

Summary
Activated macrophages and their inflammatory products play a key role in innate immunity and in pathogenesis of autoimmune/inflammatory diseases. Macrophage activation needs to be tightly regulated to rapidly mount responses to infectious challenges but to avoid toxicity associated with excessive activation. Rapid and potent macrophage activation is driven by cytokine-mediated feedforward loops, while excessive activation is prevented by feedback inhibition. Here we discuss feedforward mechanisms that augment macrophage responses to Toll-like receptor (TLR) ligands and cytokines that are mediated by signal transducer and activator of transcription 1 (STAT1) and induced by interferon-γ (IFN-γ). IFN-γ also drives full macrophage activation by inactivating feedback inhibitory mechanisms, such as those mediated by IL-10 and STAT3. Priming of macrophages with IFN-γ reprograms cellular responses to other cytokines, such as type I IFNs and IL-10, with a shift toward pro-inflammatory STAT1-dominated responses. Similar but partially distinct priming effects are induced by other cytokines that activate STAT1, including type I IFNs and interleukin-27. We propose a model whereby opposing feedforward and feedback inhibition loops crossregulate each other to fine tune macrophage activation. In addition, we discuss how dysregulation of the balance between feedforward and feedback inhibitory mechanisms can contribute to the pathogenesis of autoimmune and inflammatory diseases, such as rheumatoid arthritis and systemic lupus erythematosus.
Keywords: monocytes/macrophages, cytokine receptors, Toll-like receptors/pattern-recognition receptors, signal transduction, inflammation
Introduction
Macrophages play a key role in host defense and inflammation. Macrophage activation is a complex process involving signal transduction events from multiple inflammatory mediators, including exogenous factors such as pathogen-associated molecules and endogenous mediators such as cytokines and chemokines. Cytokines are major regulators of macrophage activation that fine tune macrophage responses to achieve effective clearance of pathogens, while limiting the amount of inflammation to avoid toxicity and tissue damage. Cytokines also regulate the qualitative nature of macrophage activation to coordinate the most effective innate and acquired immune responses to clear the invading pathogen. Cytokines that signal via the Janus kinase (Jak)-signal transduction and activator of transcription (STAT) pathway are key regulators of macrophage activation and function. Interferon-γ (IFN-γ) is a major macrophage activator via STAT1, whereas IL-10 is a strong deactivator of macrophages via STAT3 that opposes STAT1 functions.
Macrophage host defense functions need to be mobilized rapidly after infectious challenge, and it is becoming increasingly clear that this rapid mobilization is facilitated by feedforward loops that are engaged by cytokines to rapidly amplify inducing signals. The best characterized feedforward loops are mediated by induction of autocrine factors, including cytokines, that work synergistically to activate macrophages. In this review, we focus on an alternative intracellular feedforward loop that is activated by IFN-γ and mediated by STAT1 (Fig. 1). This STAT1-mediated feedforward loop amplifies not only signaling by cytokines but also enhances macrophage responses to microbial inducers such as Toll-like receptor (TLR) ligands. At the same time macrophages are activated, feedback inhibitory loops are engaged to restrain the amplitude of activation and thus prevent toxicity associated with excessive macrophage activation. A major feedback inhibitory loop induced by activating cytokines such as IFN-γ is the expression of suppressor of cytokine signaling (SOCS) proteins that are inhibitors of signal transduction (Fig. 1). Microbial products such as TLR ligands activate another well known feedback inhibitory loop that is mediated by autocrine-acting interleukin-10 (IL-10) that activates STAT3 (Fig. 2). In this review, we develop the concepts that opposing feedforward and feedback inhibition loops crossregulate each other to fine tune macrophage activation (Fig. 1) and that IFN-γ can promote full macrophage activation by TLRs by inactivating feedback inhibitory loops, such as those mediated by IL-10 and STAT3 (Fig. 2). Since regulatory mechanisms of individual signaling pathways have been extensively reviewed elsewhere, here we focus on crossregulation of cytokine-cytokine signaling and cytokine-TLR signaling, with a particular emphasis on regulation by IFN-γ and other cytokines that activate STAT1-mediated regulation, such as type I IFNs (IFN-α/β) and IL-27. In addition, we will discuss how dysregulation of the balance between feedforward and feedback inhibitory mechanisms can contribute to the pathogenesis of autoimmune and inflammatory diseases, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE).
Fig. 1.

Cytokines activate opposing negative and positive feedback loops.
Fig. 2. TLRs activate opposing negative and positive feedback loops.
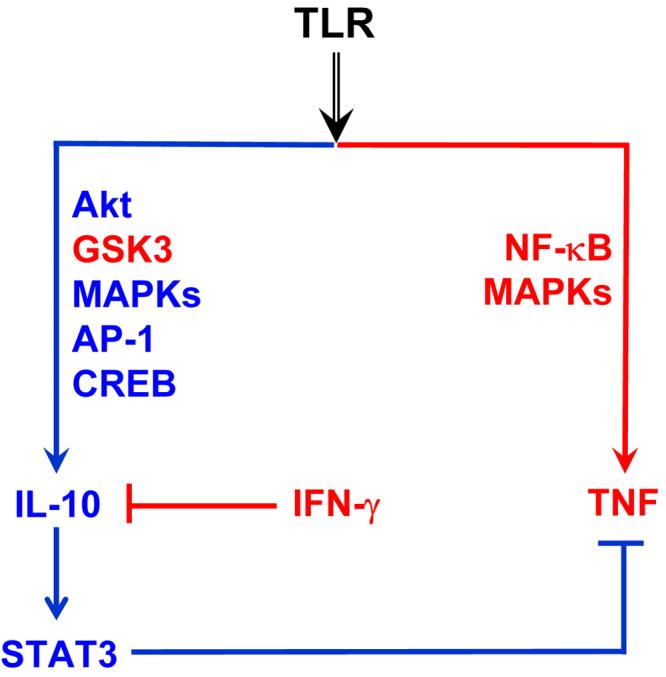
TLRs induce production of both inflammatory cytokines such as TNF and anti-inflammatory molecules such as IL-10. IL-10 inhibits production of TNF via a STAT3-dependent feedback loop. IFN-γ primes for enhanced TNF production by disruption of IL-10-STAT3 feedback loop.
Priming of Jak-STAT signaling by IFN-γ: a feedforward loop mediated by STAT1
IFNs were originally discovered as agents that interfere with viral replication and are now classified into type I and type II according to receptor specificity and sequence homology. Both types of IFNs belong to the class II cytokine family, which also includes IFN-λs (also called IL-28 and IL-29) and IL-10-related cytokines (1). Type I IFNs are comprised of IFN-α, IFN-β, IFN-ω, and IFN-τ, are induced in many cell types by viral or bacterial products, and have potent antiviral effects (2). In addition to antiviral effects, type I IFNs modulate many aspects of immune and inflammatory reactions (2).
IFN-γ is the sole type II IFN and is a key endogenous activator of macrophages. IFN-γ exerts its biological activities by binding to a specific cell surface receptor that is distinct from IFN-α/β receptor. The functional IFN-γ receptor consists of two chains of the ligand-binding subunit (IFNGR1) and two chains of the signal transducing subunit (IFNGR2). As with most cytokines that utilize the Jak-STAT pathway, two Jak protein tyrosine kinases, Jak1 and Jak2, are involved in IFN-γ signal transduction. Jak1 and Jak2 constitutively bind IFNGR1 and IFNGR2, respectively. Upon receptor engagement and Jak activation, an essential tyrosine residue on IFNGR1 (Y440) is phosphorylated and serves as docking site for STAT1. STAT1 is then activated by tyrosine phosphorylation and dissociates from receptor complex. STAT1:STAT1 homodimers are the major STAT species mediating IFN-γ transcriptional program (3).
A key aspect of IFN biology is the concept of ‘priming’, whereby type I or type II IFNs induce increased cellular responsiveness to several extracellular stimuli, including IFNs themselves, IL-6, microbial products such as lipopolysaccharide (LPS), and viruses (4). Priming can be effectively achieved by low concentrations of IFNs that do not actually activate cells. The classical function of priming is to potentiate early innate immune responses that occur when cytokine concentrations are low, thereby enhancing host defense. Priming is felt to ‘rev up’ the immune response to ensure rapid and strong responses to environmental challenges such as infections, and a role for priming has been implicated in antiviral and antibacterial responses (4, 5). However, it has become clear that priming, such as occurs during a viral infection, also results in hyper-responsiveness on subsequent exposure to microbes or environmental antigens. This hyper-responsiveness can have deleterious consequences in terms of increased tissue damage and even increased lethality secondary to excessive cytokine production (6-8). We and others (4, 9-12) have considered the possibility that priming may also occur during the course of autoimmune and inflammatory diseases, in which case enhanced inflammatory responses to cytokines and other activating factors could contribute to pathogenesis. The IFN levels expressed in the tissues and blood of RA and SLE patients are sufficient to achieve priming (13), and the evidence suggests that priming occurs in these diseases and may contribute to pathogenesis via enhanced responses to minor infections, auto-antigens, environmental antigens, components of damaged tissues, or cytokines.
Although IFN priming was first described in the 1970s, understanding of underlying mechanisms is limited (5). IFN-γ priming of tumor necrosis factor (TNF) responses is mediated, at least in part, by extended activation of nuclear factor-κB (NF-κB) in primed cells and cooperation between NF-κB and STAT1 in the activation of gene promoters. Priming for production of large amounts of type I IFNs is mediated by an autoamplification loop in which IFN-α induces expression of the transcription factor IFN regulator factor 7 (IRF7) that in turn activates IFN-α gene promoters. Recently, there has been progress in understanding how IFNs prime responses of cells to IFNs and other cytokines that use the Jak-STAT pathway, including published work from our laboratory (4, 11, 14-16). A common theme is that priming results in increased activation of STAT1, with its attendant inflammatory actions. STAT1 further amplifies cell activation by activating STAT1-mediated feedforward mechanisms and by counteracting feedback pathways such as those mediated by SOCS proteins or STAT3. However, there appear to be several mechanisms that mediate enhanced STAT1 activation that may function in a cell type-specific manner. In the following sections, we mainly discuss IFN priming of cytokine signaling and responses, which contributes to the pro-inflammatory properties of IFNs.
Autoamplification of IFN-γ signaling
It has been increasingly appreciated that intracellular signal transduction events can be positively modulated or sensitized through self-activated feedforward loops. For example, IFN-γ signaling can be amplified in a positive feedforward loop involving the induction of two IRF family proteins, IRF1 and IRF8, also termed for IFN consensus sequence-binding protein (ICSBP). IRF1 and ICSBP, when induced by IFN-γ through STAT1, in turn generate a second wave of transcription of IFN target genes, thereby contributing to full-fledged IFN-γ transcriptional responses (4, 17). Another example of such a positive self-regulatory loop is the induction of IFN-stimulated gene 15 (ISG15) by IFNs and positive regulation of IFN signaling by IFN-activated ISG15. ISG15, a small protein homologous to ubiquitin, can be conjugated to cellular proteins in a process called ISGylation, and ISGylation in turn greatly enhances IFN-activated Jak-STAT signaling (18).
Autoregulation of IFN-γ signaling by IFN-γ appears to be complex and cell-type specific, as IFN-γ suppresses IFN-γ signaling in lymphocytes by downregulating expression of the IFN-γ receptor (19, 20). In contrast to the observations in lymphocytes, our group has reported that during macrophage activation, IFN-γ signaling is actually sensitized in a positive feedforward loop by low doses of IFN-γ by a mechanism that involves increased STAT1 expression (11). In this model, low concentrations of IFN-γ do not actually activate macrophages, but instead transiently induce expression of a small subset of IFN-γ-inducible genes, including STAT1. STAT1 protein accumulates in primed cells because of ongoing gene expression and the stability of STAT1 protein, which exhibits a half life of greater than 24 h. Primed macrophages, then, appear to be quiescent but strongly activate STAT1 upon rechallenge with even very small amounts of IFN-γ, with concomitant activation of downstream STAT1-dependent genes and inflammatory functions. IFN-γ is a major activator of macrophages, and sensitization of IFN-γ signaling may be particularly important to achieve full macrophage activation early in immune responses when IFN-γ levels are low.
Regarding the mechanisms of IFN-γ signaling sensitization, several lines of evidence support a role for increased STAT1 expression in this process. First, sensitization is not accompanied by any changes in expression of IFN-γ receptors or in the levels of activation of Jak1, Jak2, or STAT3 by IFN-γ. These results indicate that IFN-γ delivers a comparable proximal signal to both non-primed and primed macrophages. Second, the rate of de-activation of STAT1 is comparable in non-primed and primed cells, indicating that priming does not inactivate a STAT1 phosphatase or suppress degradation by proteasomes. Third, sensitization of signaling is specific for STAT1 relative to STAT3 when either IFN-γ or IFN-α were used to stimulate primed cells, consistent with the relative expression levels of these STATs, and increased STAT1 activation was recapitulated by forced expression of STAT1. These data argue for a model where an increased intracellular STAT1 concentration leads to more efficient docking onto the activated IFN-γ receptor complex.
An important component of this model is that low priming doses of IFN-γ capable of activating sustained STAT1 expression only transiently and weakly activate expression of SOCS1, which is a highly unstable protein, and do not effectively activate feedback inhibition by SOCS1. STAT1 activation proceeds in primed macrophages unopposed by SOCS1. In contrast, high activating doses of IFN-γ induce sustained expression of SOCS1 and thus engage feedback inhibition that counterbalances STAT1 activation (Fig. 3). Furthermore, STAT1 and SOCS1 bind to the same docking site on the IFNGR, thus suggesting direct opposition mediated by competition for binding to a limiting number of IFNγR molecules. These results suggest a model of regulation of IFN signaling, whereby the amplitude of signaling is regulated by relative activities of positive signaling molecules (STAT1 in case of IFN-γ) versus negative signaling molecules (SOCS1 in case of IFN-γ). The net outcome of a particular signaling event is not solely determined by the strength of positive signaling but instead is determined by the balance between positive and negative arms downstream of the receptor (Fig. 1). In addition to the model proposed by Taniguchi (4) where priming can ‘rev up’ the immune response to make the system ready for secondary challenges, our model suggests a complementary scenario where IFN priming drives a positive feedforward loop and thus enables cells to elicit a strong primary response.
Fig. 3. The effects on signaling sensitization by low concentrations versus high concentrations of IFN-γ.
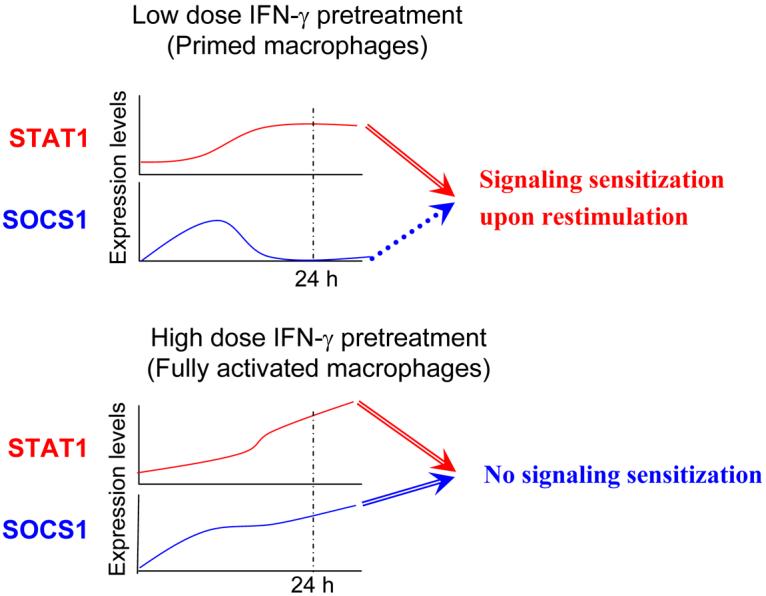
Low doses of IFN-γ engage positive but not negative feedback loops. High doses of IFN-γ result in sustained activation of feedback inhibitory molecules such as SOCS1 and thus can not prime cells upon restimulation.
STAT1 expression is dynamically regulated during the course of immune and inflammatory responses, and STAT1 expression levels thus can regulate the pattern of STAT activation by a cytokine over time. For example, elevation of STAT1 expression during the course of a viral infection results in increased STAT1 activation and diminished STAT4 activation in response to type I IFNs (21). Interestingly, elevated STAT1 mRNA and/or protein levels have been detected in several autoimmune/inflammatory conditions, including SLE, RA, and hepatitis (11, 22-26). Thus, increased Stat1 expression in these conditions will alter cellular responses to cytokines such as IFN-γ. The role for IFN-γ in RA has been controversial, and IFN-γ protein has been difficult to detect in RA joints (27, 28). Recent work demonstrating that RA synovial cells express high levels of STAT1 and also express IFN-γ-inducible genes (9, 11, 29, 30, authors' unpublished data) supports the notion that low levels of IFN-γ may activate gene expression through the above described autosensitization mechanism in RA synovium.
IFN-γ priming of IFN-α-STAT1 signaling
Type I IFNs possess many immune-activating functions. Type I IFNs can promote innate immunity by activating natural killer (NK) cells and inducing production of cytokines and chemokines, can promote the transition from innate to acquired immunity by driving the differentiation and maturation of dendritic cells (DCs), resulting in enhanced antigen presentation, and can regulate acquired immunity by suppressing apoptosis of lymphocytes, promoting T-helper 1 (Th1) responses and by enhancing antibody production, class switching, and immunological memory. Like most cytokines, type I IFNs are pleiotropic and also have suppressive effects on certain aspects of immunity and inflammation. The suppressive effects of type I IFNs can predominate in certain (patho)physiological settings, and type I IFNs are used to treat multiple sclerosis. The mechanisms underlying the suppressive effects of type I IFNs include suppression of cell proliferation, suppression of IL-12 and TNF production, and, under specific conditions, suppression of IFN-γ production. Recently, IL-27-dependent suppression of Th17 responses has emerged as another mechanism that may contribute to the beneficial effects of type I IFNs in central nervous system inflammation (31, 32).
The type I IFNs all utilize a ubiquitously expressed heterodimeric IFN-α/β receptor and generate similar, although not necessarily identical, signals. The IFN-α/β receptor consists of IFNAR1 and IFNAR2 subunits that are associated with cytoplasmic protein tyrosine kinases Tyk2 and Jak1, respectively. IFNAR2 is also constitutively associated with STAT2 (33). Ligation of the IFN-α/β receptor results in activation of Tyk2 and Jak1 and phosphorylation of tyrosine residues in the IFN-α/βR cytoplasmic domains, thus creating docking sites for STATs, including STAT2 and likely STAT1. STAT1 and STAT2 are activated at the IFN-α/βR signaling complex by tyrosine phosphorylation, with subsequent formation of STAT1:STAT2 heterodimers and STAT1:STAT1 homodimers. These STAT proteins translocate to the nucleus, where STAT1:STAT1 homodimers bind to γ-activated sequence (GAS) sites (consensus sequence TTCNNNGAA) in gene promoters and activate transcription. In contrast, STAT1:STAT2 heterodimers associate with IRF9 to form the ISGF3 complex that binds to an IFN-stimulated response element (ISRE) site (consensus sequence TTTCNNTTTC). Thus, type I IFNs activate expression of genes that have either GAS or ISRE sites or both in their promoters (33).
Type I IFNs have been shown to prime cells for strong responses to IFN-γ (11, 14). One mechanism of priming involves association of a tyrosine phosphorylated chain of the IFNα/β receptor, IFNAR1, with the IFN-γ receptor in caveolar membrane domains (14). IFNAR1:IFNGR interaction leads to enhanced dimerization of STATs that have been tyrosine-phosphorylated in response to IFN-γ stimulation. Notably, priming by this mechanism does not result in increased STAT tyrosine phosphorylation but instead results in increased dimerization of tyrosine-phosphorylated STATs and thus increased DNA binding activity and gene transcription. An alternative complementary mechanism of priming involves IFN-α-dependent increases in STAT1 expression, thus resulting in increased STAT1 interaction with IFN-γ receptor docking sites and increased levels of tyrosine phosphorylation when primed cells are stimulated with IFN-γ (11) (similar to the mechanism of autoamplification of IFN-γ signaling discussed above).
To extend the above studies, we investigated the reciprocal phenomenon, namely IFN-γ priming for IFN-α signaling. Our laboratory reported that in IFN-γ-primed macrophages that express high STAT1 levels, IFN-α-induced activation of STAT1 is amplified evidenced by increased STAT1 tyrosine phosphorylation and STAT1 DNA-binding activities (15). However, IFN-γ does not significantly alter the activation of STAT2 and STAT3 downstream of IFNAR, resulting in STAT1 being the predominant signaling molecule activated by IFN-α in IFN-γ-primed macrophages. The shift in IFN-α-induced STAT activation toward increased STAT1 relative to STAT2 and STAT3 activation results in enhanced expression of STAT1-dependent inflammatory genes such as the chemokines CXCL9 and CXCL10 and a shift to a more inflammatory phenotype. Preferential activation of STAT1 and subsequent stronger pro-inflammatory macrophage responses to IFN-α might have a role in the pathogenesis of IFN-mediated diseases such as SLE (16). In contrast to the IFN-γ signaling autosensitization where high STAT1 levels are both necessary and sufficient to mediate priming, increased STAT1 expression is necessary but not sufficient for hyperactivation of STAT1 downstream of IFNAR. Our results show that additional signaling input from immunoreceptor tyrosine-based activation motif (ITAM)-coupled receptors is required to achieve full STAT1 activation. We proposed a model whereby IFN-γ induces expression of an ITAM-coupled receptor that mediates interactions with IFNAR. In addition, ITAM-activated Syk kinase can phosphorylate STAT1 directly, providing a mechanism by which increased STAT1 levels can directly augment STAT1 activation by promoting interaction with Syk (15) (Fig. 4). Crosstalk between ITAM-dependent pathways and Jak-STAT signaling has been reviewed elsewhere (34) and is not discussed extensively here.
Fig. 4. Model for coupling Syk to IFN-α and STAT1 signaling by IFN-γ priming.

IFN-γ priming leads to increased levels of intracellular STAT1. Syk could directly phosphorylate STAT1 and thus result in enhanced STAT1 activation. Alternatively, Syk could phosphorylate potential new STAT1 docking sites on IFNAR and promote recruitment of STAT1 to the receptor.
Given the pleiotropic nature of type I IFNs and relative limited signaling pathways downstream of IFN-α/β receptor, it is intriguing how activating and suppressive effects of type I IFNs are orchestrated during immune responses. Our studies provide an example where regulation of IFN-α/β receptor signaling by other factors contributes to plasticity of IFN biology. The more inflammatory phenotype acquired after priming may also contribute to the pathogenesis of autoimmune/inflammatory disorders such as SLE, where IFNs play a prominent role. The relevance of priming with disease pathogenesis is discussed in more details in the last section of this review. In summary, IFN-γ is capable of priming for STAT1 signaling by both type I IFNs and IFN-γ itself, although the mechanisms are partially distinct (Fig. 5).
Fig. 5. Positive and negative regulation of IFN signaling.

This model describes auto- and cross-regulatory mechanisms that modulate intracellular signaling by type I and type II IFNs. (1) Positive feedback regulation of IFN-γ signaling by increased expression of STAT1. (2) Negative feedback inhibition of IFN-γ signaling by induction of SOCS. (3) IFN-γ-mediated increase in STAT1 levels enhances STAT1 activation by IFN-α. (4) IFN-γ priming couples Syk kinase to IFN-α signaling, resulting in enhanced STAT1 activation by IFN-α. (5) Constitutive subthreshold IFN-α signaling and the association of IFNGR2 and IFNAR1 facilitate activation of STAT1 by IFNGR.
Reprogramming of IL-10 signaling by IFNs
One emerging concept in cytokine signaling is that not only the quantity of signal transduction can be positively or negatively regulated but also the nature of cytokine responses can be altered or reprogrammed. Thus, the exact cellular response to a cytokine is determined by the presence of a particular stimulus as well as the microenvironment to which cells are exposed. One such example is the reprogramming of IL-10 signaling by IFN-α and IFN-γ (35, 36). IL-10 is a potent immunosuppressive and anti-inflammatory cytokine that deactivates macrophages and dendritic cells (37). Mechanisms of IL-10 action include suppression of antigen presentation and of production of cytokines and inflammatory mediators. IL-10 is produced as part of the homeostatic response to infection and inflammation and plays a critical role in limiting the duration and intensity of immune and inflammatory reactions (37). IL-10 production is tightly regulated, as excessive IL-10 leads to inability to control infectious pathogens, while insufficient IL-10 leads to pathology secondary to tissue injury. IL-10 binds to a heterodimeric receptor consisting of IL-10R1 and IL-10R2 subunits and activates receptor-associated Jak1 and Tyk2 kinases, leading to activation of STATs. IL-10 activates predominantly STAT3 in myeloid cells (38, 39), and STAT3 is required for many anti-inflammatory effects of IL-10 in these cells (40, 41). The immunosuppressive potency of IL-10 depends upon the timing of IL-10 expression, and IL-10 suppressive activity can become diminished during immune and inflammatory responses. For example, IL-10 effectively suppressed cytokine production when given before, but not when given after, LPS in experimental endotoxemia (42). Similarly, IL-10 activity became diminished during establishment of a chronic infection with the LP-BM5 retrovirus (43). Thus, modulation of IL-10 activity plays a role in regulation of the intensity and chronicity of immune and inflammatory reactions. Diminished IL-10 activity during the active phase of an infection can be beneficial to the host, as it will result in enhanced immunity and clearance of pathogens (44).
IL-10 is a weak activator of STAT1 in myeloid cells and does not normally induce STAT1 target genes as does IFN-γ. However, we showed that pre-exposure to type I IFNs reprograms STAT activation by IL-10 such that STAT1 is preferentially activated by IL-10 in these cells, resulting in induction of a group of STAT1-dependent genes and thereby a gain of inflammatory function (35). IFN-γ is also able to switch the balance of IL-10 STAT activation from STAT3 to STAT1, and IFN-γ's action is two fold. First, IFN-γ suppresses STAT3 function, with concomitant downregulation of STAT3-dependent gene expression including feedback regulator SOCS3, and partial attenuation of IL-10 anti-inflammatory function (36). In the presence of IFN-γ, IL-10 is less effective at suppressing cytokine and chemokine production and in downregulating major histocompatibility complex (MHC) class II expression. Thus, during the active phase of an immune or inflammatory response characterized by high levels of IFN-γ production, IL-10 activity would be restrained, such that pathogens could be effectively cleared. This type of block in IL-10 activity has been observed in vivo under conditions where IFN-γ is expressed (42, 45, 46). Since IL-10 is a major deactivator of macrophages that mediates a key feedback inhibitory loop (Fig. 2), this type of downregulation of STAT3 functions implies that high STAT1 levels can attenuate IL-10/STAT3-mediated feedback inhibition; this idea is further considered below in the context of TLR signaling.
Second, IFN-γ priming leads to predominant STAT1 activation by IL-10 and thus redirects IL-10 signaling from activation of STAT3, which is anti-inflammatory in macrophages, to activation of STAT1, which is pro-inflammatory. Thus, IFN-γ co-opts IL-10 to signal more like IFN-γ itself and allows IL-10 to activate STAT1 at a time when IFN-γ activation of STAT1 has been downregulated by feedback inhibition (11). Activation of STAT1 may mediate some of the pro-inflammatory functions of IL-10 that have been described during inflammation in vivo (42, 47-50) and may help to explain the lack of efficacy of IL-10 as an anti-inflammatory therapeutic agent in treatment of inflammatory disorders such as RA and Crohn's disease (51). It appears that IFNs operate a switch that regulates STAT activation by IL-10 and alters macrophage responses to IL-10. A switch in cytokine activity that is induced by an antagonistic cytokine adds an additional level of complexity to cytokine crossregulation that goes beyond simple inhibition of signaling. Despite both being manifested by strong STAT1 activation, the mechanisms of IFN-α- and IFN-γ-mediated reprogramming of IL-10 signaling may be distinct and would be interesting subjects for future investigation.
We have described so far IFN-γ-mediated priming and reprogramming for three groups of cytokines with important immune functions, namely IFN-γ itself, type I IFNs, and IL-10 (Fig. 6). One common feature of such signaling regulation is that IFN-γ priming results in strong STAT1 activation by other cytokines and make them ‘IFN-γ-like’. IFN-γ is able to prime for activation of positive signaling events without engaging negative feedback mechanisms, and such action is achieved either by passively sparing induction of inhibitory factors such as SOCS with low doses of IFN-γ or by actively suppressing functions of opposing pathways. Dynamic regulation of the activation and expression of STAT1 by IFN-γ priming contributes to the pro-inflammatory properties of IFN-γ and provides a mechanism by which cells can integrate and balance signals delivered by different cytokines. Interestingly, IL-27, a member of the IL-12 family of cytokines, which activates STAT1 and STAT1 target genes in human monocytes, induces high levels of STAT1 expression and is also capable of priming for IL-10-induced STAT1 activation and of suppressing IL-10-induced, STAT3-dependent gene induction (52). In addition, IL-27 primes human monocytes for enhanced STAT1-mediated responses when cells are restimulated with IFN-γ or IFN-α (L. Ivashkiv, unpublished observations). This observation supports a common role for elevated STAT1 in altering macrophage responses to cytokines, and argues for proinflammatory and IFN-γ-like effects of IL-27 at least in human primary monocytes. Whether IL-27 and IFN-γ employ similar mechanisms for IL-10 signaling regulation remains to be seen.
Fig. 6. Crosstalk between IFN signaling and signaling by other cytokines.

Preexposure to IFN-γ (left side) alters the signal transduction pathways to several cellular stimuli (within the box in the middle).
Regulation of endogenous inflammatory signaling by IFN-γ
Besides regulation of cytokines that utilize the Jak-STAT pathway, IFN-γ is also able to regulate signaling by cytokines that activate distinct signaling cascades. IL-1 is a multifunctional cytokine produced primarily by monocytes and macrophages in response to numerous endogenous and exogenous stimuli. IL-1 possesses a broad spectrum of bioactivities including induction of cytokines and chemokines, upregulation of inflammatory mediators, and regulation of the central nervous system (53). Another prominent function of IL-1 is its ability to promote tissue-invasive and destructive processes such as cartilage breakdown, bone erosion, and angiogenesis (53). Given the key role of IL-1 in inflammation and tissue destruction, it is important to understand interaction between IFN-γ and IL-1 receptor signaling pathways. IL-1 receptor belongs to IL-1R/Toll-like receptor (TLR) superfamily. We reported that both type I and type II IFNs suppress an array of IL-1-mediated effects in vitro and in vivo including induction of cytokines and other inflammatory mediators, production of matrix metalloproteinases (MMPs), cell migration and tissue invasion, and cartilage degradation. The underlying mechanisms involve STAT1-dependent extinction of IL-1 type I receptor (IL-1RI) expression (54).
Although being predominantly viewed as a pro-immune and pro-inflammatory cytokine, IFN-γ can act to limit inflammation under certain circumstances. Endogenous IFN-γ was shown to be protective in animal models of autoimmune arthritis (55-57) and multiple sclerosis (55, 58, 59). The mechanisms underlying the disease-limiting activities of IFN-γ in these conditions are not well understood. Regulation of IL-1 receptor expression and resultant lack of responsiveness of macrophages to IL-1 may provide a specific molecular mechanism underlying the suppressive effects of IFNs (53). Inhibition of IL-1 responses likely contributes to the previously described suppressive effects of IFN-γ and STAT1 on IL-1-dependent diseases, such as arthritis and osteolysis (55-57, 60-63). In addition, suppression of cell invasion and MMP expression may contribute to the beneficial effects of type I IFNs in the treatment of multiple sclerosis. As delineated in the signal integration model (64), the outcome of cytokine signaling is often determined by the balance between opposing signaling pathways triggered by the same cytokine. STAT1 has been established as a key mediator of IFN activation of cells and an indispensable component of IFN-dependent innate defense mechanisms against viral infections (65, 66). However, IFN-γ and IFNα/β also activate STAT1-independent signaling pathways, such as STAT3 and phosphoinositide 3-kinase (PI3K) (67, 68), that have suppressive effects on macrophage activation (41, 69) and would be predicted to mediate the homeostatic functions of IFNs. Thus, it is noteworthy that the homeostatic function that we analyzed, IFN-γ suppression of IL-1RI expression, is dependent on STAT1 (54). STAT1 mediates both activating and homeostatic functions of IFN-γ in macrophages, and these studies highlight and extend our appreciation of the pleiotropic and suppressive activities of STAT1. The exact contribution of STAT1 to disease pathogenesis will be determined by the balance between STAT1 pro- and anti-inflammatory activities in the cell types important for pathogenesis and by the effects of IFNs and STAT1 on other factors that drive disease pathogenesis.
The IL-1R and TLRs activate similar signaling pathways with many shared components. Interestingly, IL-1R and TLR4 activities are differentially regulated by IFNs. IFN-γ strongly potentiates TLR responses (5)(discussed in detail below), while both IFN-α and IFN-γ reduce cellular responsiveness to IL-1. It is logical that IFNs potentiate inflammation when pathogenic microbes that activate TLRs are present, yet limit the extent of tissue damage and inflammation in response to endogenous cytokines, as excessive activity of cytokines such as IL-1 can be deleterious to the host if left uncontrolled. IFN-mediated suppression of IL-1RI expression is observed in macrophages, osteoclasts, and T cells, and is associated with almost complete suppression of all IL-1 responses in macrophages that were tested. Thus, IFNs and STAT1 will most effectively suppress IL-1-dependent inflammation in processes and diseases in which these cells play a prominent role, such as arthritis and osteolysis. This notion is supported by a substantial body of literature documenting suppression of arthritis and osteolysis by IFNs and STAT1 (55-57, 60-63). IL-1 promotes osteoclast-mediated bone resorption (70). Therefore, IFN and STAT1 anti-bone resorptive activity may be mediated by suppression of IL-1 responses in addition to previously described mechanisms involving suppression of receptor activator of NF-κB-dependent osteoclastogenesis (61, 62).
Inhibition of TLR-engaged feedback mechanisms by IFN-γ
It has been appreciated for many decades that full inflammatory activation of macrophages requires synergistic action of exogenous factors (such as microbial products that activate TLRs) and endogenously produced cytokines (especially IFN-γ) (71). A key property of IFN-γ is its capacity to dramatically enhance macrophage responses to other inflammatory factors such as TLR ligands. Activation with TLR ligands leads to production of multiple pro-inflammatory mediators, including inflammatory cytokines such as TNF and IL-1. TNF is a prototypical proinflammatory cytokine whose induction by TLRs is thought to be dependent on NF-κB and mitogen-activated protein kinases (MAPKs). TLR activation of macrophages also induces cytokines of the IL-6/IL-12 family, namely IL-6, IL-12, IL-23, and IL-27, that regulate the transition from innate to acquired immunity (72). Given the critical and nonredundant role of IL-6/IL-12 family cytokines in driving T-cell differentiation, namely IL-12 for Th1 and IL-6 and IL-23 for Th17 cells, TLRs have been implicated in the onset of T-helper cell responses. There is accumulating evidence suggesting that engagement of distinct TLRs may trigger differential adaptive immune responses. For example, the strong inflammatory activation of DCs and macrophages by TLR4 has been linked to the development of Th1 responses (73), and activation of TLR2 can be less inflammatory and favor the development of Th2 responses (74-77). Activation of TLR2 in conjunction with Dectin-1, a pattern recognition receptor of C-type lectin family, resulted in production of ample amounts of IL-6 and IL-23 but little IL-12 and thus promoted differentiation of Th17 cells (78). Although DCs are generally believed to play a major role in regulating T-cell differentiation in lymphoid tissues, activated macrophages likely contribute to T-cell differentiation and activation at local sites of inflammation (79). In addition, activating effects of DC- and macrophage-derived cytokines on memory T-cell function have been described, for example IL-23-induced activation of IL-17 production by human memory Th17 cells (80, 81).
As a major product of adaptive immunity and Th1 responses, IFN-γ has profound effects on innate TLR signaling and dramatically increases TLR-induced expression of inflammatory mediators and immune effectors including multiple cytokines and chemokines. Investigation of the synergistic activation of macrophages by IFN-γ and TLRs has focused on enhanced activation of pro-inflammatory pathways. For example, IFN-γ enhances LPS-induced NF-κB activation and increases TLR expression (5). We reasoned that suppression of TLR-induced homeostatic responses and feedback inhibitory pathways would provide an additional mechanism by which IFN-γ synergizes with TLRs. Prior to analysis of IFN-γ's effects on TLR responses, we briefly review feedback mechanisms that negatively regulate TLR signaling and lay the ground for the following discussion pertaining their regulation by IFN-γ. Since negative regulation of TLR signaling has been extensively reviewed in recent literature (82), the sections below only provide examples of regulatory mechanisms relevant to our discussion and are not intended to serve as a comprehensive review.
Negative regulation of TLR signaling
While production of sufficient amounts of inflammatory mediators is essential for effective immune responses, unrestrained activation of TLR responses can lead to excessive inflammation and tissue damage and contribute to pathogenesis of inflammatory disorders such as septic shock. Given the potentially deleterious consequences of TLR activation, TLR signaling is subject to negative regulation and feedback inhibition at multiple levels (83). It is believed that several different homeostatic mechanisms work together to control TLR responses and prevent potentially catastrophic consequences, including lethality secondary to ‘cytokine storm’. According to their mode of action, mechanisms of negative regulation of TLR responses can be categorized into at least three types (Fig. 7).
Fig. 7. Negative regulation of TLR-induced gene expression.
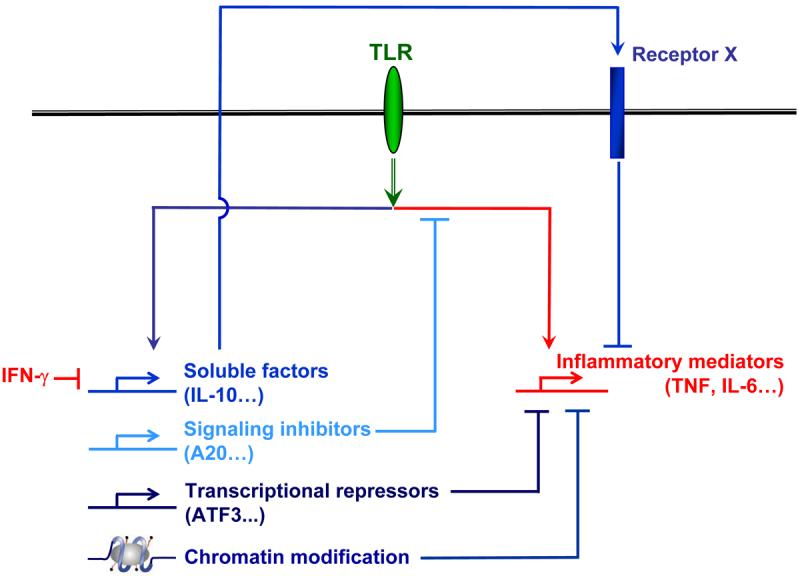
TLRs induce production of proinflammatory cytokines (depicted in red) as well as engage feedback inhibitory mechanisms (depicted in blue) that serve to limit excessive inflammation.
Soluble factors
TLRs induce secreted factors that serve to restrain the extent of inflammatory activation and thus limit toxicity and tissue damage. Among the most potent of TLR-induced homeostatic soluble factors is the cytokine IL-10 that participates in a feedback inhibitory loop that limits inflammatory cytokine production. IL-10 is predominantly an immunosuppressive and anti-inflammatory cytokine that is best known as a potent deactivator of DCs and macrophages. A key biological activity of IL-10 is to suppress inflammation by suppressing TNF and IL-1 production and by antagonizing TNF and IL-1 function (37). The suppressive effects of IL-10 on myeloid cells are dependent upon STAT3 (41). TLR induction of IL-10 is largely mediated by MAPKs and downstream transcriptional factors such as activator protein-1 (AP-1) and cyclic adenosine monophosphate (cAMP)-responsive element binding proteins (CREB). Recently, our laboratory and others identified glycogen synthase kinase 3 (GSK3) as a negative regulator of IL-10 production in macrophages (84, 85).
Signaling inhibitors
A panoply of endogenous inhibitors of TLR signaling have been identified during recent years. Some of the inhibitory molecules are expressed at baseline in the absence of TLR stimulation and act to suppress TLR signaling constitutively. Examples of constitutive inhibitors include PI3K, radioprotective 105 (RP105), myeloid differentiation factor 88 (MyD88), ST2, DAP12, β-arrestin, and single immunoglobulin IL-1R-related molecule (SIGIRR). Another group of inhibitors are themselves TLR inducible and mediate feedback inhibition of TLR responses. Examples of inducible feedback inhibitors include A20, MAPK phosphatase 1 (MKP1), IRF4, and SOCS1 (86). Signaling inhibitors often act on proximal components of TLR signaling pathways and inhibit expression of multiple downstream genes.
Selective regulation of subsets of TLR-inducible genes: transcriptional repressors and chromatin modification
As described above, signaling inhibitors often attenuate expression of multiple genes downstream of major TLR-induced signaling pathways and thus have limited capability to inhibit expression of specific subgroups of TLR-inducible genes. Recent work has demonstrated that TLRs induce expression of transcription repressors, such as activating transcription factor 3 (ATF3), that feed back and suppress expression of specific subsets of TLR-inducible genes (87). ATF3 suppresses IL-6 mRNA expression by modifying chromatin structure and therefore alters accessibility of transcription factors. Given the large number of genes induced by TLRs and the need to control gene expression in a specific manner, it is very likely that besides ATF3, there are additional transcription repressors that modulate gene activation downstream of TLRs.
Some of the most potent homeostatic mechanisms are apparent in ‘endotoxin tolerance’, where pre-exposure of cells to TLR ligands abrogates induction of inflammatory genes on subsequent rechallenge with TLR ligands. Mechanisms of endotoxin tolerance include alterations of TLR signaling and TLR-induced inhibition of inflammatory gene expression that appears to occur via epigenetic regulation of inflammatory gene loci by modification of histones and chromatin that can either activate or silence gene expression (88-90). Silencing by such epigenetic modifications during endotoxin tolerance, which is likely mediated by transcriptional repressors (88), plays a key role in specifically restraining potentially toxic inflammatory cytokine expression, while allowing beneficial expression of host defense genes. Thus, selective regulation of subsets of TLR-inducible genes allows fine-tuning of distinct biological functions induced by TLRs.
Regulation of TLR-induced feedback inhibitory loops by IFN-γ
To characterize the signaling pathways that could be potentially modulated by IFN-γ, we analyzed the effects of IFN-γ-mediated inhibition of TLR-induced gene expression (85, 91) by using microarray analysis to define the subset of TLR-inducible genes that are inhibited by IFN-γ in primary human macrophages. Analysis of microarray data showed that IFN-γ inhibits the expression of approximately 15% of TLR2-inducible genes, including multiple putative AP-1 and CREB target genes. One of the IFN-γ-suppressed genes is IL-10, and we further analyzed the mechanisms of IFN-γ-mediated inhibition of IL-10 expression. IFN-γ suppresses TLR-mediated induction of IL-10 protein and mRNA expression, downstream STAT3 activation, and induction of a STAT3-dependent gene SOCS3 (85). By inhibiting IL-10-STAT3 axis, IFN-γ interrupts a TLR2-induced feedback inhibition loop (Fig. 2) and results in increased production of the inflammatory cytokines TNF and IL-6. These findings demonstrate that abrogation of IL-10-mediated feedback inhibitory loop contributes to synergistic activation of macrophages by IFN-γ and TLR ligands.
Subsequent work revealed several clues about mechanisms by which IFN-γ inhibits IL-10 production. One mechanism involves attenuation of proximal TLR2-induced signaling by IFN-γ, which includes attenuation of TLR2-induced activation of the PI3K-Akt pathway that had been previously shown to have a rapidly induced feedback inhibitory function (69, 92). Thus, these findings fit with the concept of interruption of feedback inhibition by IFN-γ. The PI3K-Akt pathway is linked to IL-10 production via GSK3 that regulates the function of transcription factors of the AP-1/CREB families that, in turn, regulate IL-10 production. Thus, GSK3 emerges as a potential therapeutic target for anti-inflammatory therapy.
In addition to downregulating TLR2-induced PI3K-Akt signaling (previously appreciated to have suppressive properties), IFN-γ paradoxically downregulates TLR2-induced MAPK activation (previously appreciated to have activating properties, including induction of inflammatory cytokines TNF and IL-6, but also to contribute to IL-10 production). It is not clear how IFN-γ-mediated suppression of proximal signaling pathways can translate to selective or even opposite regulation of target genes that are dependent on that pathway. One intriguing potential answer to this conundrum is IFN-γ-mediated regulation of transcription factor expression or function that can selectively regulate aspects of the TLR response. One possibility, alluded to above, is that IFN-γ can attenuate induction of a subset of TLR-induced transcriptional activators, such as AP-1 and CREB proteins (85, 91, 93). This can occur by several mechanisms, including decreased AP-1/CREB mRNA expression, decreased MAPK-mediated phosphorylation of AP-1/CREB proteins, or destabilization of Fos and Jun proteins by as yet unknown mechanisms. Diminished AP-1 and CREB activity in IFN-γ-primed, TLR2-activated cells can lead to decreased expression of multiple AP-1 and CREB target genes, including IL-10 and also the CREB-dependent PAI-2 gene and the AP-1-dependent MKP-1 and MMP-3 genes (85), indicating that IFN-γ-mediated inhibition of AP-1/CREB activation has a broad impact in terms of regulation of TLR2-induced transcriptional programs. Another possibility is that IFN-γ can induce the expression of transcriptional repressors that selectively downregulate subsets of TLR-inducible genes. We have recently obtained evidence to support this latter model by finding that IFN-γ priming superinduces TLR activation of the ATF3 transcriptional repressor, which then blocks TLR induction of MMP-1 and potentially other genes (93). It is also possible that STAT1 can directly repress expression of a subset of TLR-inducible genes; however, there is no clear evidence that STAT1 can act as a transcriptional repressor at direct target genes.
Overall, it appears that one mechanism by which IFN-γ primes macrophages for enhanced TLR responses is suppression of feedback inhibitory pathways consisting of PI3K/Akt/AP-1/IL-10/STAT3 and resultant amplification of proinflammatory pathways. Interestingly, IL-27 can also strongly suppress TLR-induced IL-10 expression and prime for enhanced production of proinflammatory cytokine in human (but not murine) macrophages in a STAT1-dependent manner (52), an observation that is reminiscent of IL-27's ability to reprogram IL-10 signaling in an ‘IFN-γ-like’ fashion. Both IFN-γ and IL-27 appear to prime macrophages via STAT1, suggesting a central role for STAT in augmenting macrophage activation.
Potential role for STAT1 and priming in autoimmune/inflammatory disease pathogenesis
Cytokines have long been implicated in modulating the pathogenesis of a variety of inflammatory and autoimmune disorders, including RA and SLE. Specific roles for the pro-inflammatory cytokines TNF and IL-1 in RA pathogenesis have been demonstrated with the remarkable efficacy of targeted biological therapies, such as infliximab and etanercept, which block TNF activity, and anakinra, which inhibits the IL-1 receptor (94, 95). In addition, TNF and IL-1 blockade have proven to be effective in the treatment of a wide variety of other diseases, which have expanded to include inflammatory bowel disease, psoriatic arthritis, ankylosing spondylitis, and plaque psoriasis. Furthermore, the scope of examining the pathogenic roles played by other cytokines in rheumatic diseases has been broadened to include the type I IFNs, namely IFN-α/β, as well as IL-6 and IL-15—molecules that activate the Jak-STAT signaling pathway (12). Therapeutic blockade of IL-6 has already been demonstrated to be beneficial in treating RA (96), and in SLE, recent emphasis has been placed on investigating what pathogenic roles type I IFNs may have and their potential as therapeutic targets (97, 98). In particular, a multitude of prior studies have demonstrated a role for type I IFNs in SLE pathogenesis. One of the first described cytokine abnormalities in autoimmunity was the observation that type I IFN expression was upregulated in sera of SLE patients (99, 100). Interestingly, a recent hypothesis posited to explain the mechanism for such a marked increase in type I IFN expression has been that nucleic acid-bound immune complexes may activate immunocytes via TLR3 and TLR9, in addition to possibly TLR7 and TLR8. Further evidence has indicated that levels of IFNα expression in SLE patients' sera can be elevated comparably to those levels observed during acute viral illnesses (13). In addition to disease activity in SLE being positively correlated to serum levels of IFN-α through protein quantification (101), more recently, gene expression analyses using microarray technology of peripheral blood mononuclear cells (PBMCs) taken from SLE patients have shown upregulated expression of IFN-inducible genes and their correlation to disease activity (22, 23). Other observations have reinforced the notion that IFN-α plays a role in SLE pathogenesis, including the onset of autoantibody production and a lupus-like constellation of symptoms in some patients who receive type I IFN therapy, such as in chronic myeloid leukemia or carcinoid tumors (102). Conversely, when the host is deficient in type I IFN receptor expression, a marked attenuation in autoantibody production and disease progression occur, as observed in the murine NZB model of SLE (103). Thus, it seems abundantly clear that type I IFNs contribute to SLE pathogenesis, though the mechanisms by which this is achieved include several possibilities, such as promoting DC maturation, upregulating innate immune, Th1, and antibody responses, suppressing apoptosis, and inducing the expression of other cytokines and chemokines (12).
To further understand the above-mentioned mechanisms by which type I IFNs modulate SLE pathogenesis, several investigators, including our group, have considered the possibility that priming of myeloid cells (DCs and macrophages) and high STAT1 expression may contribute to pathogenesis (4, 9-12). As previously noted, the IFNs are expressed in a wide variety of rheumatologic diseases, including RA and SLE, and at levels that could modulate host immune priming, despite the fact that they may not directly activate immunocytes themselves. In actively priming the host response, these factors can, in turn, enhance the inflammatory cellular responses to other molecules long implicated in modulating the pathogenesis of autoimmune diseases, such as TNF, IL-1 and IL-6, IFN-γ, and TLRs. In fact, a multitude of animal models for autoimmune/inflammatory diseases have been utilized to suggest that priming does contribute to disease pathogenesis and progression (4, 10, 12, 103-105). Additionally, clinical evidence has supported the notion that exposure to IFNs and priming play a role in enhancing patient responses to cytokines and TLR ligands at various time points in autoimmune and inflammatory disease progression. Specifically, in SLE, systemic IFN levels have been shown to bolster antigen-presenting cell (APC) function of monocytes, similar to partially activated DCs (13, 101). Furthermore, leukocytes isolated from SLE patients have been found to express increased levels of STAT1, a key marker in IFN priming (12, 23, 26, 106). Interestingly, with the use of corticosteroids, suppression of STAT1 expression occurs concomitantly with demonstrated clinical improvement in SLE patients (12, 23), thereby providing a possible mechanism of action for this drug class in systemic lupus. Elevated STAT1 expression, measured both at the mRNA and protein levels, and implying priming in vivo, has been observed in other inflammatory processes, such as T-cell-mediated hepatitis, RA, and dermatomyositis (9, 11, 25, 26). In SLE patients who have not undergone treatment, isolated monocytes displayed an enhanced response to TLR activators, such as LPS and IL-1β (107), and it is thought that such a hyper-responsiveness of TLRs in vivo can be secondary to the effects of IFN priming (6-8, 108-110). Moreover, as a consequence of this hyper-responsiveness, patients with rheumatic diseases have been observed to mount exaggerated immune responses to infections (104). Lastly, despite low levels of IFN-γ expression in synovia of RA patients, RA synovial cells nevertheless strongly express an array of IFN-γ-inducible genes (9), which can be explained by the priming of synovial cells, increased expression of STAT1, and their hyper-responsiveness to cytokines that serve to activate STAT1. Thus, a substantial body of data supports the notion that priming of immunocytes readily occurs in rheumatic diseases and that its contribution to determining cellular activation and cytokine production contributes to disease pathogenesis.
However, the rate of progression of inflammatory disease and its eventual severity and morbidity are also shaped by a dynamic interface between pro- and anti-inflammatory molecules that influence whether the inflammatory response progresses or defervesces. Thus, just as type I IFNs can serve to bolster the immune response through priming, cytokines with known immunosuppressive properties, such as IL-10, are also thought to play a role in RA and SLE pathogenesis. The immunosuppressive effects of IL-10 are contingent upon the timing of its expression, and its suppressive properties can be diminished during active inflammatory responses. For instance, diminished IL-10 activity in the acute phase of infection would be beneficial to the host, e.g. in mycobacterial infection (111), as it would allow for enhanced immunity and clearance of the pathogen. The immunosuppression normally conferred by IL-10 has been shown to be compromised in RA and SLE (45, 46, 49, 112, 113). Our group has shown that IL-10 signaling and downstream gene expression are suppressed in synovial macrophages isolated from patients with RA, in addition to the observation that IL-10 responses are diminished in monocytes derived from SLE patients (45, 46, 112). Inhibition of IL-10 signaling is most effective in cells exposed to IFNs and correlates with diminished STAT3 activation and function, suggesting the potential involvement of some of the activating mechanisms of IFN-γ discussed above. In the context of autoimmune/inflammatory disorders, a loss in IL-10 activity may have adverse consequences for the host, as this would imply an inability to control and reduce the inflammatory response. With respect to cytokine expression, decreased IL-10 activity would lead to increased expression of pro-inflammatory cytokines, such as TNF, IL-1, and IL-6, thereby allowing the inflammatory response to proceed unchecked. Hence, it is clear that the potential for pathogenesis of rheumatic diseases and the severity of the inflammatory response are determined not only by the priming effects of type I IFNs and other cytokines, but also by the perturbation of IL-10 expression and signaling and its subsequent loss in regulating immunosuppression.
Acknowledgements
Xiaoyu Hu is supported by the Arthritis Foundation and Lionel B. Ivashkiv is funded by the National Institutes of Health.
References
- 1.Renauld JC. Class II cytokine receptors and their ligands: key antiviral and inflammatory modulators. Nat Rev Immunol. 2003;3:667–676. doi: 10.1038/nri1153. [DOI] [PubMed] [Google Scholar]
- 2.Biron CA. Interferons alpha and beta as immune regulators--a new look. Immunity. 2001;14:661–664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- 3.Matsumoto M, et al. Activation of the transcription factor ISGF3 by interferon-gamma. Biol Chem. 1999;380:699–703. doi: 10.1515/BC.1999.087. [DOI] [PubMed] [Google Scholar]
- 4.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 5.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 6.Doughty L, Nguyen K, Durbin J, Biron C. A role for IFN-alpha beta in virus infection-induced sensitization to endotoxin. J Immunol. 2001;166:2658–2664. doi: 10.4049/jimmunol.166.4.2658. [DOI] [PubMed] [Google Scholar]
- 7.Nansen A, Randrup Thomsen A. Viral infection causes rapid sensitization to lipopolysaccharide: central role of IFN-alpha beta. J Immunol. 2001;166:982–988. doi: 10.4049/jimmunol.166.2.982. [DOI] [PubMed] [Google Scholar]
- 8.Durbin J, Doughty L, Nguyen K, Caligiuri M, Van Deusen J, Biron C. The role of STAT1 in viral sensitization to LPS. J Endotoxin Res. 2003;9:313–316. doi: 10.1179/096805103225002575. [DOI] [PubMed] [Google Scholar]
- 9.van der Pouw Kraan TC, et al. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48:2132–2145. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]
- 10.Kono DH, Baccala R, Theofilopoulos AN. Inhibition of lupus by genetic alteration of the interferon-alpha/beta receptor. Autoimmunity. 2003;36:503–510. doi: 10.1080/08916930310001624665. [DOI] [PubMed] [Google Scholar]
- 11.Hu X, et al. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3:859–866. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 12.Ivashkiv LB. Type I interferon modulation of cellular responses to cytokines and infectious pathogens: potential role in SLE pathogenesis. Autoimmunity. 2003;36:473–479. doi: 10.1080/08916930310001605882. [DOI] [PubMed] [Google Scholar]
- 13.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 14.Takaoka A, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 15.Tassiulas I, et al. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat Immunol. 2004;5:1181–1189. doi: 10.1038/ni1126. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, et al. ‘Tuning” of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat Immunol. 2008;9:186–193. doi: 10.1038/ni1548. [DOI] [PubMed] [Google Scholar]
- 17.Contursi C, et al. IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macrophages. Proc Natl Acad Sci USA. 2000;97:91–96. doi: 10.1073/pnas.97.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malakhova OA, et al. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003;17:455–460. doi: 10.1101/gad.1056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bach EA, et al. Ligand-induced autoregulation of IFN-gamma receptor beta chain expression in T helper cell subsets. Science. 1995;270:1215–1218. doi: 10.1126/science.270.5239.1215. [DOI] [PubMed] [Google Scholar]
- 20.Pernis A, et al. Lack of interferon gamma receptor beta chain and the prevention of interferon gamma signaling in TH1 cells. Science. 1995;269:245–247. doi: 10.1126/science.7618088. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen KB, et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 2002;297:2063–2066. doi: 10.1126/science.1074900. [DOI] [PubMed] [Google Scholar]
- 22.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrt S, et al. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001;194:1123–1140. doi: 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong F, et al. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110:1503–1513. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuroiwa T, Schlimgen R, Illei GG, Boumpas DT. Monocyte response to Th1 stimulation and effector function toward human mesangial cells are not impaired in patients with lupus nephritis. Clin Immunol. 2003;106:65–72. doi: 10.1016/s1521-6616(02)00022-0. [DOI] [PubMed] [Google Scholar]
- 27.Ivashkiv LB. Cytokine expression and cell activation in inflammatory arthritis. Adv Immunol. 1996;63:337–376. doi: 10.1016/s0065-2776(08)60859-7. [DOI] [PubMed] [Google Scholar]
- 28.Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 2002;46:298–308. doi: 10.1002/art.502. [DOI] [PubMed] [Google Scholar]
- 29.Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. 2003;48:2092–2096. doi: 10.1002/art.11095. [DOI] [PubMed] [Google Scholar]
- 30.Antoniv TT, Ivashkiv LB. Dysregulation of interleukin-10-dependent gene expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum. 2006;54:2711–2721. doi: 10.1002/art.22055. [DOI] [PubMed] [Google Scholar]
- 31.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity. 2008;29:68–78. doi: 10.1016/j.immuni.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 34.Ivashkiv LB. A signal switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. Nat Rev Immunol. 2008 doi: 10.1038/nri2396. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharif MN, Tassiulas I, Hu Y, Mecklenbrauker I, Tarakhovsky A, Ivashkiv LB. IFN-alpha priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. 2004;172:6476–6481. doi: 10.4049/jimmunol.172.10.6476. [DOI] [PubMed] [Google Scholar]
- 36.Herrero C, et al. Reprogramming of IL-10 activity and signaling by IFN-gamma. J Immunol. 2003;171:5034–5041. doi: 10.4049/jimmunol.171.10.5034. [DOI] [PubMed] [Google Scholar]
- 37.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 38.Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol. 1995;155:1079–1090. [PubMed] [Google Scholar]
- 39.Weber-Nordt RM, Riley JK, Greenlund AC, Moore KW, Darnell JE, Schreiber RD. Stat3 recruitment by two distinct ligand-induced, tyrosine-phosphorylated docking sites in the interleukin-10 receptor intracellular domain. J Biol Chem. 1996;271:27954–27961. doi: 10.1074/jbc.271.44.27954. [DOI] [PubMed] [Google Scholar]
- 40.Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for antiinflammatory action. J Biol Chem. 1999;274:16513–16521. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- 41.Takeda K, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 42.Pajkrt D, et al. Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J Immunol. 1997;158:3971–3977. [PubMed] [Google Scholar]
- 43.Avdiushko R, Hongo D, Lake-Bullock H, Kaplan A, Cohen D. IL-10 receptor dysfunction in macrophages during chronic inflammation. J Leukoc Biol. 2001;70:624–632. [PubMed] [Google Scholar]
- 44.Neyer LE, Grunig G, Fort M, Remington JS, Rennick D, Hunter CA. Role of interleukin-10 in regulation of T-cell-dependent and T-cell-independent mechanisms of resistance to Toxoplasma gondii. Infect Immun. 1997;65:1675–1682. doi: 10.1128/iai.65.5.1675-1682.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mongan AE, Ramdahin S, Warrington RJ. Interleukin-10 response abnormalities in systemic lupus erythematosus. Scand J Immunol. 1997;46:406–412. doi: 10.1046/j.1365-3083.1997.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 46.Hart PH, Ahern MJ, Smith MD, Finlay-Jones JJ. Comparison of the suppressive effects of interleukin-10 and interleukin-4 on synovial fluid macrophages and blood monocytes from patients with inflammatory arthritis. Immunology. 1995;84:536–542. [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, et al. High-dose cellular IL-10 exacerbates rejection and reverses effects of cyclosporine and tacrolimus in Mouse cardiac transplantation. Transplant Proc. 1997;29:1081–1082. doi: 10.1016/s0041-1345(96)00412-5. [DOI] [PubMed] [Google Scholar]
- 48.Blazar BR, Taylor PA, Smith S, Vallera DA. Interleukin-10 administration decreases survival in murine recipients of major histocompatibility complex disparate donor bone marrow grafts. Blood. 1995;85:842–851. [PubMed] [Google Scholar]
- 49.Bussolati B, Rollino C, Mariano F, Quarello F, Camussi G. IL-10 stimulates production of platelet-activating factor by monocytes of patients with active systemic lupus erythematosus (SLE) Clin Exp Immunol. 2000;122:471–476. doi: 10.1046/j.1365-2249.2000.01392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJ, van der Poll T. Proinflammatory effects of IL-10 during human endotoxemia. J Immunol. 2000;165:2783–2789. doi: 10.4049/jimmunol.165.5.2783. [DOI] [PubMed] [Google Scholar]
- 51.O'Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223:114–131. doi: 10.1111/j.1600-065X.2008.00635.x. [DOI] [PubMed] [Google Scholar]
- 52.Kalliolias GD, Ivashkiv LB. IL-27 activates human monocytes via STAT1 and suppresses IL-10 production but the inflammatory functions of IL-27 are abrogated by TLRs and p38. J Immunol. 2008;180:6325–6333. doi: 10.4049/jimmunol.180.9.6325. [DOI] [PubMed] [Google Scholar]
- 53.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 54.Hu X, Ho HH, Lou O, Hidaka C, Ivashkiv LB. Homeostatic role of interferons conferred by inhibition of IL-1-mediated inflammation and tissue destruction. J Immunol. 2005;175:131–138. doi: 10.4049/jimmunol.175.1.131. [DOI] [PubMed] [Google Scholar]
- 55.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol. 1997;158:5507–5513. [PubMed] [Google Scholar]
- 56.Manoury-Schwartz B, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. 1997;158:5501–5506. [PubMed] [Google Scholar]
- 57.Guedez YB, et al. Genetic ablation of interferon-gamma up-regulates interleukin-1beta expression and enables the elicitation of collagen-induced arthritis in a nonsusceptible mouse strain. Arthritis Rheum. 2001;44:2413–2424. doi: 10.1002/1529-0131(200110)44:10<2413::aid-art406>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 58.Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 59.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 60.Kim S, et al. Stat1 functions as a cytoplasmic attenuator of Runx2 in the transcriptional program of osteoblast differentiation. Genes Dev. 2003;17:1979–1991. doi: 10.1101/gad.1119303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takayanagi H, et al. T-cell-mediated regulation of osteoclastogenesis by signalling crosstalk between RANKL and IFN-gamma. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 62.Takayanagi H, Kim S, Matsuo K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 63.de Hooge AS, et al. Local activation of STAT-1 and STAT-3 in the inflamed synovium during zymosan-induced arthritis: exacerbation of joint inflammation in STAT-1 gene-knockout mice. Arthritis Rheum. 2004;50:2014–2023. doi: 10.1002/art.20302. [DOI] [PubMed] [Google Scholar]
- 64.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 65.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 66.Meraz MA, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 67.Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J Biol Chem. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 68.Choudhury GG. A linear signal transduction pathway involving phosphatidylinositol 3-kinase, protein kinase Cepsilon, and MAPK in mesangial cells regulates interferon-gamma-induced STAT1alpha transcriptional activation. J Biol Chem. 2004;279:27399–27409. doi: 10.1074/jbc.M403530200. [DOI] [PubMed] [Google Scholar]
- 69.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 70.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 71.Adams DO, Hamilton TA. The cell biology of macrophage activation. Annu Rev Immunol. 1984;2:283–318. doi: 10.1146/annurev.iy.02.040184.001435. [DOI] [PubMed] [Google Scholar]
- 72.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 73.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 74.Agrawal S, et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171:4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 75.Dillon S, et al. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 76.Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. 2001;167:5067–5076. doi: 10.4049/jimmunol.167.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Redecke V, et al. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J Immunol. 2004;172:2739–2743. doi: 10.4049/jimmunol.172.5.2739. [DOI] [PubMed] [Google Scholar]
- 78.LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 79.Szekanecz Z, Koch AE. Macrophages and their products in rheumatoid arthritis. Curr Opin Rheumatol. 2007;19:289–295. doi: 10.1097/BOR.0b013e32805e87ae. [DOI] [PubMed] [Google Scholar]
- 80.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 81.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 82.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 83.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 84.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hu X, et al. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 86.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–266. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 87.Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 88.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 89.Ramirez-Carrozzi VR, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–296. doi: 10.1101/gad.1383206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 91.Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol. 2007;82:237–243. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- 92.Fukao T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 93.Ho HH, Antoniv TT, Ji JD, Ivashkiv LB. Lipopolysaccharide-induced expression of MMPs in human monocytes is suppressed by IFN-gamma via superinduction of ATF-3 and suppression of AP-1 proteins. J Immunol. 2008 doi: 10.4049/jimmunol.181.7.5089. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 95.Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- 96.Choy EH, et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002;46:3143–150. doi: 10.1002/art.10623. [DOI] [PubMed] [Google Scholar]
- 97.Crow MK. Interferon-alpha: a new target for therapy in systemic lupus erythematosus? Arthritis Rheum. 2003;48:2396–23401. doi: 10.1002/art.11226. [DOI] [PubMed] [Google Scholar]
- 98.Crow MK. Type I interferon and autoimmune disease. Autoimmunity. 2003;36:445–446. doi: 10.1080/08916930310001625961. [DOI] [PubMed] [Google Scholar]
- 99.Friedman RM, Preble O, Black R, Harrell S. Interferon production in patients with systemic lupus erythematosus. Arthritis Rheum. 1982;25:802–803. doi: 10.1002/art.1780250717. [DOI] [PubMed] [Google Scholar]
- 100.Preble OT, Black RJ, Friedman RM, Klippel JH, Vilcek J. Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science. 1982;216:429–431. doi: 10.1126/science.6176024. [DOI] [PubMed] [Google Scholar]
- 101.Bengtsson AA, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 2000;9:664–671. doi: 10.1191/096120300674499064. [DOI] [PubMed] [Google Scholar]
- 102.Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000;43:1431–1442. doi: 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 103.Santiago-Raber ML, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–788. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klinman D. Does activation of the innate immune system contribute to the development of rheumatoid arthritis? Arthritis Rheum. 2003;48:590–593. doi: 10.1002/art.10852. [DOI] [PubMed] [Google Scholar]
- 105.Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR. The role of IFN-gamma in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res. 2001;3:136–141. doi: 10.1186/ar290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baechler EC, Gregersen PK, Behrens TW. The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol. 2004;16:801–807. doi: 10.1016/j.coi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 107.Scuderi F, et al. Effect of pro-inflammatory/anti-inflammatory agents on cytokine secretion by peripheral blood mononuclear cells in rheumatoid arthritis and systemic lupus erythematosus. Autoimmunity. 2003;36:71–77. doi: 10.1080/0891693031000079275. [DOI] [PubMed] [Google Scholar]
- 108.Mohty M, Vialle-Castellano A, Nunes JA, Isnardon D, Olive D, Gaugler B. IFN-alpha skews monocyte differentiation into Toll-like receptor 7-expressing dendritic cells with potent functional activities. J Immunol. 2003;171:3385–3393. doi: 10.4049/jimmunol.171.7.3385. [DOI] [PubMed] [Google Scholar]
- 109.Paterson HM, et al. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–1483. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- 110.Krutzik SR, et al. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med. 2003;9:525–532. doi: 10.1038/nm864. [DOI] [PubMed] [Google Scholar]
- 111.Murray PJ, Wang L, Onufryk C, Tepper RI, Young RA. T cell-derived IL-10 antagonizes macrophage function in mycobacterial infection. J Immunol. 1997;158:315–321. [PubMed] [Google Scholar]
- 112.Ji JD, et al. Inhibition of interleukin 10 signaling after Fc receptor ligation and during rheumatoid arthritis. J Exp Med. 2003;197:1573–1583. doi: 10.1084/jem.20021820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gergely P, Jr., Niland B, Gonchoroff N, Pullmann R, Jr., Phillips PE, Perl A. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol. 2002;169:1092–1101. doi: 10.4049/jimmunol.169.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]