

Abstract
By using a detergent-washed membrane preparation, the interaction of the fungal natural product inhibitor aureobasidin A (AbA) with inositol phosphorylceramide synthase (IPC synthase) was studied by kinetic analysis of wild-type and mutant enzyme-catalyzed reactions. AbA inhibited the wild-type enzyme from both Candida albicans and Saccharomyces cerevisiae in an irreversible, time-dependent manner, with apparent Ki values of 183 and 234 pM, respectively. Three synthetic chemistry-derived AbA derivatives, PHA-533179, PHA-556655, and PHA-556656, had affinities 4 to 5 orders of magnitude lower and were reversible inhibitors that competed with the donor substrate phosphatidylinositol (PI). AbA was a reversible, apparently noncompetitive inhibitor, with a Ki of 1.4 μM, of the IPC synthase from an AbA-resistant S. cerevisiae mutant. The Km values for both substrates (ceramide and PI) were similar when they interacted with the mutant and the wild-type enzymes. By contrast, the Vmax for the mutant enzyme was less than 10% of that for the wild-type enzyme. A comparison of the results obtained with AbA with those obtained with two other natural products inhibitors, rustmicin and khafrefungin, revealed that while rustmicin appeared to be a reversible, noncompetitive inhibitor of the wild-type enzyme, with a Ki of 16.0 nM, khafrefungin had the kinetic properties of a time-dependent inhibitor and an apparent Ki of 0.43 nM. An evaluation of the efficiencies of these compounds as inhibitors of the mutant enzyme revealed for both a drop in the apparent affinity for the enzyme of more than 2 orders of magnitude.
Sphingolipids are important components of eukaryotic cell membranes (8). Properties like membrane dimensions and rigidity are believed to be directly influenced by the presence of sphingolipids (5, 11, 18). Moreover, it has been demonstrated that sphingolipids are important signaling molecules (8, 26), that they are involved in the transport and targeting of membrane proteins (4, 15, 27, 29), and that the downregulation of sphingolipid biosynthesis impairs the pathogenicity of the human pathogen Cryptococcus neoformans (14, 19). Fungal cellular membranes contain significant amounts of sphingolipids (33). As much as 16% of the total lipid content in the Saccharomyces cerevisiae plasma membrane and approximately 10% of the total lipid content in the Golgi membrane are sphingolipids (17, 24). The composition of fungal sphingolipids differs from that of their mammalian counterparts, primarily in the substituent at the 1-hydroxyl of the ceramide backbone. While mammalian sphingolipids may be substituted at this position with phosphocholine or, on glycosylceramides, various carbohydrates, fungal sphingolipids mainly contain phosphoinositol and, in some species, carbohydrates. The ceramide-linked phosphoinositol moiety in fungi can be further derivatized with mannose, galactose, and an additional phosphoinositol group (8). Although the reaction steps in the assembly of the ceramide portion of sphingolipids are similar in mammalian and fungal cells, the fungal biosynthetic pathway diverges at the addition of phosphoinositol. As a consequence, the enzyme catalyzing this reaction, inositol phosphorylceramide synthase (IPC synthase), has been pursued as a target in antifungal drug discovery (16, 23, 34, 36). Supporting the validity of this concept are the facts that the IPC synthase gene (AUR1) has been shown to be essential in fungi and that potent antifungal compounds that are specific inhibitors of IPC synthase have been identified (23).
A functional homolog of the fungal AUR1 gene was recently identified in the protozoan Leishmania major, and the activity of the corresponding enzyme, IPC synthase, was shown to be sensitive to the fungal IPC synthase inhibitor aureobasidin A (AbA) (7). IPC synthase activities have also been identified in Trypanosoma cruzii and Trypanosoma brucei (6, 25). Moreover, since unperturbed sphingolipid synthesis appears to be essential for protozoan parasite infectivity in trypanosomes as well as in Toxoplasma gondii, IPC synthase may represent a tractable target for the development of drugs for the treatment not only of fungal infections but also of protozoan infections (10, 25, 28).
The specific IPC synthase inhibitors identified to date are natural compounds isolated from the fermentation medium of microorganisms (20, 21, 31, 32, 35). The structures of three important inhibitor compounds, AbA, rustmicin, and khafrefungin, are shown in Fig. 1.
FIG. 1.

Structures of AbA, synthetic AbA derivatives, khafrefungin, and rustmicin.
AbA is a cyclic depsipeptide isolated from the fungus Aureobasidium pullulans (31). It is a comparatively large molecule with a number of side chains, several of which are believed to be important for activity (3, 16). Considerable efforts have been invested in attempts to map which portions of the AbA molecule contain the elements essential for the inhibitory activity, the so-called pharmacophore, but no comprehensive conclusions have been reached. Substantial efforts have also been made to create derivatives of AbA, such as the three synthetic AbA derivatives evaluated in the investigation described here, with structures that are less complex but that retain the inhibitory properties of the native molecule. Again, these efforts have largely been unsuccessful (see reference 16 and references therein). AbA has MICs in the low- and sub-μg/ml range for S. cerevisiae, Candida albicans, and C. neoformans. It is considerably less effective against Aspergillus fumigatus (37). The in vitro 50% inhibitory concentrations (IC50s) for IPC synthase activities in various Saccharomyces, Candida, and Aspergillus strains have been reported to range from 0.2 to 4.9 nM (23, 37); and a Ki value of 0.55 nM for the IPC synthase activity in S. cerevisiae has been reported (36).
Although AbA is an efficient inhibitor of IPC synthase, mutants resistant to the compound can be generated by chemical mutagenesis. Sequence analysis of these mutants has identified two positions, His 157 (13) and Phe 158 (12), in the IPC synthase (AUR1) gene sequence where mutations can generate resistant enzymes. The substituting amino acid in both reported mutants is a tyrosine. Interestingly, both sites (His 157 and Phe 158) are, to a considerable extent, removed from the amino acid residues believed to be involved in the catalytic function of the enzyme (18). Data on the kinetic properties of the mutant enzymes have not been reported.
Rustmicin (galbonolide A) is a 14-membered macrolide produced by the fungi Micromonospora chalcea and Streptomyces galbus (1, 9, 30). The molecule is a potent antifungal agent with MICs in the low-μg/ml range for both C. albicans and S. cerevisiae. It has also been reported that rustmicin is a reversible IPC synthase inhibitor and has an in vitro IC50 of about 3.8 nM (21).
Khafrefungin, an agent isolated from an unidentified fungus, is composed of an aldonic acid esterified to a linear polyketide (Fig. 1). Khafrefungin is fungicidal against both C. albicans and S. cerevisiae, and the compound reportedly inhibits the C. albicans IPC synthase with an IC50 of 0.6 nM (20).
This report presents the results of an in vitro kinetic evaluation of the inhibition of C. albicans and S. cerevisiae IPC synthases by native AbA. The time-dependent nature of AbA inhibition is documented, and an apparent Ki value is presented. In addition, a comparison of the properties of AbA with those of three synthetic derivatives, as well as those of the IPC synthase inhibitors rustmicin and khafrefungin, is presented, together with data describing the altered kinetic properties of IPC synthase from an AbA-resistant (AbAr) S. cerevisiae mutant. Constraints on the possible mechanisms of AbA inhibition are discussed.
MATERIALS AND METHODS
Cells.
Candida albicans (ATCC 38247) cells were cultured as described previously (2) Saccharomyces cerevisiae (SJ21R) cells were cultured, and an AbAr mutant was constructed as described previously (12, 13).
Enzymes.
Detergent-washed membranes from C. albicans, S. cerevisiae, and S. cerevisiae AbAr cells were prepared as described previously (2).
Inhibitors.
AbA was from TaKaRa Biomedicals. PHA-533179, PHA-556655, and PHA-556656 (Fig. 1) were synthesized at Pharmacia. Khafrefungin was isolated from Mycelia sterilia (ATCC 74305) by previously published procedures (20, 22). Rustmicin (galbonolide A) was a gift from Hans Achenbach (University of Erlangen, Erlangen, Germany).
IPC synthase assay.
IPC synthase assays were performed essentially as described previously (2). Briefly, A 28-μl enzyme mixture containing 10 μg (protein) of detergent-washed membranes, phosphatidylinositol (PI), and potassium phosphate buffer (pH 7.0) was preincubated for 30 min in a 96-well plate. The standard enzymatic reaction was started by addition of a substrate mix containing C6-NBD (7-nitro-2-1,3-benzoxadiazol-4-yl)-ceramide (Avanti Polar Lipids) in ethanol or 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) diluted to 12 μl with water to the preincubated membranes. For inhibitor studies, the substrate mix also contained 2 μl inhibitor solution in dimethyl sulfoxide (DMSO). The final assay volume was 40 μl; and the final concentrations were 50 mM potassium phosphate (pH 7.0), 0.25 mg membrane protein/ml, 100 μM PI, 5 μM C6-NBD-ceramide, 0.6 mM CHAPS, 0.3% ethanol, and 5% DMSO. Following incubation at room temperature, the reaction was stopped by addition of 200 μl of 96% methanol, and the reaction product was isolated on ion-exchange resin and quantified as outlined previously (20).
Data analysis.
The inhibition of C. albicans and S. cerevisiae wild-type enzymes by AbA was analyzed as two-step irreversible inhibition. Progress curves were consistent with first-order kinetics, and Ki and inactivation constant (kinact) values were calculated from the first-order rate constant (kobs) with the following equation:
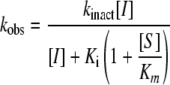 |
where [S] and [I] denote the substrate and the inhibitor concentrations, respectively
Kinetic parameters for both substrates were calculated by using the Michaelis-Menten equation.
Competitive, reversible inhibitors were analyzed by measuring the initial reaction velocities (v) and calculating Ki by using the following equation:
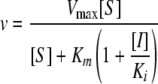 |
AbA inhibition of the S. cerevisiae AbAr enzyme was analyzed as noncompetitive, reversible inhibition by measuring the initial reaction velocities and calculating Ki by using the following equation:
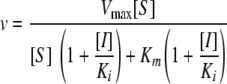 |
RESULTS AND DISCUSSION
As a first step in the analysis of the effect of AbA on IPC synthase, a determination was made as to whether the interaction between the enzyme and the inhibitor was time dependent. Product formation was plotted as a function of time at several different inhibitor concentrations. Figure 2A and B shows that both the C. albicans and S. cerevisiae enzymes clearly interact with the inhibitor in a time-dependent manner. Particularly at higher inhibitor concentrations, the product formation reaches a plateau 10 to 15 min into the reaction, indicating that product formation has ceased, that the substrate cannot compete with the inhibitor, and that the inhibitor is essentially titrating the enzyme. Figure 2C and D shows that AbA inhibited the C. albicans and S. cerevisiae enzymes with very similar apparent Ki values of 183 pM and 234 pM, respectively. The apparent kinact values for these two enzymes were 2.5 ms−1 and 1.8 ms−1, respectively, hence confirming the potency of this inhibitor observed in the experiments described above.
FIG. 2.

Inhibition of Candida albicans IPC synthase by AbA. IPC synthase assays were performed and the data were analyzed as outlined in Materials and Methods. (A and B) Progress curves generated in the absence (0) and in the presence of increasing concentrations (0.125, 0.25, 0.5, and 1 nM) of AbA. (C and D) Kinetic parameters were calculated as described in Materials and Methods. (A and C) Data generated with S. cerevisiae IPC synthase; (B and D) data generated with C. albicans IPC synthase.
To confirm the irreversible nature of the interaction between IPC synthase and AbA, an experiment was performed in which the C. albicans enzyme was preincubated with AbA prior to the initiation of the reaction. Figure 3 shows that this clearly resulted in a time-dependent inactivation of the enzyme. There was a significant difference in the amount of product formed in the reaction with preincubation compared to that formed in the reaction without preincubation.
FIG. 3.

Time dependency of AbA inhibition. IPC synthase assays were performed and the data were analyzed as outlined in Materials and Methods. Diamonds, control; squares, no preincubation with AbA and 0.2 nM AbA in assay; triangles, preincubation with 0.2 nM AbA and 0.2 nM AbA in assay; circles, preincubation with 2.0 nM AbA diluted to 0.2 nM AbA in assay.
The compounds PHA-533179, PHA-556655, and PHA-556656 (Fig. 1) are derived from the native AbA structure. The molecules are the result of a synthetic chemistry effort to generate smaller, less complicated molecules that retain at least a significant portion of the inhibitory properties of native AbA. Evaluation of the compounds with the C. albicans enzyme revealed that although these compounds retained significant inhibitory activity, they were all at least 3 orders of magnitude less potent than native AbA (Fig. 4D, inset). Figure 4A shows that, in contrast to AbA, PHA-533179 do not inhibit IPC synthase in a time-dependent manner. The amount of product formed increased with time at all inhibitor concentrations tested. Moreover, determinations of the kinetic parameters at increasing substrate concentrations in the presence of increasing concentrations of inhibitor revealed that while the Km values for PI increased significantly with increasing inhibitor concentrations, the Vmax values remained essentially constant (Fig. 4B). By contrast, no such increase was observed when the ceramide concentrations were varied (Fig. 4C). These observations suggest that PHA-533179 (as well as PHA-556655 and PHA-556656, with which similar results were obtained; data not shown) competes with the donor substrate PI. Analysis of the three compounds as reversible, competitive inhibitors resulted in Ki values of 11.8, 10.5, and 7.9 μM, respectively (Fig. 4D, inset). These values are more than 4 orders of magnitude higher than the Ki value of 183 pM determined for native AbA (compare this result with those described above), suggesting that the derivatives retain only a minor fraction of the potency of the parent molecule. It appears likely that these molecules, which in essence are fragments of the native AbA molecule, are lacking several of the binding functionalities required for high-affinity binding to the IPC synthase enzyme. Previous investigations have shown that mere manipulations of side chains on AbA can have drastic effects on the inhibitory effect of the compound (16). Conceivably, at least some of these effects may have been caused by a loss of affinity for the enzyme. Interestingly, all three AbA derivatives appeared to compete with the donor substrate PI. Since the native molecule is a time-dependent inhibitor, it was not possible to determine whether it interacts with the substrate binding site(s) on the enzyme. Nonetheless, given the observations discussed above and assuming that the three AbA derivatives still inhibit IPC synthase in a manner similar to that of native AbA, although they are less potent than native AbA, it is possible that the inhibitory effect of the native compound is the result of binding to or at the PI binding site on the enzyme. Consequently, AbA may in fact be a competitive inhibitor, albeit a very potent one. Interestingly, however, this conclusion is not consistent with the data generated for the AbAr enzyme (see below).
FIG. 4.

Inhibition of Candida albicans IPC synthase with PHA-533179. IPC synthase assays were performed and the data were analyzed as outlined in Materials and Methods. (A) Progress curves generated in the absence (0) and in the presence of increasing concentrations (5, 10, 25, and 50 μM) of the inhibitor; (B) kinetic parameters for the PI substrate determined in the absence (0) and in the presence of increasing concentrations (5, 10, and 25 μM) of the inhibitor; (C) kinetic parameters for the ceramide substrate determined in the absence (0) and in the presence of increasing concentrations (5, 10, and 20 μM) of the inhibitor; (D) Ki calculated for a competitive inhibitor.
To gain additional insight into the interaction(s) between IPC synthase and AbA, the kinetic properties of IPC synthase from an AbA-resistant S. cerevisiae mutant were studied. The IPC synthase in this strain contained the previously described F158Y mutation (12). As a first step in the characterization of this enzyme, the kinetic parameters of both substrates were determined for the wild-type S. cerevisiae enzyme. This resulted in a Km value of 3.0 μM for C6-NBD-ceramide, which is quite similar to that of the C. albicans enzyme (3.3 μM), and a Km value for PI of 555 μM, which is approximately four times higher than that of the C. albicans enzyme (Table 1). The Vmax values for the S. cerevisiae enzyme were 367 and 824 pmol min−1 mg protein−1 for C6-NBD-ceramide and PI, respectively. Both these values are a little less than 40% of the values obtained with the C. albicans enzyme (884 and 1864 pmol min−1 mg protein−1, respectively [2]). Taken together, the data indicate that slight differences in the catalytic properties of the C. albicans and S. cerevisiae enzymes may exist. However, it appears more likely, given that both enzyme preparations are made from detergent-washed membranes, that differences in the overall protein, lipid, and/or carbohydrate compositions of the two cell types, as well as in the amounts of the enzyme preparations recovered (again, likely caused by structural differences in the two cell types), are the true reasons for the observed differences. A more accurate comparison must await purification of the two enzymes. Nonetheless, the data generated strongly suggest that the catalytic properties of IPC synthase are quite similar in C. albicans and S. cerevisiae. This conclusion is further supported by the very similar sensitivities of the two enzymes to AbA (Fig. 2 and data not shown).
TABLE 1.
Kinetic properties of C. albicans wild-type, S. cerevisiae wild-type and S. cerevisiae AbAr IPC synthasea
| Organism | PI
|
Ceramide
|
||
|---|---|---|---|---|
| Km (μM) | Vmax (pmol min−1 mg protein−1) | Km (μM) | Vmax (pmol min−1 mg protein−1) | |
| C. albicans wt | 130 | 1,864 | 3.3 | 884 |
| S. cerevisiae wt | 555 | 824 | 3.0 | 367 |
| S. cerevisiae AbAr | 330 | 69 | 2.0 | 25 |
The assays were performed as outlined in the text. Ceramide, C6-NBD-ceramide.
Determination of kinetic parameters (for the two substrates) for the S. cerevisiae AbAr mutant enzyme (Table 1) yielded Km values similar to those for the wild-type enzyme: approximately 2.0 and 330 μM for C6-NBD-ceramide and PI, respectively (compare these results with those presented above). In contrast, the Vmax values, at approximately 25 and 69 pmol min−1 mg protein−1 for C6-NBD-ceramide and PI, respectively, were less than 10% of those obtained for the wild-type S. cerevisiae enzyme. Clearly, the amino acid substitution in IPC synthase conferring AbA resistance has a considerable impact on the catalytic performance of the enzyme. Nonetheless, the affinities for the two substrates appeared to be essentially unaltered for the mutant enzyme. This is in stark contrast to the affinity of the mutant enzyme for AbA, which was reduced by more than 3 orders of magnitude. A kinetic evaluation revealed that AbA is a reversible inhibitor of the S. cerevisiae AbAr mutant enzyme, with an apparent Ki of approximately 1.4 μM (Fig. 5). Determinations of the values of the kinetic parameters at several substrate concentrations in the presence of increasing concentrations of the inhibitor showed that in assays with the mutant enzyme, AbA does not appear to compete with the binding of either PI (Fig. 5B) or ceramide (Fig. 5C).
FIG. 5.

Inhibition of Saccharomyces cerevisiae AbAr IPC synthase with AbA. IPC synthase assays were performed and the data were analyzed as outlined in Materials and Methods. (A) Progress curves generated in the absence (0) and in the presence of increasing concentrations (0.5, 1, 2, 4, and 8 μM) of the inhibitor; (B) kinetic parameters for the PI substrate determined in the absence (0) and in the presence of increasing concentrations (0.1, 0.2, 0.4, 0.8, and 1.6 μM) of the inhibitor; (C) kinetic parameters for the ceramide substrate determined in the absence (0) and in the presence of increasing concentrations (0.05, 0.1, 0.2, and 0.4 μM) of the inhibitor; (D) Ki calculated for a noncompetitive inhibitor.
Although the finding that native AbA does not compete with the binding of PI in the mutant enzyme is in contrast to the mechanism of action of AbA-derived compounds PHA-533179, PHA-556655, and PHA-556656, this finding is consistent with the fact that the mutations cause a drop in the affinity of AbA for the mutant IPC synthase of more than 3 orders of magnitude. Since the mutations have virtually no effect on the affinity of AbA for either substrate (Table 1) but cause a drastic drop in the reaction velocity, the mutations may alter the enzyme such that although the substrates are still capable of binding to the enzyme with an affinity similar to that of the wild-type enzyme, their orientation or intermolecular distance(s) in the active site is compromised. AbA may bind to a site that is related to or part of the active site. Alternatively, the compound may bind to a site that is removed from the near vicinity of the active site but that still influences (by steric means or otherwise) the catalytic function of the enzyme. It is noteworthy, in this context, that the amino acid substitutions of the AbAr mutants are located at positions believed to be quite remote from the enzyme active site (12, 13, 18).
Two other natural products, rustmicin and khafrefungin, are both specific inhibitors of IPC synthase. However, they differ significantly from AbA in their interaction with the enzyme. In contrast to AbA, rustmicin is a reversible inhibitor with an apparent Ki. almost 2 orders of magnitude higher than that of AbA (Fig. 6; Table 2). The data also suggest that this inhibitor does not compete with either of the two substrates. As discussed above, since the interaction of AbA with wild-type IPC synthase is time dependent, it is difficult to determine whether this compound interacts with the enzyme in a competitive or a noncompetitive manner. Interestingly, the IC50 of rustmicin for the AbAr enzyme shows the same increase as that of AbA of approximately 3 orders of magnitude compared to that for the wild-type enzyme (Table 2). This indicates a possible overlap between the binding sites for AbA and rustmicin. It is also consistent with the possibility (supported by the data generated with the AbAr enzyme) that AbA does not compete with the substrate binding site(s) on the wild-type enzyme. Despite significant differences in the structures of khafrefungin and AbA, khafrefungin appears to be similar to AbA in its interaction with IPC synthase. The compound is a time-dependent inhibitor with an apparent Ki of approximately 425 pM (Fig. 7; Table 2). Moreover, the binding site of this compound appears, at least to some extent, to overlap with that of AbA, since the AbAr enzyme was again about 3 orders of magnitude less sensitive to this inhibitor than the wild-type enzyme (Table 2).
FIG. 6.

Inhibition of C. albicans IPC synthase by rustmicin. IPC synthase assays were performed and the data were analyzed as outlined in Materials and Methods. (A) Progress curves generated in the absence (0) and in the presence of increasing concentrations (5, 10, 20, 30, and 40 nM) of the inhibitor; (B) kinetic parameters for the PI substrate determined in the absence (0) and in the presence of increasing concentrations (10, 20, and 30 nM) of the inhibitor; (C) kinetic parameters for the ceramide substrate determined in the absence (0) and in the presence of increasing concentrations (5, 10, 20, and 30 nM) of the inhibitor.
TABLE 2.
Comparison of the inhibitory properties of AbA, khafrefungin, and rustmicina
| Organism | Aureobasidin A
|
Khafrefungin
|
Rustmicin
|
|||||
|---|---|---|---|---|---|---|---|---|
| Ki (nM) | Inh. | Ki (nM) | IC50 (nM) | Inh. | Ki (nM) | IC50 (nM) | Inh. | |
| C. albicans wild type | 0.18 | T-d | 0.43 | T-d | 16 | Non | ||
| S. cerevisiae wild type | 0.23 | T-d | 43 | ND | 13 | ND | ||
| S. cerevisiae AbAr | 1.4 × 103 | Non | 11.3 × 103 | ND | 27.2 × 103 | ND | ||
The assays were performed as outlined in the text. Abbreviations: Inh., inhibition type; T-d, time dependent; Non, noncompetitive; ND, not determined.
FIG. 7.

Inhibition of C. albicans IPC synthase by khafrefungin. IPC synthase assays were performed and the data were analyzed as outlined in Materials and Methods. (A) Progress curves generated in the absence (0) and in the presence of increasing concentrations (1, 2, 4, 8, and 16 nM) of the inhibitor; (B) Ki calculated for an irreversible inhibitor.
Taken together, the observations presented in this report suggest that AbA is an irreversible, time-dependent inhibitor of IPC synthase in both C. albicans and S. cerevisiae. Moreover, the entire, intact structure of the compound appears to be important for both the affinity and the specific inhibitory mechanism of this compound. Hence, the inhibitory activity of AbA may not be mediated by a specific portion of the structure, a pharmacophore, but rather, the entire structure appears to be essential for full biological activity. All fragments of the structure evaluated were comparatively inactive, and compounds comprising other portions of the AbA structure have even less potency (data not shown). The data are consistent with previously published data that suggest that the range of structural alterations that can be made to AbA but that result in the retention of its inhibitory activity is quite limited (summarized in reference 16). Two other inhibitors, rustmicin and khafrefungin, show some similarities to AbA in their modes of action with the IPC synthase, with the most significant difference being their lower affinity for the enzyme (particularly for rustmicin). Evidence for a competitive mechanism was not found for any of the three inhibitors.
Acknowledgments
We thank Joyce Cialdella, Ray Zielinski, and Ming-Shang Kuo, at Pfizer, Inc., for the preparation and purification of khafrefungin.
Footnotes
Published ahead of print on 1 December 2008.
REFERENCES
- 1.Achenbach, H., A. Muhlenfeld, U. Fauth, and H. Zahner, H. 1988. The galbonolides. Novel, powerful antifungal macrolides from Streptomyces galbus ssp. eurythermus. Ann. N. Y. Acad. Sci. 544:128-140. [DOI] [PubMed] [Google Scholar]
- 2.Aeed, P. A., A. E. Sperry, C. L. Young, M. M. Nagiec, and Å. P. Elhammer. 2004. Effect of membrane perturbing agents on the activity and phase distribution of C. albicans inositol phosphorylceramide synthase; development of a novel assay. Biochemistry 43:8483-8493. [DOI] [PubMed] [Google Scholar]
- 3.Awazu, N., K. Ikai, J. Yamamoto, K. Nishimura, S. Mizutani, K. Takesako, and I. Kato. 1994. Structures and antifungal activities of new aureobasidins. J. Antibiot. 48:525-527. [DOI] [PubMed] [Google Scholar]
- 4.Bagnat, M., S. Keränen, A. Shevchenko, A. Shevchenko, and K. Simons. 2000. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc. Natl. Acad. Sci. USA 97:3254-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, D., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221-17224. [DOI] [PubMed] [Google Scholar]
- 6.Denny, P. W., D. Goulding, M. A. Ferguson, and D. F. Smith. 2004. Sphingolipid-free Leishmania are defective in membrane trafficking, differentiation and infectivity. Mol. Microbiol. 52:313-327. [DOI] [PubMed] [Google Scholar]
- 7.Denny, P. W., H. Shams-Eldin, H. P. Price, D. F. Smith, and R. T. Schwarz. 2006. The protozoan inositol phosphorylceramide synthase: a novel drug target that defines a new class of sphingolipid synthase. J. Biol. Chem. 281:28200-28209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickson, R. C., and R. L. Lester. 1999. Yeast sphingolipids. Biochim. Biophys. Acta 1426:347-357. [DOI] [PubMed] [Google Scholar]
- 9.Fauth, U., H. Zahner, A. Muhlenfeld, and H. Achenbach. 1986. Galbonolides A and B, two non-glycosidic antifungal macrolides J. Antibiot. (Tokyo) 39:1760-1764. [DOI] [PubMed] [Google Scholar]
- 10.Figueiredo, J. M., W. B. Dias, L. Mendonoca-Previato, J. O. Previato, and N. Heise. 2005. Characterization of the inositolphosphorylceramide synthase activity from Trypanosoma cruzi. Biochem. J. 387:519-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillas, I., P. A. Kirchman, R. Chuard, M. Pfefferli, J. C. Jiang, S. M. Jazwinski, and A. Conzelmann. 2001. C26-CoA-dependent ceramide synthesis of Saccharomyces cerevisiae is operated by Lag1p and Lac1p. EMBO J. 20:2655-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashida-Okado, T., A. Ogawa, M. Endo, R. Yasumoto, K. Takesako, and I. Kato. 1996. AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: a study of defective morphologies in Aur1p-depleted cells. Mol. Gen. Genet. 25:236-244. [DOI] [PubMed] [Google Scholar]
- 13.Heidler, S. A., and J. A. Radding. 1995. The AUR1 gene in Saccharomyces cerevisiae encodes a dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 39:2765-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heung, L. J., C. Luberto, A. Plowden, Y. A. Hannun, and M. Del Poeta. 2004. The sphingolipid pathway regulates Pkc1 through the formation of diacylglycerol in Cryptococcus neoformans. J. Biol. Chem. 279:21144-21153. [DOI] [PubMed] [Google Scholar]
- 15.Kübler, E., H. G. Dohlman, and M. P. Lisanti. 1996. Identification of Triton X-100 insoluble membrane domains in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 271:32975-32980. [DOI] [PubMed] [Google Scholar]
- 16.Kurome, T., and K. Takesako. 2000. SAR and potential of the aureobasidin class of antifungal agents. Curr. Opin. Anti-Infect. Investig. Drugs 2:375-386. [Google Scholar]
- 17.Leber, A., C. Hrastnik, and G. Daum. 1995. Phospholipid-synthesizing enzymes in the Golgi membranes of the yeast Saccharomyces cerevisiae. FEBS Lett. 337:271-274. [DOI] [PubMed] [Google Scholar]
- 18.Levine, T. P., C. A. R. Wiggins, and S. Munro. 2000. Inositol phosphorylceramide synthase is located in the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell 11:2267-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luberto, C., D. L. Toffaletti, E. A Willis, S. C. Tucker, A. Casadevall, J. R. Perfect, Y. A. Hannun, and M. Del Poeta. 2001. Roles for inositol-phosphoryl ceramide synthase 1 (IPC1) in pathogenesis of C. neoformans. Genes Dev. 15:201-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandala, S. M., R. A. Thornton, M. Rosenbach, J. Milligan, M. Garcia-Calvo, H. G. Bull, and M. B. Kurtz. 1997. Khafrefungin, a novel inhibitor of sphingolipid synthesis. J. Biol. Chem. 272:32709-32714. [DOI] [PubMed] [Google Scholar]
- 21.Mandala, S. M., R. A. Thornton, J. Milligan, M. Rosenbach, M. Garcia-Calvo, H. G. Bull, G. Harris, G. K. Abruzzo, A. M. Flattery, C. J. Gill, K. Bartizal, S. Dreikorn, and M. B. Kurtz. 1998. Rustmicin, a potent antifungal agent, inhibits sphingolipid synthesis at inositolphosphorylceramide synthase. J. Biol. Chem. 273:14942-14949. [DOI] [PubMed] [Google Scholar]
- 22.Mandala, S. M., and G. H. Harris. 2000. Isolation and characterization of novel inhibitors of sphingolipid synthesis: australifungin, viridiofungins, rustmicin, and khafrefungin. Methods Enzymol. 311:335-348. [DOI] [PubMed] [Google Scholar]
- 23.Nagiec, M. M., E. E. Nagiec, J. A. Baltisberger, G. B. Wells, R. L Lester, and R. C. Dickson. 1997. Sphingolipid synthesis as a target for antifungal drugs. J. Biol. Chem. 272:9809-9817. [DOI] [PubMed] [Google Scholar]
- 24.Patton, J. L., and R. L. Lester. 1991. The phosphoinositol sphingolipids of Saccharomyces cerevisiae are highly localized in the plasma membrane. J. Bacteriol. 173:3101-3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salto, M. L., L. E. Bertello, M. Vieira, R. Docampo, S. N. Moreno, and R. M. de Lederkremer. 2003. Formation and remodeling of inositolphosphoceramide during differentiation of Trypanosoma cruzi from trypomastigote to amastigote. Eukaryot. Cell 2:756-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneiter, R. 1999. Brave little yeast, please guide us to Thebes: sphingolipid function in Saccharomyces cerevisiae. Bioessays 21:1004-1010. [DOI] [PubMed] [Google Scholar]
- 27.Simons, K., and E. Ilkonen. 1997. Functional rafts in cell membranes. Nature 387:569-572. [DOI] [PubMed] [Google Scholar]
- 28.Sonda, S., G. Sala, R. Ghidoni, A. Hemphill, and J. Pieters. 2005. Inhibitory effect of aureobasidin A on Toxoplasma gondii. Antimicrob. Agents Chemother. 49:1794-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sütterlin, C., T. L. Doering, F. Schimmöller, S. Schröder, and H. Reizman. 1997. Specific requirements for the ER to Golgi transport of GPI-anchored proteins in yeast. J. Cell Sci. 110:2703-2714. [DOI] [PubMed] [Google Scholar]
- 30.Takatsu, T., H. Nakayama, A. Shimazu, K. Furihata, K. Ikeda, K. Furihata, H. Seto, and N. J. Otake. 1985. Rustmicin, a new macrolide antibiotic active against wheat stem rust fungus. J. Antibiot. (Tokyo) 3:1806-1809. [DOI] [PubMed] [Google Scholar]
- 31.Takesako, K., K. Ikai, F. Haruna, M. Endo, K. Shimanaka, E. Sono, T. Nakamura, and I. Kato. 1991. Aureobasidins, new antifungal antibiotics. Taxonomy, fermentation, isolation, and properties. J. Antibiot. 44:919-924. [DOI] [PubMed] [Google Scholar]
- 32.Takesako, K., H. Kuroda, T. Inoue, F. Haruna, Y. Yoshikawa, and I. Kato. 1993. Biological properties of aureobasidin A, a cyclic depsipeptide antifungal antibiotic. J. Antibiot. 46:1414-1420. [DOI] [PubMed] [Google Scholar]
- 33.van der Rest, M. E., A. H. Kamminga, A. Nakano, Y. Anraku, B. Poolman, and W. N. Konings. 1995. The plasma membrane in Saccharomyces cerevisiae: structure, function and biogenesis. Microbiol. Rev. 59:304-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wills, E. A., M. R. Redinbo, J. R. Perfect, and M. Del Poeta. 2000. New potential targets for antifungal development. Emerg. Ther. Targets 4:1-32. [Google Scholar]
- 35.Yano, T., A. Aoyagi, S. Kozuma, Y. Kawamura, I. Tanaka, Y. Suzuki, Y. Takamatsu, T. Takatsu, and M. Inukai. 2007. Pleofungins, novel inositol phosphorylceramide synthase inhibitors from Phoma sp. SANK 13899. I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 60:136-142. [DOI] [PubMed] [Google Scholar]
- 36.Zhong, W., D. J. Murphy, and N. H. Georgopapadakou. 1999. Inhibition of yeast inositol phosphorylceramide synthase by aureobasidin A measured by a fluorometric assay. FEBS Lett. 463:241-244. [DOI] [PubMed] [Google Scholar]
- 37.Zhong, W., M. W. Jeffries, and N. H. Georgopapadakou. 2000. Inhibition of inositol phosphorylceramide synthase by aureobasidin A in Candida and Aspergillus species. Antimicrob. Agents Chemother. 44:651-653. [DOI] [PMC free article] [PubMed] [Google Scholar]