 1% osmium tetroxide, dehydrated in an ascending acetone series, and embedded in Epon. Cochleae were sectioned in the mid-modiolar plane at 4.0 µm, then stained with toluidine blue for bright field viewing with a Nikon Optiphot™ light microscope using a 100x oil objective and a calibrated grid ocular. Typically 50 sections were collected and examined from each cochlea, covering a linear sequence over 200 µm centered on the modiolar core.
Morphometry
Estimates of hair cell density, afferent neuronal density, and strial thickness were obtained from mid-modiolar sections from left cochleae. Separate measurements were made for lower basal turn, upper basal turn, and lower apical turn. For each animal, every fourth section was analyzed through a distance of 80 µm, for a total 5 sections per animal, and each metric was averaged across sections to yield a single estimate for each animal at a given cochlear location.
Hair cell loss was estimated semi-quantitatively by simple count of partial or complete hair cell profiles in the organ of Corti at each cochlear location. Both inner and outer hair cells (IHCs, OHCs) were counted together, to yield a single number at each location. We observed that OHCs nearly always disappeared first at each location, and that more than one IHC profile rarely appeared in the same section. Thus a value near ‘1.0’ for a given animal and location (see Fig. 3) indicates that only inner hair cells remained in most sections. Values recorded in the youngest animals (Fig. 3) generally ranged from 4–6, as might be expected from a canonical radial section of the organ of Corti (1 IHC + 3 OHCs), and given that OHCs were generally captured in tangential section. Spiral ganglion cell density was estimated by counting nucleated neuronal profiles within a 3,600 µm2 grid roughly centered on Rosenthal’s canal at each location.
Figure 3.

A–C. Average total hair cell profiles versus age in three cochlear regions, as viewed in mid-modiolar section. All complete and incomplete hair cell profiles were included in counts. Values of 4–6 in young mice represent normal values, as expected if the organ of Corti is viewed in near-radial plane. Gray area (Y≤1) denotes survival of primarily inner hair cells, based on the typical observation that outer hair cells were lost first at any location (See Methods).
Strial thickness was measured orthogonal to the strial midpoint at each location. Images taken for illustration were captured using a Diagnostic Instruments Model 1.4.0 digital camera controlled by Openlab™ software, then further processed using Canvas™.
Results
From the earliest age tested (1.5 mos) the NOD congenics showed elevated hearing thresholds above 5 kHz (Fig. 1). By 6–9 mos of age, few mice produced detectable CAP responses above 5 kHz. Most animals still retained some response at 5 kHz by 26 mos (Fig. 1, inset).
Figure 1.
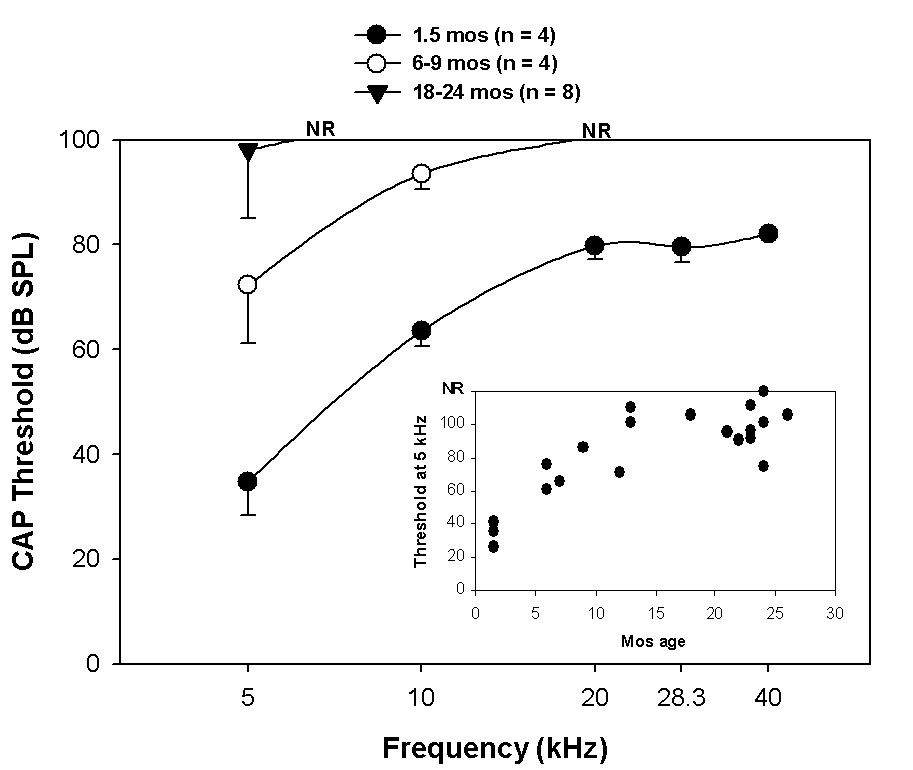
CAP thresholds versus age in NOD.NON-H2nb1/LtJ for three age brackets. Inset shows progression of threshold increase with age at 5 kHz. ‘NR’ indicates that the majority of animals in a set showed no response at all higher frequencies.
Early and broad pathology of sensory cells and stria vascularis provided multiple bases for threshold elevation. Figure 2 illustrates the types and extent of degeneration that typified older mice in our sample. Older mice (here a 23 month-old female) reliably showed extensive spiral ganglion cell loss (Fig. 2A), hair cell loss (Fig. 2C, arrows), and strial degeneration (Fig. 2E). This animal had little stria remaining in the lower apex, while in the lower base only a monolayer of cells remained (arrows in Fig. 2E). Within the upper basal turn, an organized strial epithelium remained (Fig. 2B), but some capillaries were missing (white arrow in Fig. 2B) and some intermediate cells were darkly stained and shrunken (black arrows). The EP was accordingly low in this animal (45 mV). Adjacent to the stria in the upper base, fibrocytes of the spiral ligament appeared normal. However, in the lower base the cells of the ligament showed unusually small and round nuclei (Fig. 2E). The only consistent pathology of the ligament across the sample was loss of Type IV fibrocytes adjacent to the organ of Corti in each turn (Fig. 2A, white arrowheads).
Figure 2.

A. Low power mid-modiolar view of cochlea from a 23 mo female mouse. Unfilled arrows mark location of missing Type IV fibrocytes. B. Expanded view of inset ‘B’ in panel A showing lateral wall in the upper base. Unfilled arrow shows track of degenerated capillary. Black arrowheads show shrunken intermediate cells. C. Expanded view of inset ‘C’ in panel A showing organ of Corti in the upper base. Arrows mark location of missing hair cells. D. Expanded view of inset ‘D’ in panel A showing dense inclusion within perivascular cells of the modiolus. E. Expanded view of inset ‘E’ in panel A. Arrows mark absence of stria vascularis in the lower base. UA: Upper apex; LA: Lower apex; UB: Upper base; LB: Lower base; SpG: Spiral ganglion; SMV: Spiral modiolar vein; SMA: Spiral modiolar artery; TI: Type I fibrocytes; StV: Stria vascularis; B: Strial basal cells; M: Strial marginal cells.
The animal in Figure 2 also possesses another feature that was common among the older NOD congenic mice, but rare among inbred strains that have been well characterized. Amid the perivascular cells adjacent to the spiral modiolar artery (‘SMA’ in Fig. 2A) there often appeared structures that stained heavily with toluidine, and sometimes were sufficiently dense to cause the sections to flake or wrinkle. Perivascular anomalies in the modiolus took several forms, and are further described in later sections.
Hair cell loss in the congenics was early and dramatic throughout the cochlea, but appeared to follow a basal-to-apical progression pattern. As shown in Figure 3, few hair cells remained in the lower basal turn after 6 mos (Fig. 3A). More apically, inner hair cells (denoted by the shaded areas in Fig. 3) were retained in some animals to about 1 yr of age (Fig. 3B,C). Neuronal loss generally followed the temporal and spatial pattern of hair cell loss, and appeared to be a secondary event. Most animals retained some neurons by 26 mos (Fig. 4). The earliest counts of both hair cells and neurons corresponded to essentially normal numbers when compared with our archival material. Thus, in terms of cell numbers, the cochlea develops normally into early adulthood, and hearing loss precedes overt sensory cell loss.
Figure 4.

A–C. Average number of nucleated spiral ganglion cell profiles per 3,600 µm2 in mid-modiolar sections versus age in three cochlear regions.
EP and strial thickness versus age
Figure 5 shows how basal turn EP in the congenics varied with age. The youngest mice uniformly showed EPs that match normal values (100–120 mV) in other strains (Ohlemiller et al., 2006). Some decline from normal values was evident by 6 mos, while values below 40 mV were recordable by 1 yr. By 26 mos the sample was marked by large variance, ranging from near-normal values to near 30 mV. Two interpretations of the overall trend are possible, each represented by a regression line in Figure 5. The trend across all ages examined suggests a progressive decline in EP (solid line). However, linear regression only to EP values measured after 6 mos yields a virtually flat relation (dashed line). If the latter characterization is accurate, then two processes may be operating: an invariant process that reduces the EP in all animals after 2 mos, and a stochastic process that promotes further EP decline in only some animals.
Figure 5.

Basal turn EP versus age in NOD.NON-H2nb1/LtJ. Solid line denotes linear correlation to all data. Dashed line denotes linear correlation for only animals older than 6 mos.
The clear correlate of age-related EP reduction was loss of strial epithelium, beginning at the basal and apical ends of the cochlea and progressing toward the upper base (Fig. 6). Linear regression supported progressive thinning of the stria at the two ends of the cochlea (Fig. 6A,C), but a nearly flat relation in the upper base (Fig. 6B). Most older animals showed strial thicknesses in the base and apex below 10 µm, roughly corresponding to loss of two thirds of strial functional volume at those locations, and potentially over most of the cochlea. Analysis of the relation between the EP and strial thickness (Fig. 7) suggested a direct causal relation between strial loss and EP reduction, with a slope significantly different from zero in only the lower base and apex.
Figure 6.

A–C. Average strial thickness in mid-modiolar sections versus age in three cochlear regions. Solid lines denote linear correlation.
Figure 7.

A–C. Basal turn EP versus average strial thickness in mid-modiolar sections in three cochlear regions. Solid lines denote linear correlation.
Microvascular pathology and strial degeneration
Survey by light microscopy suggested that strial degeneration and loss was preceded by occlusion and degeneration of strial capillaries. The sequence of capillary and strial loss was rarely associated with strial swelling or necrotic cells, but rather seemed to reflect a chronic and progressive condition. Often, an otherwise normal strial profile showed a mix of normal-appearing patent capillaries, occluded capillaries, and traces of capillaries that had partially degenerated (Fig. 8A). In such cases, the epithelium was usually composed of well-defined layers comprising basal, intermediate, and marginal cells, although some pyknotic cells could be found (Fig. 8A, 2B). Among strial cell types, basal cells were least likely to appear abnormal. In a few cases (Fig. 8B) dramatic degeneration was observed that suggested acute and severe capillary occlusion, followed by rapid cell loss. In the example shown in Figure 8B, capillaries do not seem to have degenerated, but virtually every capillary is occluded (arrows). Both the intercellular space and the luminal cells are swollen, and no intermediate or marginal cell nuclei appear, suggesting rapid necrotic loss of these cells.
Figure 8.

Examples of strial degeneration in NOD.NON-H2nb1/LtJ. A. Strial profile from upper base in a 9 mo male showing patent capillaries (c), occluded capillaries (unfilled arrow), and partially degenerated capillary profiles (black arrows). B. Strial profile from upper base of 21 mo female showing all occluded capillaries (arrows). Larger view (inset) shows that the occluding material consists of red blood cells suspended in gray (osmium-stained) material that may be principally lipid. The cell surrounding the upper capillary, possibly a pericyte, contains dark gray vesicles (arrow) that may also have high lipid content. C. Moderately degenerated strial profile in the lower base of a 22 mo male shows several patent capillaries (bottom), actively degenerating capillaries (black arrows), and complete absence of stria proceeding toward Reissner’s membrane (white arrowheads). D.–H. Sequential 4 µm sections (proceeding apically) of stria from the upper base of an 18 mo female showing merging of some capillaries (dashed arrows) over a distance of 20 µm. At the bottom, two darkly stained degenerating capillaries merge and end blindly, leaving patches of debris (arrows in G,H). In H, a large vacuole that may be a macrophage process appears continuous with some debris (arrowhead). Within the avascular regions in G and H, the only recognizable cells are basal cells (B). UB: Upper base; LB: Lower Base; TI: Type I fibrocytes; TII: Type II fibrocytes; B: Basal cells; I: Intermediate cells; M: Marginal cells.
In this exploratory study of these mice, we applied ‘standard’ methods not necessarily optimized for study of the stria. Thus ‘capillary occlusion’ as reported here means material sufficiently concentrated or adherent to capillary walls to remain in place during transcardial perfusion with aldehydes. For this reason, some capillary occlusion may have been missed. The material within occluded capillaries was typically composed of tightly balled red blood cells, or sometimes primarily platelets. These were suspended in a translucent material that typically stained lightly with toluidine, but sometimes was gray and stained only by osmium, suggesting a primarily lipid composition. Examples of the latter are shown in Figure 8A (white arrow) and in Figure 8B (inset). Notably, the inset of Figure 8B shows one capillary surrounded by a multivesiculated cell that is probably a pericyte (arrow). Although possibly less apparent in a non-color image, the dark gray vacuoles in this cell were also stained only by osmium, and thus primarily lipid in content. Pericytes can serve many functions, including the role of macrophage and regulation of capillary permeability (Hirschi and D'Amore, 1996; Edelman et al., 2006), so that the one shown could be phagocytosing lipid and other material either from, or bound for, the capillary. Such examples provide indirect evidence that the material contributing to capillary occlusion was substantially lipid. Since aldehyde fixatives remove free lipids, any remaining lipid deposits seem likely to represent only very high local concentration, and only a fraction of those present in the intact animal.
Circumstantial support for a microvascular origin of strial degeneration derived in part from many instances like that shown in Figure 8C, which is suggestive of both a spatial and temporal trajectory of strial degeneration subsequent to capillary degeneration. At its basal end, this strial profile shows a normal capillary distribution, and all strial cell types are present. Moving upward toward Reissner’s membrane, however, one encounters an occluded capillary, then two densely stained, actively degenerating capillaries. It was not possible to determine at the level of the light microscope whether this dense material was composed only of capillary debris, or whether it also included macrophage processes or an ‘activated’ state of surrounding pericytes (Farkas and Luiten, 2001). Moving still more in the direction of Reissner’s membrane, the stria—which should extend all the way to Reissner’s—is completely missing (white arrows).
Apparent capillary degeneration did not always resemble the example shown in Fig. 8C, and did not always clearly follow occlusion. Many instances were noted like one depicted in Figure 8D–H, which tracks the degeneration of seemingly patent capillaries proceeding apically from the upper base over a 20 µm distance. Following this sequence from left to right, two clusters of capillaries are seen to merge. Toward the bottom of the panels, two merging capillaries are densely stained and appear to be degenerating. These join to form a closed loop, with only debris extending more apically. Figure 8G shows four patches of debris that may mark the former tracks of capillaries (also marked by arrows in Fig. 8H). In Figure 8H, a large clear vacuole that may be a macrophage process merges with one patch of debris (arrowhead). The capillary at the bottom of panels D–H, while also appearing to degenerate, continues through this sequence and did not end in the sections obtained. The densely stained material appeared to be degenerating pericytes, although this could not be distinguished with certainty from macrophage or activated pericyte processes. The latter may account for the beaded appearance of the material surrounding the capillary, particularly in Figure 8F. Note that the upper region of the stria in panels Figure 8D–H appears fairly normal, and includes all characteristic cell layers and types, while the avascular region in panels G–H includes only a basal cell layer (see labels), and few other nuclei are visible.
Strial abnormalities did not include any signs of inflammatory infiltrate or invading inflammatory cells within the stria or strial capillaries. Multivesiculated cells or other unusual cell types within the stria were rare. Since we did not test for the presence of immunoglobulins adherent to capillary endothelial cells or basement membrane, we do not know whether these were present, or whether the NOD congenics may resemble other mouse autoimmune models in this regard (Trune, 1997; Ruckenstein and Hu, 1999)
In no case were clearly occluded or degenerating vessels observed in the spiral ligament or spiral limbus, and the density of vasculature in ligament and limbus seemed unremarkable. Spiral ligament fibrocyte density and appearance did not correspond to any particular state of the adjacent stria. In a few cases, Type II fibrocytes appeared vacuolated and Type I fibrocytes were shrunken. In several cases, especially in the oldest animals, these were clearly reduced in number. However, as highlighted in Figure 2E, even when present, cells that populated the ligament could not be assumed to represent specific and functional fibrocyte types. Only Type IV fibrocytes appeared reliably reduced in number in both cochlear base and apex, independent of the state of the stria.
Anomalies within the modiolus
The majority of older animals (11 of 18 mice aged 6 mos and older) showed unusual structures among the perivascular cells of the cochlear modiolus (Fig. 9). Most often, these took the form of densely toluidine-stained inclusions that appeared limited to the extracellular space. They were usually globular (Fig. 9A, arrow), but sometimes appeared composed of large granules (Fig. 2D, 9C). One animal also featured an additional, more lightly stained, fibrotic mass (Fig. 9A, dashed enclosure) that invaded about one third of the width of the modiolus. Two animals showed extensive hyperplasia of fibrocytes that extended across the modiolus, giving rise to a spherical structure over 100 µm in diameter (Fig. 9B). In the example shown, the sphere possesses a lumen filled with lightly stained acellular material. In the second animal (not shown), the lumen contained darkly-staining globular material resembling that in Figure 9A (arrow).
Figure 9.

Dense inclusions and other unusual structures observed in the modiolus. A. Densely-stained globular material (arrow) and fibrosis (dashed enclosure) among perivascular cells. B. Hyperplasia of fibroblasts featuring large sphere with lumen containing lightly-stained acellular material (unfilled arrow). C. Variation of perivascular inclusions showing granular material. SMA: Spiral modiolar artery; Bn: Bone.
Lipid accumulation
The putatively lipid-rich osmium-stained material that appeared to contribute to capillary occlusion in Figure 8B was not specific to strial capillaries, or to NOD.NON-H2nb1 mice. It was sometimes also found in other cochlear vessels of all sizes, as well as bone marrow spaces of the cochlear capsule. It was also frequently found in cochlear vessels and marrow spaces of 10 old albino BALB/cJ (19–22 mos) and 8 old C57BL/6-Tyrc-2J albino congenic mice (18–24 mos) examined for comparison. However, in the comparison strains it was not observed in strial capillaries. The NOD congenics were also unique in the extravascular appearance of this material within the cochlea. Figure 10A shows an extracellular deposit (arrow) surrounded by an unusual hyperplasia of fibrocytes on the medial side of the spiral limbus. Figure 10B shows gray unstained material surrounding a perivascular inclusion (arrow) in the modiolus. Figure 10C shows a particularly dense gray deposit immediately adjacent to the spiral modiolar artery. Perivascular cells include both fibrocytes and adipocytes, so that it could not always be determined at the level of the light microscope whether such deposits were located between fibrocytes or within fat cells.
Figure 10.

Distribution of gray (osmium-stained), putatively lipid, material (see Fig. 8C) in locations other than within capillaries. A. Gray deposit within unusual cell mass on the medial aspect of spiral limbus (arrow). B. Gray material (arrow) surrounding perivascular inclusion similar to those in Figure 9. C. Particularly dense gray deposits (arrows) that may either lie between fibroblasts or within adipocytes. SpL: Spiral limbus; SMA: Spiral modiolar artery.
Strial vascularity versus strial composition
Irrespective of how it arises, the dynamic capillary loss in NOD.NON-H2nb1 mice offers an opportunity to track changes in strial function and appearance as blood vessels withdraw, as well as how the vessels withdraw. Figure 11 shows how the stria of the upper basal turn changes in composition over a progressively apical 80 µm span in which a normal complement of capillaries disappears, leaving the stria completely avascular. Because the stria in the lower apex was completely absent, it is likely that this sequence captures the end of vascularized stria, proceeding apically. The sequence begins with a normal appearing stria, possessing all cell layers and characteristic types (Fig. 11A). Over the next 32 µm, capillaries are seen to merge and end in blind loops, leaving the epithelium progressively less well supplied. At 36 µm (Fig. 11G) the imprint of the last capillary loop can be seen (arrow). At this point, the stria is composed both of regions still retaining three cell layers (inset a) and regions showing hyperplasia of poorly specified cells (inset b). At 80 µm distance (Fig. 11H), only abnormal cells are found (inset c), yet surprisingly the area of the epithelium is only modestly reduced. Note that the tracks of degenerated blood vessels can still be seen (inset d, arrows), suggesting a rapid withdrawal of the supporting vasculature. The ligament in this region appears normal, with several patent capillaries.
Figure 11.

A.–H. Sequential 4 µm sections proceeding apically from the upper base of a 24 mo male showing progressive merging of strial capillaries, leaving the stria completely avascular over a span of 80 µm. Dashed arrows in A.–F. show how capillaries progressively merge, ultimately forming a blind loop. The first completely avascular segment (G) features well-organized regions (inset a), as well as regions with increased numbers of poorly differentiated cells (inset b). The arrow in G shows the wall of the last capillary loop. By 80 µm (H), the stria is still present but thin, and shows hyperplasia of undefined cell types over most of its length (inset c). Traces of degenerated capillaries can still be seen (inset d, arrows). The ligament at this location appears normal. TI: Type I fibrocytes; TII: Type II fibrocytes; B: Basal cells; I: Intermediate cells; M: Marginal cells.
Discussion
From the present and previous studies (Johnson and Zheng, 2002), NOD.NON-H2nb1/LtJ mice show rapidly progressing hearing loss whose origins could lie in sensory cell loss, as well as in EP reduction caused by strial degeneration. Of two alleles known to be carried by these mice, Cdh23ahl and Ahl2, the former is known to promote hair cell loss (Li and Hultcrantz, 1994; Spongr et al., 1997), while the cell pathology caused by the latter has not yet been determined. Although Ahl2, whose gene product remains unidentified, could conceivably exert its primary effects on the stria, hearing loss associated with this allele on a Cdh23ahl homozygous background is severe by 6 mos, that is, before marked EP reduction is consistently found. Ahl2 also shows epistasis with Cdh23ahl suggesting that both loci affect hair cells. Figure 12 summarizes the progression of sensory cell loss, strial degeneration, and EP decline in the lower basal turn of the NOD congenics. Here, data from each animal have been normalized to the maximum value measured, and then fitted with a single exponential decay. Note that spiral ganglion cell loss lags hair cell loss, as would be expected if this loss were secondary to hair cell loss. Both EP decline and strial degeneration follow a slower time course, supporting the interpretation that sensory cell loss dominates the observed hearing loss, and that EP decline and strial loss reflect genetic influences other than Cdh23ahl and Ahl2.
Figure 12.
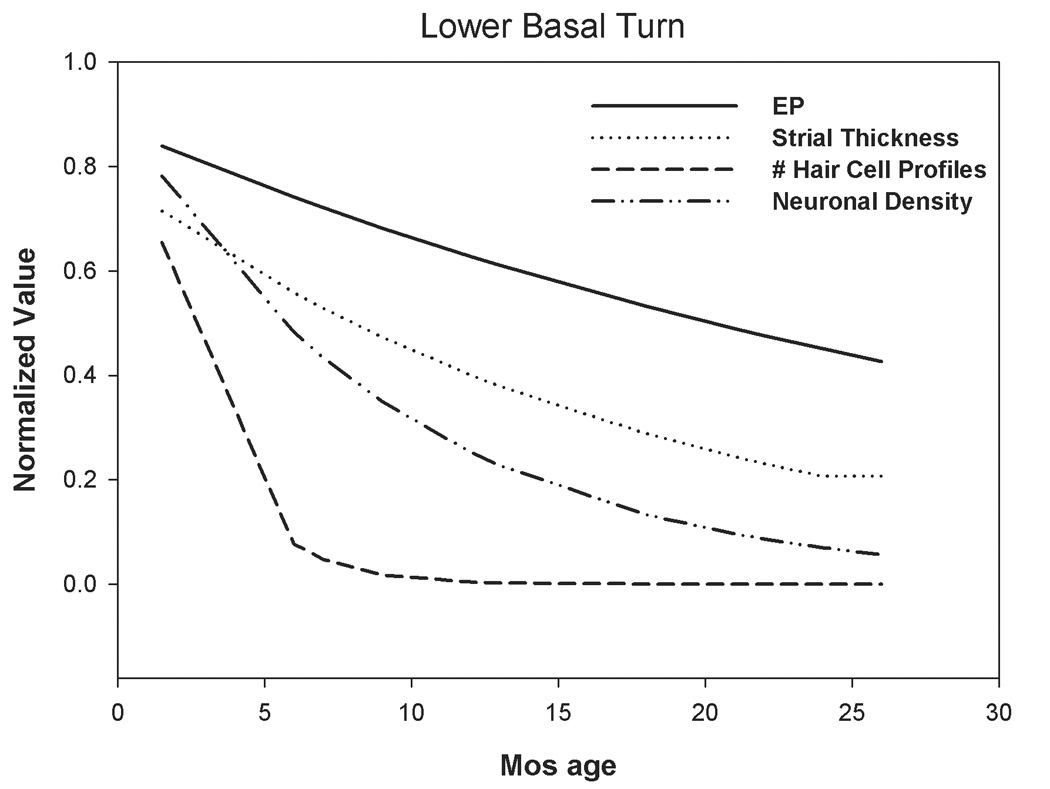
Age versus basal turn EP, strial thickness, and sensory cell loss in the lower base of NOD.NON-H2nb1/LtJ. Basal turn data from Figure 3–Figure 6 have been normalized to the maximum value recorded and fit to single exponential curves. Few hair cells remain after 6 mos. Hearing loss (see Fig. 1) closely follows hair cell loss. Spiral ganglion cell loss somewhat lags hair cell loss, and is likely to be a secondary event. Strial thinning and EP decline are similar to each other in their time course, as expected, but progress more slowly than hair cell and neural loss.
Among other candidate processes, immune dysfunction emerges as a possible contributor, given that NOD.NON-H2nb1/LtJ mice retain diabetogenic, autoimmune-promoting, alleles (see below), and that strial vascular pathology is a feature of mouse autoimmune models (Trune et al., 1991; Ruckenstein et al., 1999a; Ruckenstein et al., 1999b; Trune, 2002). Yet there are substantial differences between the pathology of autoimmune mouse stria and that described here. Other contributing processes are suggested, including impaired lipid metabolism. Our evidence for this is highly anecdotal, and based on tissue preparation methods that leach most free lipids from tissues. Using fixatives and stains better suited to lipids, even more marked lipid accumulation might be found to distinguish the NOD congenics from other strains, and perhaps within strial capillaries that appeared unobstructed (Fig. 8D–H). NOD mice also carry other alleles that could potentially influence the stria by altering the melanin-related reactions, including Tyrc (albinism) and A (agouti). We have shown, however (Ohlemiller et al., 2006), that these do not produce age-associated pathology like that in NOD. We posit that the broad pathology in NODs reflects independent genetic influences on hair cells and neurons versus stria. Using appropriate genetic crosses, it should be possible to segregate alleles in NOD.NON-H2nb1/LtJ that impart sensory cell loss versus strial degeneration, and then further to isolate probable multiple genes that promote strial pathology.
Strial degeneration and EP reduction
Few studies have directly demonstrated a relationship between the EP and any anatomic metric for strial degeneration or injury. Work in gerbils (Schulte and Schmiedt, 1992) has supported the assertion based on human morphometric data (Pauler et al., 1988) that nearly half of strial functional volume may be lost without EP reduction. In cases of severe strial degeneration, strial volume—and reasonable proxy measures for strial volume—presumably offer informative metrics for strial functional capacity. Thus it was possible to establish correlations between strial thickness in the cochlear base and apex and the EP in the NOD congenics (Fig. 7). In cases of subtle degeneration, like that in aging BALB/c mice (Ohlemiller et al., 2006), it has turned out that strial cellular composition is a better predictor of EP than strial spatial dimensions. However, models such as connexin 30 (Cx30) and Claudin-11 knockout mice, which show EP reduction but little strial degeneration (Gow et al., 2004; Cohen-Salmon et al., 2007), make it clear that there may not always be any obvious correlate of reduced EP detectable at the light microscope level. Figure 7 suggests a simple linear correlation between strial thickness and EP in the NOD congenics. If initially healthy stria possesses functional capacity in excess, this relation would be expected to be flat over some range. It seems likely that some characteristic not captured by spatial dimensions contributes to the EP reduction that precedes marked strial loss. This is consistent with a two factor interpretation of the age-related trend in EP values (Fig. 5), whereby all of the congenics undergo some EP reduction by ~6 mos of age, but more substantial declines over the next 20 mos seem to involve a probabilistic element that interacts with the genetic background.
Strial capillary occlusion and capillary loss
Our contention that strial degeneration in the NOD congenics was triggered by microvascular dysfunction, while circumstantial, was supported by a host of instances in which the only pathology noted was restricted to capillaries. Conversely, markedly abnormal strial profiles overwhelmingly featured missing, degenerating, or occluded capillaries. Less clear, however, was whether capillary degeneration was always preceded by occlusion. Often, apparently actively degenerating capillaries like those shown in Figure 8D–H did not seem occluded. This could be an artifact of processing. Alternatively, it may reflect degenerative processes unrelated to occlusion. At the level of light microscopy, capillary degeneration seemed to affect principally pericytes and adjacent basement membrane; Normal-appearing endothelial cells could often be found lining affected capillaries. Other interpretations of the densely-stained capillary profiles, such as invading macrophages or activated pericytes playing a macrophage role, cannot presently be discounted.
Comparison of NOD.NON-H2nb1/LtJ to mouse autoimmune models
NOD.NON-H2nb1/LtJ mice retain diabetogenic alleles from the NOD/ShiLtJ parent strain. Some of these likely promote immune dysfunction. Mapped loci other than H2 where NODs are believed to carry disease-promoting alleles include Idd10, Idd17, Idd18, Idd3 (for which interleukins 2 and 21 are candidates), and Idd16 (for which TNFα is a candidate) (Ikegami et al., 2003). These alleles are retained in the congenics, and thus represent candidate genes for the strial pathology described here. In mouse autoimmune models such as MRL-Faslpr and Palmerston North, the main effect on hearing is thought to be exerted via strial dysfunction (Trune et al., 1991; Ruckenstein et al., 1999a; Ruckenstein et al., 1999b; Trune, 2002). Similarities of the NOD congenics to other mouse autoimmune models are therefore possible, and immune dysfunction emerges as a likely contributor to strial pathology. The major autoimmune diseases modeled by NOD, MRL-Faslpr, and Palmerston North collectively include lupus, Sjogren’s disease, and Type I diabetes, all of which involve inflammatory infiltration and destruction of organs and connective tissue. By contrast, cochlear pathology in these mice does not involve inflammation. Instead, immunoglobulins (Ig) shown to bind to endothelial cells and capillary basement membrane (Trune, 1997; Ruckenstein and Hu, 1999) may increase capillary permeability to the point of ‘shorting out’ the EP (Lin and Trune, 1997; Trune, 2002). Since we did not test for Ig deposits, we have no information as to whether a similar process is operating in the NOD congenics. However, immunoglobulins can be found in the strial pericapillary space under other conditions, including aging (Sakaguchi et al., 1997), in Cx30 knockouts (Cohen-Salmon et al., 2007), and in mice lacking strial melanocytes (Fujimura et al., 2005), and it is not clear when their presence heralds a significant ‘shorting out’ process. MRL-Faslpr and Palmerston North mice, in which strial pathology is best studied, show strial ultrastructural pathology and reduced strial volume (Trune et al., 1991; Ruckenstein et al., 1999a; Trune and Kempton, 2001), but have not been proven to undergo capillary loss or irreversible loss of strial epithelium. In fact, the marked loss of strial volume in MRL-Faslpr can be largely reversed by application of steroids (Trune et al., 2000; Trune and Kempton, 2001), suggesting that the loss of strial volume in these mice occurs through retraction of cell processes, not cell loss. Palmerston North mice show perivascular anomalies like those we noted in the cochlear modiolus (Hertler and Trune, 1990; Khan et al., 2000). These anomalies presumably do not promote hearing loss directly, although they could compress the axons of cochlear nerve fibers. Instead they may reflect a process that, in parallel, also promotes strial dysfunction. Trune and colleagues (Trune et al., 1990) showed that similar dense inclusions in Palmerston North mice contain high levels of calcium, and further proposed that these represent fibrosis and abnormal bone remodeling characteristic of autoimmune models. Evidence for unusual lipid accumulation such as that presented here has not been reported as a prominent feature of autoimmune mice. However, the lacrimal glands of NOD/ShiLtJ mice show intracellular lipid accumulation (Ding et al., 2006), and this Sjogren’s-like manifestation has been compared to a lysosomal storage disease affecting lipid metabolism.
Relevance to strial ARHL
Although cochlear autoimmune pathology is distinct from most descriptions of age-related cochlear pathology, age-related pathology may often involve autoimmune phenomena (Brod, 2000; Hasler and Zouali, 2005), and there are reasons to suspect a role for immune dysfunction in the strial pathology of NOD.NON-H2nb1/LtJ. However, the striking irreversible loss of first strial capillaries, then stial cells, in the congenics has no counterpart in the best described mouse autoimmune models. Whereas mouse autoimmune models examined show early and reportedly invariant EP reduction (Ruckenstein et al., 1999b), NOD.NON-H2nb1/LtJ mice show delayed and highly variable EP reduction more characteristic of animal models of strial ARHL. Our observation that vascular pathology in the stria may begin with pericyte degeneration is echoed by observations in microvascular disease associated with aging, Alzheimer’s disease, and delayed-onset diabetes (Hirschi and D'Amore, 1996; Farkas and Luiten, 2001; Edelman et al., 2006). This possibility could place NOD mice into the larger context of age-related microvascular disease, and loci that may contribute to their phenotype (Ikegami et al., 2003) may merit testing as candidate loci for some strial ARHL. Even if these mice are shown not to model strial ARHL in a mechanistically direct way, they offer a presently unparalleled opportunity to study the process of strial capillary loss, and of the relation between capillary loss and progressive strial dysfunction and metaplasia.
Acknowledgements
Supported by NIH R01 DC008321 (KKO), R01 DC03454 (KKO), P30 DC04665 (R. Chole), P30 NS057105 (D. Holtzman), and Washington University Medical School Department of Otolaryngology. Thanks to Dr. Dennis Trune for comments on the manuscript.
Funding Agencies: NIH R01 DC008321 (KKO), R01 DC03454 (KKO), P30 DC04665 (R. Chole), Washington University Medical School Department of Otolaryngology
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brod SA. Unregulated inflammation shortens human functional longevity. Inflamm. Res. 2000;49:561–570. doi: 10.1007/s000110050632. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon M, Regnault B, Cayet N, Caille D, Dermuth K, Hardelin J-P, Janel N, Meda P, Petit C. Connexin30 deficiency causes intrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc. Nat. Acad. Sci. 2007;105:6229–6234. doi: 10.1073/pnas.0605108104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz RC, Vazquez AE, Dou H, Wei D, Cardell EL, Lingrel J, Shull GE, Doyle KJ, Yamoah EN. Conservation of hearing by simultaneous mutation of Na,K-ATPase and NKCCl. J. Asso. Res. Otolaryngol. 2007;8:422–434. doi: 10.1007/s10162-007-0089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C, MAcVeigh M, Pidgeon M, daCosta SR, Hamm-Alvarez SF, Schechter JE. Unique ultrastructure of exorbital lacrimal glands in male NOD and BALB/c mice. Curr. Eye Res. 2006;31:13–22. doi: 10.1080/02713680500428613. [DOI] [PubMed] [Google Scholar]
- Edelman DA, Jiang Y, Tyburski J, Wilson RF, Steffes C. Pericytes and their role in microvasculature homeostasis. J. Surg. Res. 2006;135:305–311. doi: 10.1016/j.jss.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Farkas E, Luiten PGM. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog. Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- Frisina ST, Mapes F, Kim S, Frisina DR, Frisina RD. Characterization of hearing loss in aged type II diabetics. Hear. Res. 2006;211:103–113. doi: 10.1016/j.heares.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura T, Suzuki H, Shimizu T, Tokui N, Kitamura T, Udaka T, Doi Y. Pathological alterations of strial capillaries in dominant white spotting W/WV mice. Hear. Res. 2005;209:53–59. doi: 10.1016/j.heares.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Gates GA, Couropmitree NN, Myers RH. Genetic associations in age-related hearing thresholds. Arch. Otolaryngol. Head Neck Surg. 1999;125:654–659. doi: 10.1001/archotol.125.6.654. [DOI] [PubMed] [Google Scholar]
- Geesaman BJ. Genetics of aging: Implications for drug discovery and development. Am J. Clin. Nutr. 2006;83:466S–469S. doi: 10.1093/ajcn/83.2.466S. [DOI] [PubMed] [Google Scholar]
- Gow A, Davies C, Southwood CM, Frolenkov G, Chrustowski M, Ng L, Yamauchi D, Marcus DC, Kachar B. Deafness in claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. 2004;24:7051–7062. doi: 10.1523/JNEUROSCI.1640-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton MA, Schmiedt RA, Schulte BA. Age-related decreases in endocochlear potential are associated with vascular abnormalities in the stria vascularis. Hear. Res. 1996;102:181–190. doi: 10.1016/s0378-5955(96)90017-9. [DOI] [PubMed] [Google Scholar]
- Hasler P, Zouali M. Immune receptor signaling, aging, and autoimmunity. Cell. Immunol. 2005;233:102–108. doi: 10.1016/j.cellimm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Hawkins JE, Johnsson L-G, Preston RE. Cochlear microvasculature in normal and damaged ears. Laryngoscope. 1972;82:1091–1104. doi: 10.1288/00005537-197207000-00001. [DOI] [PubMed] [Google Scholar]
- Hertler CK, Trune DR. Otic capsule bony lesions in the Palmerston North autoimmune mouse. Otolaryngol. Head Neck Surg. 1990;103:713–718. doi: 10.1177/019459989010300509. [DOI] [PubMed] [Google Scholar]
- Hibino H, Kurachi Y. Molecular and physiological bases of the K+ circulation in the mammalian inner ear. Physiology. 2006;21:336–344. doi: 10.1152/physiol.00023.2006. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, D'Amore PA. Pericytes in the microvasculature. Cardiovascular Research. 1996;32:687–698. [PubMed] [Google Scholar]
- Ikegami H, Fujisawa T, Makino S, Ogihara T. Congenic mapping and candidate sequencing of susceptibility genes for type 1 diabetes in the NOD mouse. Ann. N.Y. Acad. Sci. 2003;1005:196–204. doi: 10.1196/annals.1288.026. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY. Ahl2, a second locus affecting age-related hearing loss in mice. Genomics. 2002;80:461–464. [PMC free article] [PubMed] [Google Scholar]
- Johnsson L-G, Hawkins JE. Vascular changes in the human ear associated with aging. Ann. Otol. 1972;81:364–376. doi: 10.1177/000348947208100307. [DOI] [PubMed] [Google Scholar]
- Khan DC, DeGagne JM, Trune DR. Abnormal cochlear connective tissue mineralization in the Palmerston North autoimmune mouse. Hear. Res. 2000;142:12–22. doi: 10.1016/s0378-5955(99)00221-x. [DOI] [PubMed] [Google Scholar]
- Li H-S, Hultcrantz M. Age-related degeneration of the organ of corti in two genotypes of mice. Oto Rhin. Laryngol. 1994;56:61–67. doi: 10.1159/000276611. [DOI] [PubMed] [Google Scholar]
- Lin DW, Trune DR. Breakdown of stria vascularis blood-labyrinth barrier in C3H/lpr autoimmune disease mice. Otolaryngol. Head Neck Surg. 1997;117:530–534. doi: 10.1016/S0194-59989770026-3. [DOI] [PubMed] [Google Scholar]
- McQueen CT, Baxter A, Smith TL, Raynor EM, Yoon SM, Prazma J, Pillsbury HC. Non-insulin-dependent diabetic microangiopathy in the inner ear. J. Laryngol. Otol. 1999;113:13–18. doi: 10.1017/s0022215100143051. [DOI] [PubMed] [Google Scholar]
- Mouadeb DA, Ruckenstein MJ. Antiphospholipid inner ear syndrome. Laryngoscope. 2005;115:879–883. doi: 10.1097/01.MLG.0000158666.15447.37. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Lett JM, Gagnon PM. Cellular correlates of age-related endocochlear potential reduction in a mouse model. Hear. Res. 2006;220:10–26. doi: 10.1016/j.heares.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Pallis M, Hopkinson N, Lowe JS, Powell R. An electron microscopic study of muscle capillary wall thickening in systemic lupus erythematosus. Lupus. 1994;3:401–407. doi: 10.1177/096120339400300507. [DOI] [PubMed] [Google Scholar]
- Pauler M, Schuknecht HF, White JA. Atrophy of the stria vascularis as a cause of sensorineural hearing loss. Laryngoscope. 1988;98:754–759. doi: 10.1288/00005537-198807000-00014. [DOI] [PubMed] [Google Scholar]
- Pillsbury HC. Hypertension, hyperlipoproteinemia, chronic noise exposure: Is there synergism in cochlear pathology? Laryngoscope. 1986;96:1112–1138. doi: 10.1288/00005537-198610000-00012. [DOI] [PubMed] [Google Scholar]
- Rothe H, Ito Y, Kolb H. Disease resistant, NOD-related strains reveal checkpoints of immunoregulation in the pancreas. J. Mol. Med. 2001;79:190–197. doi: 10.1007/s001090000182. [DOI] [PubMed] [Google Scholar]
- Ruckenstein MJ, Hu L. Antibody deposition in the stria vascularis of the MRL-Faslpr mouse. Hear. Res. 1999;127:137–142. doi: 10.1016/s0378-5955(98)00189-0. [DOI] [PubMed] [Google Scholar]
- Ruckenstein MJ, Keithley EM, Bennett T, Powell HC, Baird S, Harris JP. Ultrastructural pathology in the stria vascularis of the MRL-Faslpr mouse. Hear. Res. 1999a;131:22–28. doi: 10.1016/s0378-5955(99)00018-0. [DOI] [PubMed] [Google Scholar]
- Ruckenstein MJ, Milburn M, Hu L. Strial dysfunction in the MRL-Faslpr mouse. Otolaryngol. Head Neck Surg. 1999b;121:452–456. doi: 10.1016/S0194-5998(99)70236-6. [DOI] [PubMed] [Google Scholar]
- Saito T, Sato K, Saito H. An expermental study of auditory dysfunction associated with hyperlipoproteinemia. Arch. Otorhinolaryngol. 1986;243:242–245. doi: 10.1007/BF00464438. [DOI] [PubMed] [Google Scholar]
- Sakaguchi N, Spicer SS, Thomopoulos GN, Schulte BA. Immunoglobulin deposition in thickened basement membranes of aging strial capillaries. Hear. Res. 1997;109:83–91. doi: 10.1016/s0378-5955(97)00048-8. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF. Pathology of the Ear. Philadelphia: Lea and Febiger; 1993. [Google Scholar]
- Schuknecht HF, Watanuki K, Takahashi T, Belal AA, Kimura RS, Jones DD. Atrophy of the stria vascularis, a common cause for hearing loss. Laryngoscope. 1974;84:1777–1821. doi: 10.1288/00005537-197410000-00012. [DOI] [PubMed] [Google Scholar]
- Schulte BA, Schmiedt RA. Lateral wall Na,K-ATPase and endodochlear potentials decline with age in quiet-reared gerbils. Hear. Res. 1992;61:35–46. doi: 10.1016/0378-5955(92)90034-k. [DOI] [PubMed] [Google Scholar]
- Sikora MA, Morizono T, Ward WD, Paparella MM, Leslie K. Diet-induced hyperlipidemia and auditory dusfunction. Acta Otolaryngol. 1986;102:372–381. doi: 10.3109/00016488609119420. [DOI] [PubMed] [Google Scholar]
- Spencer JT. Hyperlipoproteinemia in the etiology of inner ear disease. Laryngoscope. 1973;83:639–378. doi: 10.1288/00005537-197305000-00002. [DOI] [PubMed] [Google Scholar]
- Spicer SS, Schulte BA. Pathologic changes of presbycusis begin in secondary processes and spread to primary processes of strial marginal cells. Hear. Res. 2005;205:225–240. doi: 10.1016/j.heares.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Spongr VP, Flood DG, Frisina RD, Salvi RJ. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life span. J. Acoust. Soc. Am. 1997;101:3546–3553. doi: 10.1121/1.418315. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kaneko M, Murai K. Influence of serum lipids on auditory function. Laryngoscope. 2000;110:1736–1738. doi: 10.1097/00005537-200010000-00033. [DOI] [PubMed] [Google Scholar]
- Trune DR. Cochlear immunoglobulin in the C3H/lpr mouse model for autoimmune hearing loss. Otolaryngology-Head and Neck Surgery. 1997;117:504–508. doi: 10.1016/s0194-5998(97)70022-6. [DOI] [PubMed] [Google Scholar]
- Trune DR. Mouse models for immunologic diseases of the auditory system. In: Willott JF, editor. Handbook of mouse auditory research: From behavior to molecular biology. New York: CRC Press; 2002. pp. 505–531. [Google Scholar]
- Trune DR, Hertler CK, Haun DKZ, Sauter RW. Histochemistry of otic capsule sclerotic lesions in Palmerston North autoimmune strain mice. Hear. Res. 1990;48:241–246. doi: 10.1016/0378-5955(90)90064-v. [DOI] [PubMed] [Google Scholar]
- Trune DR, Kempton JB. Aldosterone and prednisolone control of cochlear function in MRL/MPJ -Faslpr autoimmune mice. Hear. Res. 2001;155:9–20. doi: 10.1016/s0378-5955(01)00240-4. [DOI] [PubMed] [Google Scholar]
- Trune DR, Kempton JB, Kessi M. Aldosterone (mineralocorticoid) equivalent to prednisone (glucocorticoid) in reversing hearing loss in MRL/MpJ-Faslpr autoimmune mice. Laryngoscope. 2000;110:1902–1906. doi: 10.1097/00005537-200011000-00025. [DOI] [PubMed] [Google Scholar]
- Trune DR, Morton JI, Craven JP, Traynor SJ, Mitchell CR. Inner ear pathology in the Palmerston North autoimmune strain mouse. Am. J. Otolaryngol. 1991;12:259–266. doi: 10.1016/0196-0709(91)90003-x. [DOI] [PubMed] [Google Scholar]
- van Blokland SCA, Versnel MA. Pathogenesis of Sjogren's Syndrome: Characteristics of different mouse models for autoimmune exocrinopathy. Clin. Immunol. 2002;103:111–124. doi: 10.1006/clim.2002.5189. [DOI] [PubMed] [Google Scholar]
- Wangemann P. K+ recycling and the endocochlear potential. Hear. Res. 2002;165:1–9. doi: 10.1016/s0378-5955(02)00279-4. [DOI] [PubMed] [Google Scholar]