

Abstract
The nuclear hormone receptor, peroxisome proliferator-activated receptor (PPAR)-γ, originally identified as a key mediator of adipogenesis, is expressed widely and implicated in diverse biological responses. Both natural and synthetic agonists of PPAR-γ abrogated the stimulation of collagen synthesis and myofibroblast differentiation induced by transforming growth factor (TGF)-β in vitro. To characterize the role of PPAR-γ in the fibrotic process in vivo, the synthetic agonist rosiglitazone was used in a mouse model of scleroderma. Rosiglitazone attenuated bleomycin-induced skin inflammation and dermal fibrosis as well as subcutaneous lipoatrophy and counteracted the up-regulation of collagen gene expression and myofibroblast accumulation in the lesioned skin. Rosiglitazone treatment reduced the induction of the early-immediate transcription factor Egr-1 in situ without also blocking the activation of Smad2/3. In both explanted fibroblasts and skin organ cultures, rosiglitazone prevented the stimulation of collagen gene transcription and cell migration elicited by TGF-β. Rosiglitazone-driven adipogenic differentiation of both fibroblasts and preadipocytes was abrogated in the presence of TGF-β; this effect was accompanied by the concomitant down-regulation of cellular PPAR-γ mRNA expression. Collectively, these results indicate that rosiglitazone treatment attenuates inflammation, dermal fibrosis, and subcutaneous lipoatrophy via PPAR-γ in a mouse model of scleroderma and suggest that pharmacological PPAR-γ ligands, widely used as insulin sensitizers in the treatment of type-2 diabetes mellitus, may be potential therapies for scleroderma.
Excessive collagen accumulation in the skin and lungs, the hallmark of systemic sclerosis (SSc), can lead to organ dysfunction, failure, and death.1 The pathogenesis of fibrosis remains incompletely understood.2 Although inflammation is a prevalent early feature, its precise role in fibrosis remains controversial, and anti-inflammatory therapies are generally ineffective in reversing or slowing the progression of the process.3 Therefore, there is an urgent need for anti-fibrotic therapies. The fibroblast is the key effector cell driving the fibrotic process in SSc. In response to extracellular cues such as transforming growth factor (TGF)-β, fibroblasts become activated with increased collagen production, expression of cell surface receptors for growth factors, secretion of cytokines and chemokines, resistance to apoptosis induction, and myofibroblast differentiation.4 In light of the key role of TGF-β in the pathogenesis of fibrosis, therapeutic strategies to block its production, activity, or intracellular signaling are under investigation.5 However, because the potent anti-inflammatory and immunosuppressive activities of TGF-β are physiologically important, global TGF-β blockade could be complicated by spontaneous autoimmunity. Indeed, mice lacking TGF-β die at an early age from severe inflammation.6 Therefore, an ideal anti-fibrotic strategy targeting TGF-β must selectively abrogate fibrotic responses without disrupting its important immunosuppressive activities.
Rosiglitazone is an insulin-sensitizing agent widely in type 2 diabetes mellitus that exerts its biological effects in part via the peroxisome proliferator activated receptor (PPAR)-γ.7 Originally identified in adipose tissue, PPAR-γ is one of a family of closely related nuclear receptors and ligand-activated transcription factors with a primary role in adipogenesis.8 In addition to adipocytes, PPAR-γ is expressed in macrophages, vascular endothelial and smooth muscle cells, and fibroblasts. The PPAR-γ receptor acts as a lipid sensor that can be activated by fatty acids, eicosanoids, and related endogenous products of metabolism.9 A majority of experimental studies of PPAR-γ have used the natural ligand 15-deoxy-Δ12,14 prostaglandin J2 (15d-PGJ2), or synthetic ligands such as rosiglitazone. In the absence of ligand, PPAR-γ is complexed to the retinoid X receptor (RXR) and co-repressors, preventing its binding to DNA. Upon receptor ligation, co-repressors are displaced from the PPAR-γ/RXR complex and co-activators such as p300 are recruited, allowing sequence-specific binding to conserved PPAR-γ response elements (PPREs) in target gene promoters.10 Ligands of PPAR-γ exert a broad range of proliferative, anti-inflammatory, and repair activities in addition to adipogenesis and insulin sensitization.11 Recent studies have shown that PPAR-γ inhibits basal and stimulated collagen synthesis in vitro and in vivo, suggesting a novel biological role in connective tissue homeostasis.12,13,14 Ligand inhibition of TGF-β-dependent fibrotic responses was mediated via the PPAR-γ receptor and involved antagonistic cross talk with the intracellular TGF-β/Smad signal transduction pathway.12
Because fibroblast activation by TGF-β is a key pathogenetic event in SSc, blockade of TGF-β signaling by PPAR-γ could be a novel therapeutic approach to pathological fibrogenesis. In the present studies, therefore, we investigated the effect of rosiglitazone, the most potent pharmacological PPAR-γ agonist, in bleomycin-induced scleroderma. The results indicate that rosiglitazone ameliorated the development of fibrosis in this mouse model of scleroderma. The anti-fibrotic effect of rosiglitazone involved blockade of TGF-β-induced fibroblast activation, as well as attenuation of the inflammatory response. Furthermore, rosiglitazone counteracted the development of subcutaneous adipose atrophy and loss of local PPAR-γ expression. Together, these findings indicate that ligands of PPAR-γ, already in clinical use for the treatment of type 2 diabetes mellitus, block fibrotic TGF-β responses without triggering aberrant immunity, suggesting their potential as novel anti-fibrotic agents in SSc.
Materials and Methods
Animals and Experimental Protocols
Six- to eight-week-old female BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) were used in these studies. The protocols were institutionally approved by the Northwestern University Animal Care and Use Committee. Mice were housed in autoclaved cages and fed sterile food and water. Filter-sterilized bleomycin [20 μg/mouse dissolved in phosphate-buffered saline (PBS)] (Mayne Pharma, Paramus, NJ) or PBS was administered by daily subcutaneous injections into the shaved backs of mice using a 27-gauge needle.15 Rosiglitazone (5 mg/kg) (GlaxoSmithKline, King of Prussia, PA) and the irreversible PPAR-γ antagonist GW9662 (Biomol International, Plymouth Meeting, PA) were administrated by daily intraperitoneal injection. At the end of each experiment, mice were sacrificed and lesional skin was processed for analysis. Each experimental group consisted of at least five mice.
Histochemical Analysis
Lesional skin tissue was embedded in paraffin, and consecutive 4-μm serial sections were stained with hematoxylin and eosin (H&E). To evaluate collagen content and organization in the lesional skin, deparaffinized sections were stained with Picrosirius Red and viewed under a polarized light microscope.16 Dermal thickness, defined as the distance between the epidermal-dermal junction and the dermal-adipose layer junction, and the adipose layer, defined as the distance between the dermal-adipose junction and the muscle, was determined in H&E-stained sections at ×100 microscopic magnification. For localizing tissue lipids, samples immersed in freezing medium (Triangle Biomedical Sciences, Durham, NC) were sectioned (7 μm) using a CM 1900 UV cryostat (Leica Microsystems, Bannockburn, IL) and stained with Oil Red O.17 Mast cells were identified by Astra blue staining and quantified by counting in six random fields under high magnification.15 To detect apoptosis, terminal dUTP nick-end labeling (TUNEL) assays were performed on paraffin-embedded tissues using an in situ cell death detection kit (Roche Molecular Biochemicals, Indianapolis, IN).
Immunohistochemistry and Immunofluorescence
Four-μm sections from lesional skin were deparaffinized, rehydrated, and immersed in TBS-T buffer (Tris-buffered saline and 0.1% Tween 20), and treated with target retrieval solution (DAKO, Carpinteria, CA) at 95°C for 10 minutes. Tissue embedded in freezing medium (Triangle Biomedical Sciences) was sectioned (7 μm) in a CM 1900 UV cryostat. Primary antibodies against CD3 (BD Pharmingen, San Diego, CA); Mac-3 (BD Pharmingen), α-smooth muscle actin (Sigma-Aldrich, St. Louis, MO), TGF-β1 and Egr-1 (both from Santa Cruz Biotechnology, Santa Cruz, CA), phospho-Smad2 (Cell Signaling, Danvers, MA), PPAR-γ (Santa Cruz), or I-Ad (BD Pharmingen) were used. Bound antibodies were detected using secondary antibodies from the Histomouse-Max kit (Zymed Laboratories, San Francisco, CA) and the DakoCytomation Envision + System-HRP (3,3′-diaminobenzidine tetrahydrochloride) according to the manufacturers’ instructions. Substitution of the primary antibody with isotype-matched irrelevant IgG served as negative controls. After counterstaining with hematoxylin, sections were mounted with Permount (Fisher Scientific, Pittsburgh, PA) and viewed under an Olympus BH-2 microscope (Olympus, Tokyo, Japan). Fibroblasts were identified by their spindle-shaped morphology. The number of immunopositive cells was determined at ×400 magnification by two blinded observers. Immunofluorescence intensity was quantitated by image analysis.
Quantification of Tissue Levels of Collagen and TGF-β1
Lesional skin tissues were homogenized in 0.5 mol/L glacial acetic acid and centrifuged at 10,000 rpm for 15 minutes. Supernatant were then assayed for soluble collagen using Sircol colorimetric assays (Biocolor, Newton Abbey, UK), and TGF-β1 was quantitated by enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction 9 (qPCR)
Total RNA was isolated from frozen skin tissue using Trizol reagent (Invitrogen, Carlsbad, CA) and purified with RNeasy mini spin columns (Qiagen, Valencia, CA). Reverse transcription was performed in a DNA thermal cycler (2720 Thermal Cycler; Applied Biosystems, Foster City, CA) using a reverse transcription system kit (Promega, Madison, WI). Real-time qPCR was performed using SYBR Green Master Mix (Applied Biosystems) in an ABI Prism 7700 sequence detection system (Applied Biosystems). The oligonucleotide primers used for qPCR are listed in Table 1. The results of real-time qPCR analysis are expressed as -fold change in mRNA levels relative to 18S RNA or GAPDH levels in each sample.
Table 1.
Primers Used for Quantitative Real-Time PCR
| Gene | cDNA sequence F, forward; R, reverse |
|---|---|
| COL1A1 | F: 5′-CCTGAGTCAGCAGATTGAGAA-3′; R: 5′-ACTGAACTTGACCGTACACCAGTACTCTCCGCTCTTCAA-3′ |
| COL1A2 | F: 5′-CCGTGCTTCTCAGAACATCA-3′; R: 5′-CTTGCCCCATTCATTTGTCT-3′ |
| α-SMA | F: 5′-CAGCGGGCATCCACGAA-3′; R: 5′-GCCACCGATCCAGACAGA-3′ |
| TGF-β1 | F: 5′-TACAGCAAGGTCCTTGCCCT-3′; R: 5′-GCAGCACGGTGACGCC-3′ |
| PPAR-γ1 | F: 5′-GAGTGTGACGACAAGATTTG-3′; R: 5′-GGTGGGCCAGAATGGCATCT-3′ |
| PPAR-γ2 | F: 5′-TCTGGGAGATTCTCCTATTGA-3′; R: 5′-GGTGGGCCAGAATGGCATCT-3′ |
| Egr-1 | F: 5′-TTTGCCTCCGTTCCACCTGC-3′; R: 5′-TGCCAACTTGATGGTCATGCGC-3′ |
| 18S rRNA | F: 5′-TTCGAACGTCTGCCCTATCA-3′; R: 5′-ATGGTAGGCACGGCGACTA-3′ |
Flow Cytometry and Determination of Adiponectin, Cytokine, and Chemokine Levels
Peripheral blood was obtained at day 5 of bleomycin injections via cardiac puncture with citrate solution (100 mmol/L Na Citrate, 130 mmol/L glucose, pH 6.5) as anticoagulant, and mononuclear cells were purified by density gradient centrifugation. Spleens were removed and triturated with the end of a 3-ml syringe, and filtered through 70-μm nylon mesh (BD Bioscience, Bedford, MA). Single cell suspensions of peripheral blood mononuclear cells or spleen cells were analyzed by flow cytometry using antibodies against I-Ad (MHC-II)-FITC, CD86-PE, or CD-11b-APC, along with mouse IgG1-PE, IgG2a/2b-FITC, and IgG-APC (all from BD Pharmingen) as isotype controls. Events were collected on a DakoCytomation (DAKO, Fort Collins, CO) by gating for live cells based on forward and side scatter, and data were analyzed using Summit software V4.3 (DAKO). Serum levels of adiponectin, and the inflammatory mediators interleukin (IL)-1β and MCP-1 were determined at 7 or 28 days after the start of bleomycin injection by ELISA (R&D).
Collagen Transcription Assays
Cultures of dermal fibroblasts were established by explantation of skin biopsies from newborn Col1a2-luc transgenic mice. These mice harbor a construct of the mouse proα2(I) collagen gene promoter containing 17 kb 5′ of the transcription start site, fused to a luciferase reporter gene, that drives high levels of reporter gene expression in fibroblasts.18 When the fibroblasts reached confluence, cultures were incubated in serum-free media containing 0.1% bovine serum albumin and indicated concentrations of rosiglitazone, followed 30 minutes later by 10 ng/ml of TGF-β1 (Amgen, Thousand Oaks, CA). At the end of a further 48 hours of incubation, cultures were harvested and cell lysates were assayed for their luciferase activities.19
Skin Organ Cultures
Triplicate 3-mm punch biopsies were obtained from the back skin of 12-week-old female C57BL/6 mice. The tissues were incubated in 96-well plates (one tissue piece per 150 μl of culture medium) in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 1% vitamins, 100 U/ml penicillin/streptomycin, 2 mmol/L l-glutamine with rosiglitazone (10 μmol/L) in presence or absence of TGF-β1 (10 ng/ml). After 48 hours, conditioned media were collected and newly synthesized soluble collagens (types I to IV) were quantified by Sircol colorimeteric assays. Total RNA was isolated from skin homogenates using Trizol reagent and subjected to real-time qPCR analysis.
In Vitro Adipogenic Differentiation Assays
To assess adipogenic differentiation, mouse 3T3L1 preadipocyte (Zen-Bio, Research Triangle Park, NC) were incubated in either preadipocyte maintenance medium or DM2 adipogenic differentiation medium (both from Zen-Bio). Low-passage (three to four) dermal fibroblasts established from the skin of newborn BALB/c mice were seeded in 12-well plates (105 cells/well), and allowed to attach overnight. Cultures were then incubated in Dulbecco’s modified Eagle’s medium or DM2 in the presence or absence of rosiglitazone (10 μmol/L) and/or TGF-β2 (5 ng/ml) (Genzyme, Framingham, MA) for up to 5 days. To assess intracellular lipid accumulation, cells were stained with Oil Red O (0.5% Oil Red O dye in 60% isopropanol) for 30 minutes followed by gentle rinse before microscopic visualization. Lipid-containing cells were enumerated in 10 random microscopic fields for each experimental condition, and experiments were repeated at least three times.
For immunocytochemistry, cultures were washed in PBS and fixed in 4% paraformaldehyde for 10 minutes followed by methanol for permeabilization, and stained with antibodies specific for leptin (Santa Cruz Biotechnology), fatty acid binding protein-4 (FABP4) (R&D), and α-smooth muscle actin (Sigma-Aldrich). Slides were overlaid with secondary horseradish peroxidase-conjugated anti-rabbit IgG for 30 minutes, and 3,3′-diaminobenzidine tetrahydrochloride was used for chromogenic localization of antibody. Sections were counterstained with hematoxylin, and images were obtained by digital capture. Fluorescein isothiocyanate (FITC)-labeled anti-mouse IgG was used as secondary antibody of α-smooth muscle actin. Nuclei were identified with 4′-6-diamidino-2-phenylindole. Images were obtained by digital capture in a Nikon Eclipse TE200 microscope (Fryer Co., Huntley, IL) and viewed under ×200 magnification.
In Vitro Cell Migration Assays
The modulation of cell migration in vitro was analyzed by monolayer wound-healing assays.14 Briefly, primary skin fibroblasts from C57BL/6 mice were grown to early confluence. Scratch wounds were induced in monolayers using standard p1000 pipette tips, followed by incubation of the cultures in media with 10 ng/ml of mitomycin C (Sigma-Aldrich) to block cell proliferation. Rosiglitazone (2 to 20 μmol/L) and TGF-β1 (10 ng/ml) were added and incubation continued for a further 24 hours. The wounds were monitored at intervals by phase contrast microscopy. Wound gap length was measured at six different sites in each sample at indicated times, and experiments were repeated multiple times with similar results.
Statistical Analysis
Results are expressed as the means ± SD or ± SEM. Mann-Whitney’s U-test (in vivo studies) or Student’s t-test (in vitro studies) was used for comparison between two groups. Values <0.05 were considered statistically significant.
Results
Effects of Rosiglitazone on Skin Inflammation and Fibrosis
To evaluate the effects of rosiglitazone in a mouse model of scleroderma, BALB/c mice given daily injections of subcutaneous bleomycin for up to 28 days were treated with intraperitoneal rosiglitazone. Bleomycin induced an early and transient accumulation of inflammatory cells in the skin that peaked on day 7 after initiation of treatment, and was most prominent in the deep dermis and subcutaneous adipose tissue (Figure 1A, and data not shown). Concurrent treatment with rosiglitazone significantly reduced the inflammatory cell infiltration (Figure 1A, bottom). Pretreatment of the mice with the selective PPAR-γ antagonist GW9662 abrogated the effects of rosiglitazone, indicating that anti-inflammatory response was mediated via activation of cellular PPAR-γ. Immunohistochemical analysis showed that mice treated with rosiglitazone plus bleomycin for 7 days had reduced accumulation of Mac-3-positive cells in the lesional skin (Figure 1B), whereas the numbers of infiltrating CD3-positive cells and mast cells were primarily unchanged (data not shown). The enhanced local expression of MHC class-II (I-Ad), a hallmark of classical monocyte/macrophage activation, was substantially attenuated by administration of rosiglitazone (Figure 1C).
Figure 1.

Rosiglitazone attenuates bleomycin-induced skin inflammation. Female BALB/c mice received subcutaneous bleomycin alone, or bleomycin plus rosiglitazone or GW9662. Lesional skin was examined (day 7). A, Top: H&E stain. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone; e, GW9662; f, bleomycin plus rosiglitazone plus GW9662. A, Bottom: The numbers of inflammatory cells in the lesional dermis were determined. Bars are the means ± SD from six determinations. B: Sections were immunostained with antibodies to Mac-3. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone; e, GW9662; f, bleomycin plus rosiglitazone plus GW9662. C: Immunofluorescence with antibodies to I-Ad. Top: a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. Red arrows indicate I-Ad-positive mononuclear cells; yellow arrows indicate hair follicles. Bottom: Cutaneous I-Ad expression was quantitated as described in the Materials and Methods. The results indicate the means ± SD from three mice in each group. *P < 0.05. Scale bars: 50 μm (B, C); 20 μm (B, inset). Original magnifications: ×100 (A); ×400 [A (inset), B].
After 28 days of bleomycin injections, a considerable increase in dermal thickness and accumulation of densely packed collagen bundles was evident in the lesional skin (Figure 2A, top). At the same time, the subcutaneous adipose layer showed striking atrophy, and was virtually replaced by acellular densely packed connective tissue. Mice treated with rosiglitazone showed significant expansion of the subcutaneous adipose layer with accumulation of intracellular lipids, as demonstrated by strong staining with Oil Red O (Figure 2B). When rosiglitazone was administered together with bleomycin, dermal fibrosis was attenuated and collagen bundles were loosely packed and randomly oriented. Quantitative evaluation showed that whereas dermal thickness of the dermis was >50% increased in bleomycin-injected mice compared with PBS-injected control mice, rosiglitazone attenuated the increase (P < 0.05). Furthermore, the integrity of the subcutaneous adipose layer was partially preserved in these mice (Figure 2A, bottom).
Figure 2.

Rosiglitazone prevents skin fibrosis and subcutaneous adipose atrophy. Lesional skin was examined after 28 days. A: H&E stain. Top: a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. Representative photomicrographs. Arrows indicate the extent of the subcutaneous adipose layer. Bottom: Thick ness of the dermis and subcutaneous adipose tissue. Results represent the means ± SD from five mice per group, *P < 0.05. B: Oil Red O stain. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone; Scale bar = 100 μm. Original magnifications: ×100 (B, insets); ×400 (B).
Staining with Picrosirius Red was used to investigate the deposition and organization of collagenous matrix in the skin. In bleomycin-injected mice, the dermis and subcutaneous tissue showed dense collagen accumulation with strong red birefringence (indicative of highly cross-linked mature fibers), whereas mice given rosiglitazone together with bleomycin showed weaker birefringence (Figure 3A). To examine the effects of rosiglitazone on collagen gene expression in vivo, mRNA in the lesional skin was quantified by real-time PCR. The results showed a threefold to sixfold increase in the levels of COL1A1 and COL1A2 mRNA in mice treated with bleomycin compared with PBS-treated control mice (Figure 3B). Concurrent treatment with rosiglitazone markedly attenuated the up-regulation of collagen mRNA (P < 0.01). The expression of α-smooth muscle actin, a marker for identifying myofibroblasts that play crucial roles in pathological fibrogenesis, was determined by immunohistochemistry. After 28 days of bleomycin, a twofold increase in α-smooth muscle actin was noted in the lesional dermis and subcutaneous layers (Figure 3C, top). Concurrent treatment of the mice with rosiglitazone significantly reduced the number of α-smooth muscle actin-positive fibroblastic cells (Figure 3C, bottom). Plasma levels of adiponectin, an adipocyte-specific circulating cytokine, were increased by ∼40% in rosiglitazone-treated mice compared with controls (5.3 ± 0.4 μg versus 3.7 ± 0.4 μg, P < 0.05). Detection of in situ cell death demonstrated a modest increase in the number of TUNEL-positive fibroblastic cells in the lesional dermis at 28 days after bleomycin injections were started; the increase in apoptotic cells was abrogated in rosiglitazone-treated mice (data not shown).
Figure 3.

Rosiglitazone attenuates collagen deposition and myofibroblast accumulation. Lesional skin (dermis plus subcutaneous adipose tissue) was examined after 28 days. A: Picrosirius Red stain (viewed under polarized microscopy). a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone; e, GW9662; f, bleomycin plus rosiglitazone plus GW9662. Representative photomicrographs. Arrows indicate the extent of the dermis. B: Real-time qPCR analysis. Results are normalized for 18S RNA and represent the means ± SD of duplicate determinations from three mice per group, *P < 0.01. C, Top: α-Smooth muscle actin immunohistochemistry. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. Bottom: The proportion of α-smooth muscle actin-positive fibroblastic cells was quantified from at least six separate fields from five mice per group. The results indicate the means ± SD. *P < 0.05. Scale bars: 100 μm (A); 50 μm (C); 20 μm (C, inset).
Reduced Mononuclear Cell Activation
Peripheral blood and spleen mononuclear cells were obtained on day 5 after treatment was initiated, and analyzed by flow cytometry. The results indicated that both the frequency and the absolute numbers of I-Ad-positive mononuclear cells were modestly increased in the circulation of bleomycin-injected mice compared with control mice, and rosiglitazone treatment was associated with a nearly 30% reduction (Figure 4A). Similar results were seen when spleen cells were analyzed (Figure 4B). Circulating CD-11b-positive monocytes expressed very low levels of CD86, which was not significantly affected by rosiglitazone (data not shown). The enhanced serum levels of the inflammatory mediators IL-1β and MCP-1 in bleomycin-treated mice at day 7 were markedly reduced by treatment with rosiglitazone (Figure 4C).
Figure 4.

Rosiglitazone attenuates mononuclear cell activation. Peripheral blood mononuclear cells (A) or spleen cells (B) were collected at day 5 of treatment. Single cell suspensions were analyzed by flow cytometry using antibodies against CD-11b and I-Ad. The results represent the means from three mice per group. C: Serum levels of IL-1β and MCP-1 at day 7 were determined by ELISA. The results represent the means of duplicate determinations from four mice per group. *P < 0.05.
Reduced PPAR-γ Expression in Lesional Skin
Because the biological activities of rosiglitazone are mediated primarily through the nuclear PPAR-γ receptor, the expression level of PPAR-γ governs the intensity of cellular responses. Immunohistochemical analysis to examine the modulation of PPAR-γ in the fibrotic response showed that in normal dermis, PPAR-γ was detectable principally in fibroblastic cells, where it appeared to be distributed in both nucleus and cytoplasm (Figure 5A). Whereas bleomycin treatment of the mice was associated with progressive loss of PPAR-γ expression in fibrotic dermis, concurrent administration of rosiglitazone partially prevented this inhibition. Because immunohistochemistry is neither highly sensitive nor quantitative for PPAR-γ, mRNA levels in lesional skin were determined by real-time qPCR analysis. The qPCR results confirmed the decrease in PPAR-γ mRNA expression associated with fibrosis, and showed significantly increased expression in mice that had received concurrent rosiglitazone (Figure 5B).
Figure 5.
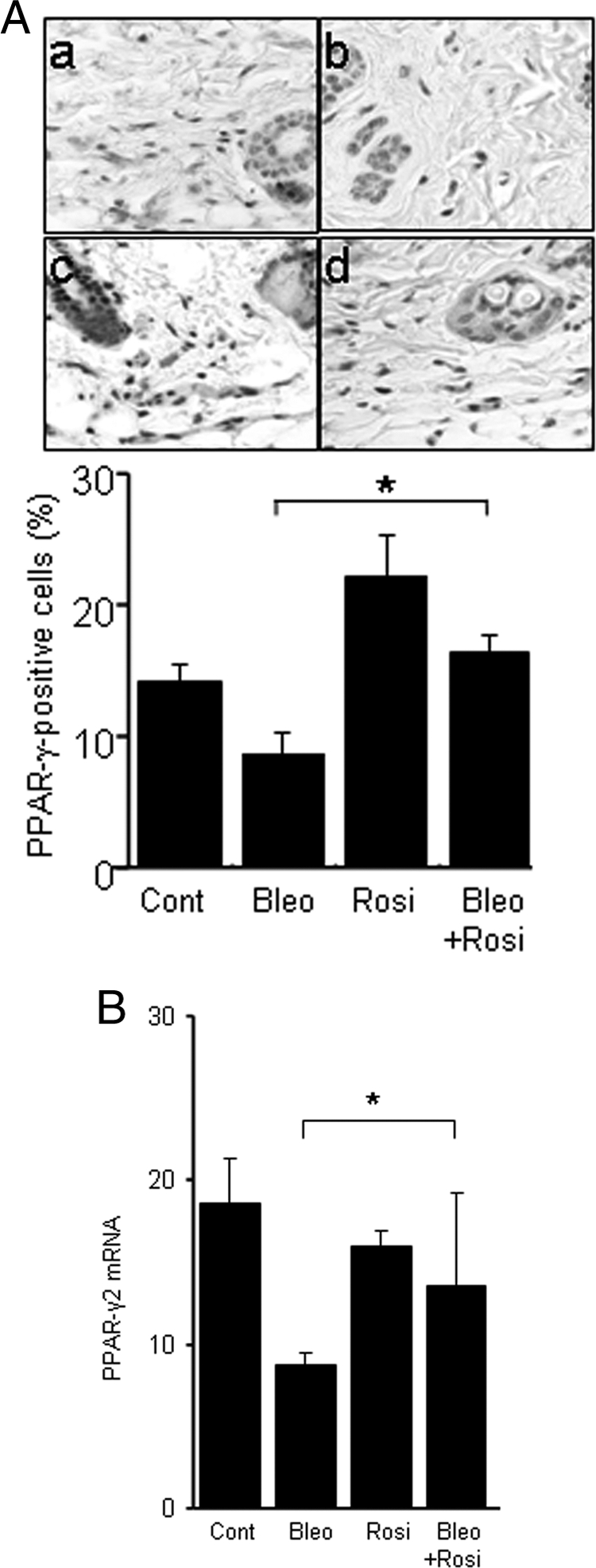
Modulation of PPAR-γ expression in the lesional skin. After 28 days, lesional skin was examined. A: Immunohistochemistry. Top: a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. Representative images. Bottom: The proportion of immunopositive fibroblastic cells was quantified. The results represent the means ± SD from five mice per group. *P < 0.05. B: Real-time qPCR analysis. The results, normalized for 18S RNA, represent the mean ± SD of duplicate determinations from three to five mice per group. *P < 0.05. Scale bars: 100 μm (A).
Effects of Rosiglitazone on TGF-β1 Expression and Activity
Transforming growth factor is a key mediator of fibrosis in a variety of fibrotic disorders, as well as in bleomycin-induced animal models of fibrosis. Secreted by infiltrating leukocytes or by activated fibroblasts, or liberated in situ from its matrix-bound sequestered latent form, active TGF-β induces both Smad-dependent and Smad-independent intracellular signaling in resident fibroblasts. To evaluate the modulation of the TGF-β by rosiglitazone in vivo, TGF-β1 mRNA and protein expression were examined in lesional skin. Concurrent administration of rosiglitazone substantially prevented the up-regulation of TGF-β1 mRNA. The accumulation of TGF-β protein in lesional skin detected by immunohistochemistry (Figure 6A) and TGF-β mRNA determined by qPCR (Figure 6B) were reduced by rosiglitazone treatment. To assess TGF-β signaling activity in situ, expression of phosphorylated Smad2, a highly sensitive and specific marker of TGF-β activity was examined. The results of immunohistochemistry showed that bleomycin by itself caused a marked up-regulation of phospho-Smad2 in fibroblastic cells in the dermis, reflecting their activation in situ by TGF-β (Figure 6C). Rosiglitazone treatment failed to attenuate this response. In contrast, rosiglitazone abrogated the up-regulation of Egr-1, an early-immediate response transcription factor that is an intracellular mediator of Smad-independent TGF-β signaling and has been implicated in the fibrotic response (Figure 6, D and E).
Figure 6.

TGF-β1 expression and signaling in the lesional skin. At 28 days after injections were started, lesional skin was examined. A: TGF-β1 immunohistochemistry. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. B: Real-time qPCR analysis. The results, normalized for 18S RNA, represent the means ± SD of duplicate determinations from three mice per group, *P < 0.05. C: Immunohistochemistry for phospho-Smad2, or D: Egr-1. Top: Representative photomicrographic images. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. Bottom: The proportion of immunopositive fibroblasts was quantified. The results represent the means ± SD from five mice per group. *P < 0.05. E: Real-time qPCR analysis. The results, normalized for 18S RNA, represent the means ± SD of duplicate determinations from five mice per group. *P < 0.05. Scale bars: 50 μm (D); 20 μm (D, inset). A, C, Original magnification ×100; inset, ×400.
Rosiglitazone Abrogates the Stimulation of Collagen Synthesis and Fibroblast Migration
To explore the mechanistic basis underlying the antifibrotic effects of rosiglitazone, ex vivo TGF-β responses were investigated by two complementary approaches. First, organ culture experiments were performed to examine the regulation of collagen production by rosiglitazone. Tissues obtained from the back skin from C57BL/6 mice were incubated in media with TGF-β and rosiglitazone for 48 hours. The results of Sircol colorimetric assays showed that TGF-β induced a 40% increase (P < 0.05) in soluble collagen secreted into the culture media (Figure 7A). Concurrent rosiglitazone treatment of the organ cultures abrogated this stimulation. Stimulation of COL1A1 and COL1A2 mRNA expression in the skin tissue were similarly abrogated by rosiglitazone (Figure 7B).
Figure 7.

Rosiglitazone abrogates collagen stimulation in vitro. A and B: Skin organ cultures were established from C57BL/6 mice and incubated with TGF-β with or without rosiglitazone for 48 hours. A: Soluble collagen secreted into the media was quantitated by Sircol colorimetric assays. Results are the means ± SD of triplicate determinations from a representative experiment. B: Total RNA was isolated and analyzed by real-time qPCR. Results are the means ± SD of triplicate determination from a representative experiment. C: Dermal fibroblasts explanted from Col1a2-luc transgenic mice or wild-type mice were incubated with rosiglitazone and TGF-β for 48 hours. Cell lysates were assayed for their luciferase activities; luciferase activity in nontransgenic control fibroblasts was undetectable. Results are the means ± SD of triplicates determination from a representative experiment. *P < 0.05.
Next, the regulation of type I collagen synthesis by rosiglitazone was investigated in monolayer cultures of dermal fibroblasts explanted from Col1a2 transgenic mice. As reported previously,13 these fibroblasts showed sustained Col1a2-luc expression in vitro even in the absence of stimulation, as determined by luciferase assays. Incubation of the cultures with TGF-β for 24 hours resulted in a more than twofold increase in luciferase activity, reflecting stimulation of the COL1A2 upstream enhancer; the response was abrogated by rosiglitazone (Figure 7C). At the range of concentrations used in these experiments, rosiglitazone had no effect on cellular toxicity as determined by trypan blue dye exclusion. Together, these results indicate that rosiglitazone abrogated TGF-β-induced stimulation of collagen production and Col1a2 promoter activity in skin organ cultures and monolayer fibroblasts in vitro. Blockade of these TGF-β responses in rosiglitazone-treated fibroblasts did not interfere with Smad2 activation (data not shown).
Migration of dermal fibroblasts plays a critical role in both normal wound healing and pathological fibrogenesis. We hypothesized that the anti-fibrotic effects of rosiglitazone were associated with reduced stimulations of fibroblast migration. To determine the effect of rosiglitazone on the wound healing response in vitro, scratch assays were performed with confluent fibroblasts. Cell proliferation was blocked with mitomycin C. A pipette tip was used to make a liner scratch in the monolayers, and fibroblast migration was monitored for up to 24 hours after mechanical injury. As shown in Figure 8, rosiglitazone significantly attenuated the stimulation of fibroblast migration and wound closure elicited by TGF-β, with a >50% reduction at 24 hours (P < 0.05).
Figure 8.

Top: Rosiglitazone abrogates stimulation of fibroblast migration. Confluent monolayers of dermal fibroblasts were scratched with a p-1000 pipette tip to induce mechanical injury, followed by incubation in media with TGF-β (10 ng/ml) with or without rosiglitazone (2 μmol/L or 20 μmol/L) for up to 24 hours. Fibroblast migration was monitored by phase contract microscopy. Representative micrographs taken at 24 hours. a, control; b, rosiglitazone 2 μmol/L; c, rosiglitazone 20 μmol/L; d, TGF-β; e, TGF-β plus rosiglitazone 2 μmol/L; f, TGF-β plus rosiglitazone 20 μmol/L. Bottom: Quantification of fibroblast migration. The width of the scratch was measured at six different sites in each sample. The results, shown as mean ± SD, were reproducible in three independent experiments. *P < 0.05.
Rosiglitazone Induces Adipocytic Differentiation of Explanted Dermal Fibroblasts
We consistently noted that the development of dermal fibrosis was accompanied by progressive atrophy of the subcutaneous adipose layer and its replacement by fibrous tissue (Figure 2). This pattern recapitulates the progression of histopathological changes in the skin seen in patients with SSc. To investigate the molecular basis underlying loss of adipose tissue that accompanies the onset of skin fibrosis, and its rescue by treatment of the mice with PPAR-γ ligand, we focused on the adipogenic modulation of fibroblast and adipocyte phenotypes in vitro. Under standard monolayer culture conditions, dermal fibroblasts retained their spindle-shaped morphology and growth characteristics for up to 1 week (data not shown). However, when incubated in media containing PPAR-γ ligand, these fibroblasts gradually acquired a rounded shape and accumulated prominent intracellular lipid droplets (Figure 9A). By day 7, up to 50% of cells in each culture were Oil Red O-positive (Figure 9B). Furthermore, rosiglitazone induced strong up-regulation of the hallmark adipogenic markers PPAR-γ, FABP4, and leptin (Figure 9, C and D), whereas mRNA levels for type I collagen and α-smooth muscle actin were decreased (data not shown). Subsequent exposure of adipocytic cells (as judged by abundant accumulation of cytoplasmic lipid droplets) to TGF-β (5 ng/ml) for 7 days resulted in recovery of the fibroblastic spindle-shaped morphology, and was associated with loss of cellular leptin and FABP4 expression, suggesting reversion to a fibroblast phenotype (Figure 9C). Under identical culture conditions, mouse 3T3-L1 subcutaneous preadipocytes were readily induced to undergo adipogenic differentiation in media containing rosiglitazone, as expected (Figure 9E). Remarkably, the adipogenic differentiation of preadipocytes was also prevented by TGF-β. Together, these results indicate that PPAR-γ ligands drive differentiation of mature dermal fibroblasts as well as preadipocytes to adipocytes, and this process is reversed by TGF-β. Rosiglitazone in these experiments had no effect on cell viability or proliferation (data not shown).
Figure 9.

Rosiglitazone induces adipogenic differentiation of skin fibroblasts. Primary cultures of confluent dermal fibroblasts from newborn BALB/c mouse (A--D), or 3T3L1 mouse preadipocytes (E) were incubated in standard media with rosiglitazone (10 μmol/L), or in differentiation media (DM2), in the presence or absence of TGF-β2 (5 ng/ml) for 7 days. A: Cells were examined by phase contrast microscopy. B Top: Cell stained with Oil Red O. Bottom: The proportion of Oil Red O-positive cells was quantitated. The results indicate the means ± SD from three separate experiments, *P < 0.05. C: Immunocytochemistry with antibodies against FABP4 (a--d) or leptin (e--h); hematoxylin counterstain. D: Real-time qPCR analysis. The results, normalized for 18S RNA, represent the means ± SD of duplicate determinations from three different experiments. *P < 0.05. E: 3T3L1 preadipocytes examined by phase contrast microscopy. Representative images. Original magnifications, ×200.
Rosiglitazone Ameliorates Skin Fibrosis When Started after Onset of Inflammation
To determine whether the anti-fibrotic effects of rosiglitazone were primarily attributable to the prevention of the inflammatory response elicited by bleomycin, or to its direct effects of TGF-β-mediated activation of fibroblasts, rosiglitazone was administered beginning on day 7 after bleomycin injections were started, and mice were sacrificed 21 days later. At day 7, significant cutaneous inflammation was evident in bleomycin-injected mice. Mice treated subsequently with rosiglitazone showed reduced skin fibrosis compared with mice treated with PBS in parallel (Figure 10A). The thickness of the dermis was 235.9 ± 50.7 μm (mean ± SD) in bleomycin-treated mice, compared with 198.6 ± 33.5 μm in bleomycin plus rosiglitazone-treated mice. Furthermore, when initiated on day 7, rosiglitazone was associated with reduced TGF-β and collagen accumulation, and reduced COL1A1 mRNA expression in the lesional skin (Figure 10, B–D). These results indicate that the anti-fibrotic effect of rosiglitazone in the bleomycin model of mouse scleroderma was at least in part independent of its potent inhibitory effect on inflammation, and may reflect a direct anti-fibrotic activity.
Figure 10.

Rosiglitazone ameliorates bleomycin-induced skin fibrosis. Rosiglitazone treatment was initiated beginning on day 7 of bleomycin injections. Lesional skin was examined at 28 days. A: H&E stain. a, control; b, bleomycin; c, rosiglitazone; d, bleomycin plus rosiglitazone. B: Soluble collagen in lesional skin was quantitated by Sircol colorimetric assays. Results are the means ± SD of five mice per group. C: Total RNA was isolated and analyzed by real-time qPCR. Results are the means ± SD of triplicate determination from a representative experiment. D: Levels of TGF-β1 in lesional skin were determined by ELISA. The results represent the means ± SD of duplicate determinations from three mice per group. *P < 0.05. Original magnifications, ×100.
Discussion
In light of the pivotal role of TGF-β in fibrosis, inhibiting TGF-β signaling represents a novel approach to targeted anti-fibrotic therapy. The intensity of TGF-β signaling is physiologically modulated by redundant and complementary mechanisms that govern ligand availability and biological activity and the magnitude and duration of responses. Molecules such as Smad7, caveolin-1, and the TGF-β pseudoreceptor BAMBI constitute an intracellular regulatory network that normally restricts the magnitude and duration of cellular responses to TGF-β.5 The present findings suggest that the nuclear receptor PPAR-γ may fulfill a similar regulatory role in the context of TGF-β-mediated fibrogenesis.
Bleomycin-induced scleroderma was used as an animal model to examine whether the in vitro suppression of TGF-β responses by PPAR-γ demonstrated in cultured fibroblasts translates into anti-fibrotic activity in vivo. Although there is no animal model that fully recapitulates all key aspects of human scleroderma, subcutaneous administration of bleomycin is increasingly popular as an experimental approach, because bleomycin elicits a predictable sequence of pathological changes in mice that parallel the evolution of fibrosis in scleroderma, with early and transient cutaneous inflammation followed by progressive fibrosis.20 Within the lesional dermis, fibroblastic cells show evidence of TGF-β activation and Smad2/3 phosphorylation, and fibrosis is attenuated in mice with targeted deletion of Smad3.15,16 Bleomycin-induced scleroderma thus permits an exploration of the complex relationship between the inflammatory and fibrotic processes, and their modulation by endogenous molecules and pharmacological agents.
The present results show that rosiglitazone attenuated the severity of dermal fibrosis and local collagen deposition, and reduced the tissue accumulation of myofibroblasts in vivo. Rosiglitazone also down-regulated the levels of TGF-β in lesional skin. The mechanisms accounting for these potent anti-fibrotic effects are complex. We observed that rosiglitazone suppressed the early inflammatory response, with reduced activation and tissue accumulation of mononuclear leukocytes. TGF-β is known to play a direct role in monocyte recruitment and activation in this animal model. 21 Although inflammation is a prominent early finding in fibrosing conditions that might contribute to both triggering and sustaining the fibrotic process, therapeutic agents targeting the inflammatory response such as corticosteroids generally fail to reverse or slow the progression of fibrosis, and are primarily ineffective in SSc.22 In the present studies, rosiglitazone attenuated the early cutaneous inflammation induced by bleomycin, consistent with the recognized inhibitory effects of PPAR-γ on immune and inflammatory responses.23 The mechanistic basis for the anti-inflammatory effects of PPAR-γ remains incompletely understood. In monocytes and macrophages, PPAR-γ ligands have been shown to suppress the production of tumor necrosis factor-α, IL-1, IL-6, and Cox, in part through blocking NF-κB activation.24,25,26,27,28 Indeed, accumulating evidence suggests that PPAR-γ represses a rage of signal-dependent transcription factors that activate proinflammatory genetic programs.
The attenuation of skin fibrosis in the bleomycin scleroderma model by rosiglitazone appears to be attributable to the direct anti-fibrotic effects of PPAR-γ in addition to suppression of the inflammatory response. Ligands of PPAR-γ blocked the stimulation of collagen production and collagen gene transcription elicited by TGF-β in vitro, and prevented myofibroblast differentiation of human lung and skin fibroblasts.12,13 Because the irreversible PPAR-γ antagonist GW9662 counteracted the anti-fibrotic effects of rosiglitazone in the present studies, we propose that these effects are mediated primarily via cellular PPAR-γ. Indeed, murine embryonic fibroblasts lacking PPAR-γ failed to show abrogation of TGF-β responses when treated with rosiglitazone (A. Ghosh and J. Varga, unpublished data). However, we cannot fully exclude a role for PPAR-γ-independent responses, such as induction of hepatocyte growth factor, a growth factor with potent anti-fibrotic activity.29,30 Because hepatocyte growth factor inhibits collagen synthesis and abrogates TGF-β signaling,31,32 its stimulation by rosiglitazone may play a role in attenuation of bleomycin-induced scleroderma. Induction of the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) by PPAR-γ may be an additional anti-fibrotic mechanism.33,34 PTEN inhibits fibroblast proliferation, collagen synthesis, and myofibroblast differentiation,35 and suppresses TGF-β production.36 Acting primarily as a lipid phosphatase, PTEN regulates signal transduction pathways mediated through PI3 kinase and Akt. The significance of PTEN as an endogenous inhibitor of fibrosis is underlined by the dramatic sensitivity of PTEN-null mice to bleomycin-induced fibrosis.35 Thus, PTEN induction by rosiglitazone could contribute to the blockade of TGF-β-induced profibrotic responses.
In agreement with our previous findings,15 we detected enhanced phospho-Smad2 expression in lesional fibroblasts in bleomycin-treated mice, indicative of their activation by TGF-β in situ. Interestingly, up-regulation of phospho-Smad2 was unaltered in rosiglitazone-treated mice. Failure to block Smad2 phosphorylation suggests that PPAR-γ ligands abrogated TGF-β-induced responses independent of Smad activation,12,13 and further indicates that the anti-fibrotic effects were not because of desensitization to TGF-β as would be the case if rosiglitazone suppressed TGF-β receptor expression, as shown for instance in hepatic stellate cells.37 In marked contrast to Smad2 activation however, up-regulation of Egr-1 was substantially reduced by rosiglitazone. Egr-1 is a TGF-β-inducible early immediate gene that functions as a transcription factor mediating Smad-independent TGF-β signal transduction in fibroblasts.38 Whereas Egr-1 is rapidly and transiently induced by inflammatory and stress signals, normally its expression is barely detectable in most tissues. In contrast, sustained Egr-1 expression was noted in lesional tissue from mice with bleomycin-induced scleroderma,39 suggesting that Egr-1 may contribute to the progression of the fibrotic response. The present results show that Egr-1 up-regulation in lesional skin was prevented by rosiglitazone, identifying Egr-1 as a target of inhibition by PPAR-γ, and implicating this transcription factor as a potentially important mediator of the anti-fibrotic effects of rosiglitazone.
The development of dermal fibrosis was accompanied by progressive loss of subcutaneous adipose layer, and its replacement by fibrous tissue. In marked contrast to PBS-treated mice that had abundant subcutaneous fat, lipoatrophy was consistently observed in the bleomycin-treated mice. Subcutaneous adipose atrophy is a characteristic histopathological feature of human scleroderma,40 as well as of TGF-β-driven scleroderma in transgenic mice.41 The basis for the loss of adipose tissue subjacent to dermal fibrosis is unclear. Potential mechanisms include loss of local adipocytes through apoptosis, impaired adipogenic differentiation of mesenchymal progenitor cells, or simply replacement and effacement of the soft adipose tissue by expansion of stiff connective tissue. It is of interest that subcutaneous adipose atrophy is prominent in, and generally precedes, cancer cachexia in animal models.42 In this setting, adipose atrophy is attributed to the loss of PPAR-γ and related adipogenic transcription factors in the local milieu, with consequent failure of adipogenic differentiation, rather than loss of adipocytes.42 The critical role of PPAR-γ in the maintenance of subcutaneous adipocyte homeostasis is further supported by the results of adipocyte-specific deletion of PPAR-γ. These mice develop subcutaneous lipoatrophy and concomitant accumulation of collagen-rich fibrous connective tissue at the same location.43,44 Similarly, in an animal model of cancer-associated lipoatrophy, the shrinking adipose layer is replaced by collagen-rich extracellular matrix.42 Cumulatively, these observations suggest that an inverse correlation may exist between adipogenesis and fibrogenesis, possibly mediated via the PPAR-γ-dependent commitment of mesenchymal precursor cells to the adipogenic or the fibrogenic lineage. In the present studies, bleomycin-induced dermal fibrosis was associated with loss of subcutaneous adipose tissue, but the process was primarily prevented when mice were pretreated with rosiglitazone. Because TGF-β is a potent inhibitor of both the expression and function of PPAR-γ (J. Wei and J. Varga, unpublished data),45,46,47,48 enhanced TGF-β signaling in bleomycin-treated mice could account for locally reduced PPAR-γ expression and function.
These observations also highlight a reciprocally antagonistic relationship between the TGF-β and PPAR-γ signaling pathways. On the one hand, PPAR-γ activation in fibroblasts by natural or synthetic ligands, and even ectopic expression in the absence of exogenous ligand, blocks TGF-β responses such as stimulation of collagen synthesis, α-smooth muscle actin expression, and cell migration.12,13 Antagonism by PPAR-γ involves blockade of Smad2/3-dependent transcriptional responses without preventing ligand-induced Smad activation and nuclear accumulation (A. Ghosh and J. Varga, unpublished data). Moreover, PPAR-γ suppresses the synthesis and secretion of TGF-β in vitro and in vivo by multiple mechanisms, including induction of PTEN,14,36,49,50 and rosiglitazone treatment reduced the accumulation of TGF-β in the lesional skin. Thus, PPAR-γ ligands exert dual antagonistic effects on TGF-β in the context of fibrogenesis by directly disrupting TGF-β signal transduction, and by suppressing TGF-β production. On the other hand, TGF-β itself negatively regulates both the expression and function of PPAR-γ, thereby desensitizing fibroblasts to PPAR-γ ligands. Our present results suggest that whereas PPAR-γ receptor expression is diminished in lesional tissues in bleomycin-induced mouse scleroderma, concurrent administration of rosiglitazone prevents PPAR-γ loss, thereby preserving rosiglitazone sensitivity even in fibrosis. The complex TGF-β/PPAR-γ relationship is likely to have important consequences for physiological connective tissue homeostasis, and tissue fibrosis.51 The balance between TGF-β and PPAR-γ signaling may be highly amenable to therapeutic manipulation using currently available pharmacological ligands such as rosiglitazone, or selective PPAR-γ agonists that are under development. These approaches merit further investigation for the treatment of fibrotic disorders.
Acknowledgments
We thank Robert Schleimer and Warren Tourtellotte and the members of the Varga Laboratory for helpful suggestions.
Footnotes
Address reprint requests to John Varga, Section of Rheumatology, Northwestern University Feinberg School of Medicine, 240 E. Huron St., Chicago IL 60611. E-mail: j-varga@northwestern.edu.
Supported by the National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin Diseases grants AR-42309 and AR-49025) and the Scleroderma Research Foundation.
References
- Jimenez S, Derk C. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–50. [PubMed] [Google Scholar]
- Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles C, Clements P, Furst DE. Systemic sclerosis: hypothesis-driven treatment strategies. Lancet. 2006;367:1683–1691. doi: 10.1016/S0140-6736(06)68737-0. [DOI] [PubMed] [Google Scholar]
- Abraham D, Eckes B, Rajkumar V, Krieg T. New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma. Curr Rheumatol Rep. 2007;9:136–143. doi: 10.1007/s11926-007-0008-z. [DOI] [PubMed] [Google Scholar]
- Trojanowska M, Varga J. Molecular pathways as novel therapeutic targets in systemic sclerosis. Curr Opin Rheumatol. 2007;19:568–573. doi: 10.1097/BOR.0b013e3282e6f495. [DOI] [PubMed] [Google Scholar]
- Dang H, Geiser A, Letterio J, Nakabayashi T, Kong L, Fernandes G, Talal N. SLE-like autoantibodies and Sjögren’s syndrome-like lymphoproliferation in TGF-beta knockout mice. J Immunol. 1995;155:3205–3212. [PubMed] [Google Scholar]
- Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman B. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Heikkinen S, Auwerx J, Argmann CA. PPARgamma in human and mouse physiology. Biochim Biophys Acta. 2007;1771:999–1013. doi: 10.1016/j.bbalip.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J Clin Invest. 2006;116:598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50:1305–1318. doi: 10.1002/art.20104. [DOI] [PubMed] [Google Scholar]
- Burgess H, Daugherty L, Thatcher T, Lakatos H, Ray D, Redonnet M, Phipps R, Sime P. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol. 2005;288:L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- Milam J, Keshamouni V, Phan S, Hu B, Gangireddy S, Hogaboam C, Standiford T, Thannickal V, Reddy R. PPAR-{gamma} agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol. 2008;294:L891–L901. doi: 10.1152/ajplung.00333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagawa S, Lakos G, Mori Y, Yamamoto T, Nishioka K, Varga J. Sustained activation of fibroblast transforming growth factor-beta/Smad signaling in a murine model of scleroderma. J Invest Dermatol. 2003;121:41–50. doi: 10.1046/j.1523-1747.2003.12308.x. [DOI] [PubMed] [Google Scholar]
- Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, Wang XJ, DiPietro LA, Varga J. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165:203–217. doi: 10.1016/s0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Zacarías J, Castro-Muñozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97:493–497. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
- Bou-Gharios G, Garrett LA, Rossert J, Niederreither K, Eberspaecher H, Smith C, Black C, Crombrugghe B. A potent far-upstream enhancer in the mouse pro alpha 2(I) collagen gene regulates expression of reporter genes in transgenic mice. J Cell Biol. 1996;134:1333–1344. doi: 10.1083/jcb.134.5.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Bhattacharyya S, Varga J. The tumor suppressor p53 abrogates Smad-dependent collagen gene induction in mesenchymal cells. J Biol Chem. 2004;279:47455–47463. doi: 10.1074/jbc.M403477200. [DOI] [PubMed] [Google Scholar]
- Wu M, Varga J. In perspective: mouse models of scleroderma. Curr Rheumatol Rep. 2008;10:173–182. doi: 10.1007/s11926-008-0030-9. [DOI] [PubMed] [Google Scholar]
- Li M, Wan Y, Sanjabi S, Robertson A, Flavell R. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Wollheim F. Treatment of pulmonary fibrosis in systemic sclerosis: light at the end of the tunnel? Arthritis Rheum. 2007;56:9–12. doi: 10.1002/art.22315. [DOI] [PubMed] [Google Scholar]
- Henson P. Suppression of macrophage inflammatory responses by PPARs. Proc Natl Acad Sci USA. 2003;100:6295–6296. doi: 10.1073/pnas.1232410100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus D, Pascual G, Li M, Welch J, Ricote M, Hsiang C, Sengchanthalangsy L, Ghosh G, Glass C. 15-Deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro M. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans R. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li A, Willson T, Kelly C, Glass C. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting A, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Li Y, Wen X, Spataro B, Hu K, Dai C, Liu Y. Hepatocyte growth factor is a downstream effector that mediates the antifibrotic action of peroxisome proliferator-activated receptor-gamma agonists. J Am Soc Nephrol. 2006;17:54–65. doi: 10.1681/ASN.2005030257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogatkevich G, Ludwicka-Bradley A, Highland K, Hant F, Nietert P, Singleton C, Silver R. Down-regulation of collagen and connective tissue growth factor expression with hepatocyte growth factor in lung fibroblasts from white scleroderma patients via two signaling pathways. Arthritis Rheum. 2007;56:3468–3477. doi: 10.1002/art.22874. [DOI] [PubMed] [Google Scholar]
- Wu M, Yokozeki H, Takagawa S, Yamamoto T, Satoh T, Kaneda Y, Katayama I, Nishioka K. Hepatocyte growth factor both prevents and ameliorates the symptoms of dermal sclerosis in a mouse model of scleroderma. Gene Ther. 2004;11:170–180. doi: 10.1038/sj.gt.3302165. [DOI] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Mimura Y, Asano Y, Yamane K, Tamaki K. Effects of hepatocyte growth factor on the expression of type I collagen and matrix metalloproteinase-1 in normal and scleroderma dermal fibroblasts. J Invest Dermatol. 2005;124:324–330. doi: 10.1111/j.0022-202X.2004.23601.x. [DOI] [PubMed] [Google Scholar]
- Patel L, Pass I, Coxon P, Downes C, Smith S, Macphee C. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Lee K, Park S, Hwang P, Yi H, Song C, Chai O, Kim J, Lee M, Lee Y. PPAR-gamma modulates allergic inflammation through up-regulation of PTEN. FASEB J. 2005;19:1033–1035. doi: 10.1096/fj.04-3309fje. [DOI] [PubMed] [Google Scholar]
- White E, Atrasz R, Hu B, Phan S, Stambolic V, Mak T, Hogaboam C, Flaherty K, Martinez F, Kontos C, Toews G. Negative regulation of myofibroblast differentiation by PTEN (phosphatase and tensin homolog deleted on chromosome 10). Am J Respir Crit Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yang E, Kim S. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. 2006;70:415–425. doi: 10.1124/mol.106.022954. [DOI] [PubMed] [Google Scholar]
- Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384:149–157. doi: 10.1042/BJ20040928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Ning H, Ishida W, Sodin-Semrl S, Takagawa S, Mori Y, Varga J. The early-immediate gene EGR-1 is induced by transforming growth factor-beta and mediates stimulation of collagen gene expression. J Biol Chem. 2006;281:21183–21197. doi: 10.1074/jbc.M603270200. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Chen SJ, Wu M, Warner-Blankenship M, Ning H, Lakos G, Mori Y, Chang E, Nihijima C, Takehara K, Feghali-Bostwick C, Varga J. Smad-independent transforming growth factor-β regulation of early growth response-1 and sustained expression in fibrosis. Implications for scleroderma. Am J Pathol. 2008;173:1085–1099. doi: 10.2353/ajpath.2008.080382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder DE, editor. Philadelphia: Lippincott Williams & Wilkins,; Lever’s Histology of the Skin, (ed 9.) 2005:pp 310–311. [Google Scholar]
- Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, Vanpelt CS, Geng YJ, Deng JM, Behringer RR, de Crombrugghe B. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007;56:334–344. doi: 10.1002/art.22328. [DOI] [PubMed] [Google Scholar]
- Bing C, Russell S, Becket E, Pope M, Tisdale MJ, Trayhurn P, Jenkins JR. Adipose atrophy in cancer cachexia: morphologic and molecular analysis of adipose tissue in tumour-bearing mice. Br J Cancer. 2006;95:1028–1037. doi: 10.1038/sj.bjc.6603360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky J, Evans R. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci USA. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Ivashchenko C, Whitesall S, D'Alecy L, Duquaine D, Brosius F, Gonzalez F, Vinson C, Pierre M, Milstone D, Mortensen R. Hypotension, lipodystrophy, and insulin resistance in generalized PPARgamma-deficient mice rescued from embryonic lethality. J Clin Invest. 2007;117:812–822. doi: 10.1172/JCI28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KM, Burdick MD, Phillips RJ, Heber D, Strieter RM. Characterization of human fibrocytes as circulating adipocyte progenitors and the formation of human adipose tissue in SCID mice. FASEB J. 2005;19:2029–2031. doi: 10.1096/fj.05-4295fje. [DOI] [PubMed] [Google Scholar]
- Fu M, Zhang J, Zhu X, Myles DE, Willson TM, Liu X, Chen YE. Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J Biol Chem. 2001;276:45888–45894. doi: 10.1074/jbc.M105490200. [DOI] [PubMed] [Google Scholar]
- Zheng S, Chen A. Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-gamma in rat hepatic stellate cells. Am J Physiol. 2007;292:G113–G123. doi: 10.1152/ajpgi.00200.2006. [DOI] [PubMed] [Google Scholar]
- Coras R, Holsken A, Seufert S, Hauke J, Eyupoglu IY, Reichel M, Trankle C, Siebzehnrubl FA, Buslei R, Blumcke I, Hahnen E. The peroxisome proliferator-activated receptor-gamma agonist troglitazone inhibits transforming growth factor-beta-mediated glioma cell migration and brain invasion. Mol Cancer Ther. 2007;6:1745–1754. doi: 10.1158/1535-7163.MCT-06-0763. [DOI] [PubMed] [Google Scholar]
- Weigert C, Brodbeck K, Bierhaus A, Häring H, Schleicher E. c-Fos-driven transcriptional activation of transforming growth factor beta-1: inhibition of high glucose-induced promoter activity by thiazolidinediones. Biochem Biophys Res Commun. 2003;304:301–307. doi: 10.1016/s0006-291x(03)00599-0. [DOI] [PubMed] [Google Scholar]
- Honda K, Marquillies P, Capron M, Dombrowicz D. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol. 2004;113:882–888. doi: 10.1016/j.jaci.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Lakatos H, Thatcher T, Kottmann R, Garcia T, Phipps R, Sime P. The role of PPARs in lung fibrosis. PPAR Res. 2007;2007:71323. doi: 10.1155/2007/71323. [DOI] [PMC free article] [PubMed] [Google Scholar]