

Abstract
Preclinical animal models informing antimalarial drug development are scarce. We have used asexual erythrocytic Plasmodium cynomolgi infections of rhesus macaques to model Plasmodium vivax during preclinical development of compounds targeting parasite phospholipid synthesis. Using this malaria model, we accumulated data confirming highly reproducible infection patterns, with self-curing parasite peaks reproducibly preceding recrudescence peaks. We applied nonlinear mixed-effect (NLME) models, estimating treatment effects in three drug studies: G25 (injected) and the bisthiazolium prodrugs TE4gt and TE3 (oral). All compounds fully cured P. cynomolgi-infected macaques, with significant effects on parasitemia height and time of peak. Although all three TE3 doses tested were fully curative, NLME models discriminated dose-dependent differential pharmacological antimalarial activity. By applying NLME modeling treatment effects are readily quantified. Such drug development studies are more informative and contribute to reduction and refinement in animal experimentation.
Malaria is caused by protozoan parasites, of which Plasmodium falciparum and P. vivax are the most important. P. falciparum causes up to 2 million deaths annually in sub-Saharan Africa; victims are mainly children under the age of 5 years (8, 21). P. vivax is prevalent in eastern and central Africa and in more temperate climates outside Africa and has enormous socioeconomic impact, particularly in South America and Asia (11). Malaria is a poverty-related disease; although antimalarial drugs are powerful therapeutic and prophylactic tools, their use in endemic regions is highly cost sensitive. Drug resistance to the cheapest and most commonly available antimalarials (chloroquine and anti-folate drugs) has spread worldwide for P. falciparum and is an increasing problem in the treatment of P. vivax infections (1). New pharmacological approaches for developing molecules with novel mechanisms of action are urgently required. Compounds effective against both P. falciparum and P. vivax are required for the large regions of the world where both parasites are prevalent.
Screening for activity against P. vivax during development adds valuable information to the compound dossier. However, the complex biology of P. vivax complicates such screening. In vitro cultivation of the parasite is severely restricted due to its requirement for reticulocytes. Like all malaria parasites, it has a highly restricted host range. Although P. vivax blood stages can be adapted to grow in certain New World monkey species (principally Saimiri and Aotus sp.) and chimpanzees, the limited availability of these experimental nonhuman primate hosts is a complicating factor.
P. cynomolgi, a malaria parasite of macaques that can also infect humans (3), is phylogenetically closely related to P. vivax (6) and provides a close biological model of P. vivax (10, 13, 23) where important characteristics (e.g., duration of blood-stage maturation, preference for young red blood cells, occurrence of recrudescences, and hypnozoite formation) are shared (3). Blood-stage parasitemia in rhesus macaques follows a characteristic pattern of first a self-curing peak parasitemia, followed by a recrudescence about a week later. Macaques, the natural host of P. cynomolgi, have a close phylogenetic and biological relationship with humans; thus, P. cynomolgi infections of macaques provide an infection model closely resembling P. vivax in humans.
We have used the P. cynomolgi-macaque model during the preclinical development of a new class of antimalarials interfering with phospholipid metabolism (2, 22, 24) to assess in vivo activity against P. vivax-type blood-stage parasites. The effects of compounds on the first peak parasitemia in this model demonstrate general antiparasitic activity of the test compounds. The relatively quick development of recrudescence in this model (i.e., a second peak parasitemia 6 to 8 days after the first peak) allows evaluation of the curative effect of test compounds. If compounds only suppress parasitemia, this will become evident by a reduced first peak parasitemia, followed by the appearance of recrudescent parasitemia within a limited time frame (that may be influenced by the in vivo half-life of the compounds). Assessment of these important compound characteristics is facilitated by the P. cynomolgi-macaque model.
The compounds evaluated here are bisquaternary ammonium salts that mimic choline structure, designed to target malaria parasite membrane biogenesis by interrupting the biosynthesis of essential phosphatidylcholine. High accumulation inside infected red blood cells (RBC) provides both potency and selectivity. Their reported dual mechanism of action (17, 22) holds promise for activity against polypharmacoresistant malaria and should reduce the risk of cross-resistance emerging. The activity against two independent targets mediated by one compound gives similar advantages to the use of combinations of two drugs in terms of restricting the development of resistance. This is because two targets must then in principle mutate in parallel to provide selective advantage and allow resistance to evolve (22). It has an added theoretical advantage over drug combinations in that there is no concern about different half-lives of the two compounds (and thus no potential for one to be exerting antiparasitic effects in the absence of the other and thereby facilitating development of resistance). G25 was the lead compound of extremely potent, yet also toxic, choline analogs; TE4gt and TE3 were developed subsequently as orally deliverable bioprecursors of the less-toxic bisthiazolium salts.
Here we report on the statistical analysis of drug efficacy using nonlinear mixed-effect models (NLME) (12, 14) to model parasitemia development in the monkeys, allowing quantitation of treatment effects. NLME were selected for statistical analysis because (i) multiple observations were made on each separate animal in the study and this (pseudo)replication with autocorrelation must be accounted for in the analysis; (ii) no data transformation is available to linearize the relationship between parasitemia and time, but a meaningful nonlinear description is possible; and (iii) the course of parasitemia through time varies between animals due to differences between individuals (random effect). The main focus of this research is on the effect of treatment (fixed effects), and the between-animal variation can be incorporated as a random effect. Mixed models use fixed and random effects for its parameters; fixed effects can be interpreted at the population level (treatment effects), and random effects can be interpreted at the individual level. The latter means that each individual can have its own level of a certain parameter. We applied this analysis to estimate treatment effects in two previously published and one new data set from studies where we tested G25 administered intramuscularly (i.m.) (24) and TE4gt and TE3 (converted in vivo into T4 and T3, respectively) (22) administered orally.
MATERIALS AND METHODS
Parasites and primates.
P. cynomolgi M strain (4, 19) was used during all studies. This parasite was originally kindly provided by W. E. Collins, Centers for Disease Control and Prevention, Atlanta, GA, and has been maintained in our labs by occasional serial passage of blood-stage parasites through the rhesus macaque, Macaca mulatta. Ring stage parasites (stored at −80°C) are routinely frozen and recovered according to published methods (16). All animal work was carried out under protocols approved by the Animal Ethics Committee (DEC) according to Dutch laws.
Drugs.
The following choline analogues were used: the bis-thiazolium salt G25 solubilized in saline (24), the thioester prodrug TE4gt solubilized in saline (22), and the thiocarbonate prodrug TE3 solubilized in dimethyl sulfoxide (DMSO) (22).
In vivo drug efficacy.
To initiate experiments, a rhesus monkey was infected by intravenous (i.v.) injection with P. cynomolgi M strain blood-stage parasites obtained from cryopreserved stock. At a parasitemia (expressed as the percentage of RBC infected with malaria parasites, as determined on Giemsa-stained thin blood films) of ca. 1%, 10 ml of blood was drawn into a heparinized tube, RBC were washed with RPMI 1640 supplemented with HEPES and l-glutamine (Gibco, Paisley, United Kingdom). After precise determination of parasitemia, dilutions were made in RPMI to infect experimental monkeys by i.v. injection of 106 infected RBC in a 1-ml final volume. Parasitemia development was monitored in each animal by microscopic examination of Giemsa-stained thin blood films prepared from finger prick blood starting day 4 postinfection. No correlation was observed throughout the studies between peak 1 parasitemia levels and the age of the monkeys (data not shown). For G25 (24), 10 infected monkeys of either sex, 3 to 6 years of age at the start of the experiment and weighing 3.5 to 8 kg, were randomly assigned to two groups of five. Drug treatment was started when parasitemia in all monkeys ranged from 0.2 to 0.5%. G25, solubilized in saline, was given twice daily for 8 consecutive days by i.m. injection to group 1 animals. Group 2 control animals received saline only, according to the same schedule. Parasitemia was monitored until day 14 posttreatment, when the recrudescence peak parasitemia had resolved in group 2 animals and these animals were cured with pyrimethamine (1 mg/kg, oral administration for 3 consecutive days). A final blood smear was checked at day 28 posttreatment to assure that all monkeys were free from parasites.
For TE4gt, seven monkeys of either sex, 4 to 5.5 years of age at the start of the experiment and weighing 3.1 to 4.6 kg, were age, weight, and sex matched and assigned to two groups (three in experimental group 1 and four in control group 2). Drug treatment was started when parasitemia in all monkeys ranged from 0.5 to 1.5%. TE4gt (40 mg/kg, solubilized in saline) was given by gavage during mild ketamine sedation of the monkeys once daily for 8 consecutive days (days 0 to 7). The monkeys were fasted overnight before each drug administration. Group 2 animals received saline only, according to the same schedule. Parasitemia was monitored until day 18 posttreatment, when the recrudescence of group 2 animals had resolved and animals were cured with pyrimethamine. A final blood smear was checked at day 24 posttreatment to assure all monkeys were free from parasites (all monkeys were negative; data not shown).
For TE3 (22), 16 rhesus monkeys of either sex, 6 to 7 years of age at the start of the experiment and weighing 4.3 to 7.3 kg, were age, weight, and sex matched and assigned to four groups of four animals. When all monkeys had parasitemias between 0.01 and 0.2%, TE3 treatment was initiated. TE3 was given by gavage (2.5 ml of TE3 in DMSO, followed by 10 ml of tap water) to sedated monkeys that had been fasted overnight, once daily for 4 consecutive days (group 1 received TE3 at 27 mg/kg, group 2 at 9 mg/kg, and group 3 at 3 mg/kg; control group 4 received DMSO only). Parasitemia was monitored until day 19 posttreatment (end of recrudescence in controls), when control animals were cured with chloroquine (Rhone-Poulenc, Paris, France) at 5 mg/kg by i.m. injection for 3 consecutive days. A final blood smear was checked at day 30 posttreatment to assure that all monkeys were free from parasites (all monkeys were negative; data not shown).
To avoid bias, all parasitemia determinations were performed on coded slides. Complete cure was defined in all experiments as nondetectable parasitemia in thin films counting a minimum of 5,000 RBC and no recurrence of detectable parasitemia through the follow-up period.
Statistical methods.
Parasitemia development over time was modeled by using NLME (14). The use of linear mixed-effects models was previously suggested by Paterson et al., and similar principles apply to NLME (12). Statistical analysis was performed using S-PLUS 6.1 for Windows (Insightful Corp. Seattle, WA) and/or R (R Foundation for Statistical Computing, Vienna, Austria).
The parameterizations applied are presented below. Briefly, parasitemia development over time was measured by three parameters: peak parasitemia height, time (days) at which the peak parasitemia was achieved, and width of the peak parasitemia (at the base of the peak, with a lower detection level of 0.0002%). Start values for modeling were estimated from numerical summaries and graphical representations. In the case of a two-peak parasitemia, the height of the second peak and the time difference between the first and second peaks were also included in the model. A data set and an analysis script for the TE3 experiment is provided in the supplemental material; this script also explains the critical steps in the calculations. Modeling was initiated by using a simple model with only fixed effects for the parameters and was subsequently refined in a stepwise manner by modeling between-animal variation by the addition of random effects for peak height and peak time parameters and modeling the treatment effects in the fixed effects for peak height and peak time. A correction for heteroscedasticity was applied in the models by modeling variance as a power of the fitted values (varPower or varConstPower [14]). Model fits were evaluated by comparing the Aikake information criterion obtained by maximum-likelihood estimation (ML), because differences in ML-log likelihoods are chi-squared distributed. ML models with the lowest Aikake information criteria were selected as final models. Parameters for the best fitting models were then estimated by using restricted ML, because restricted ML yields more “conservative” error estimates and unbiased estimates of the random and fixed parameters. The estimates from the final model are presented as the estimated parameter values in the text and as the difference in parameters between control and treatment groups in Tables 1 and 2.
TABLE 1.
Parameter estimates from NLME for TE3 control group (controls) versus all TE3 dosages combined (treated)a
| Parameter | Estimate | SE | df | t-value | P | Lower 95% CL | Upper 95% CL |
|---|---|---|---|---|---|---|---|
| Peak 1 ht for controls | 3.02 | 0.42 | 310 | 7.2 | 6.24 × 10−12 | 2.2 | 3.9 |
| Peak 1 ht difference for treated | -2.46 | 0.48 | 310 | -5.1 | 6.38 × 10−07 | -3.4 | -1.5 |
| Peak 2 ht for controls | 0.72 | 0.19 | 310 | 3.9 | 0.000125 | 0.4 | 1.1 |
| Peak 2 ht difference for treated | -0.72 | 0.22 | 310 | -3.3 | 0.001037 | -1.1 | -0.3 |
| Peak 1 time for controls | 3.87 | 0.05 | 310 | 75.7 | 5.9 × 10−202 | 3.8 | 4.0 |
| Peak 1 time difference for treated | -2.02 | 0.11 | 310 | -18.8 | 4.12 × 10−53 | -2.2 | -1.8 |
| Time difference between peaks 1 and 2 | 7.61 | 0.23 | 310 | 32.9 | 5.3 × 10−103 | 7.2 | 8.1 |
| Peak width | 1.33 | 0.09 | 310 | 14.6 | 4.19 × 10−37 | 1.2 | 1.5 |
Parameterizations are shown in Materials and Methods (two-peak model). The peak 2 time is the difference between first and second peak parasitemias. The estimate obtained from the NLME for “controls” is given as the actual parameter estimate and for “treated” is given as the difference between treated and controls. t-value, t distribution value; P, probability at the given t-value and df, The lower and upper 95% CL are the lower and upper 95% confidence limits for the estimates.
TABLE 2.
Estimates from NLME for the first peak in TE3 dose-range treatmentsa
| Parameter | Estimate | SE | df | t-value | P | Lower 95% CL | Upper 95% CL |
|---|---|---|---|---|---|---|---|
| Peak time for 3-mg/kg group | 2.03 | 0.06 | 154 | 32.4 | 9.92 × 10−71 | 1.9 | 2.2 |
| Peak time for 9-mg/kg group (difference from 3) | -0.29 | 0.10 | 154 | -2.8 | 0.005092 | -0.5 | -0.1 |
| Peak time for 27-mg/kg group (difference from 3) | -0.40 | 0.11 | 154 | -3.6 | 0.000509 | -0.6 | -0.2 |
| Peak 1 ht for treatment groups | 0.63 | 0.12 | 154 | 5.1 | 1.03 × 10−6 | 0.4 | 0.9 |
| Peak width for all treatment groups | 0.96 | 0.08 | 154 | 11.8 | 3.04 × 10−23 | 0.8 | 1.1 |
Parameterizations are shown in Materials and Methods (one-peak model). The estimate obtained from the NLME for “controls” is given as the actual parameter estimate and for “treated” is given as the difference between treated (9 and 27 mg/kg) versus 3 mg/kg. t-value, t distribution value; P, probability at given t-value and df, Lower and upper 95% CL are the lower and upper 95% confidence limits for the estimates.
Parameterization for one-peak model.
Parameterization for the one-peak model was calculated as follows:
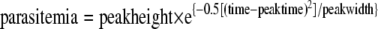 |
where “peakheight” is the maximum parasitemia (% infected RBC), “time” is the time in days in the experiment relative to start of treatment, “peaktime” is the time in days at which the first peak occurs, and “peakwidth” is the square of the width of the first parasitemia peak, describing the width at the base of the curve.
Parameterization for two-peak model.
Parameterization for the two-peak model was calculated as follows:
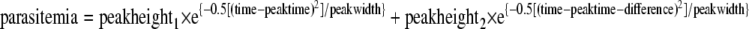 |
where “peakheight1” is the maximum parasitemia of the first peak, “time” is the time in days in the experiment relative to start of treatment, “peaktime” is the time in days at which the first peak occurs, “peakwidth” is the square of the width of the parasitemia peak(s), describing the width at the base of the curve(s), “peakheight2” is the maximum parasitemia of the second peak, and “peaktime-difference” is the time delay in days between the two peak parasitemias.
RESULTS
P. cynomolgi M strain yields a highly reproducible blood-stage infection pattern in M. mulatta.
The experiments were performed over a 5-year period. During this time, infection of rhesus macaques with 106 infected RBC derived from infected parasite donor monkeys resulted in reproducible parasitemia development (Fig. 1 and 2). All 13 control monkeys used in the studies had an initial self-limiting peak parasitemia of between 1 and 10% at day 9 to 12 postinfection (data for individual animals not shown). Parasitemia then dropped to undetectable levels until a recrudescence became evident 5 to 8 days after the first peak with maximum parasitemias between 0.2 and 4.5%. This recrudescence was also self-limiting and was followed by a period of low-level chronic parasitemia that was cured by oral pyrimethamine or i.m. chloroquine administration. Because of small between-experiment fluctuations in parasitemia peak height and time (which are inherent to biological experiments, e.g., different parasite donor animals used), we used a control group in every experiment. The parasitemia development can be modeled per animal by using biologically relevant parameterization as described in Materials and Methods. NLME modeling accurately describes the observed course of parasitemia, with the proviso that peak height is estimated at lower values than actually observed (Fig. 1). The inclusion of per animal random effects for peak time and height, allowing each animal to have a different peak parasitemia at a slightly different time point, enabled a more realistic estimate of peak height. This random effect allows for modeling of animals within one treatment group that may have small differences in the timing of peak parasitemia. Experimental design for the different compounds evolved throughout the study period in response to accumulating data on the high level of reproducibility of infection, with group sizes necessary for given outcomes, being adjusted to use the minimal numbers of animals commensurate with expected outcomes. Following the three experiments described here, using five, three to four, and four animals per group in experiments 1, 2, and 3, respectively, we currently use a standard of four animals per group in drug studies (unless very small intergroup differences are anticipated).
FIG. 1.

Observed and predicted (NLME) parasitemias in blood-stage P. cynomolgi-infected M. mulatta. Observed (black dots) and predicted (solid lines) parasitemias are shown for five individual control monkeys from the G25 experiment (A to E) that were treated as described in Materials and Methods. Parasitemia was determined by counting parasitized RBC on blinded Giemsa-stained thin films and is expressed as a percentage of total RBC. The parasitemia predicted by NLME was determined as described in Materials and Methods. Day 0 is the time when G25 treatment was started in the treatment group.
FIG. 2.

Average parasitemia in P. cynomolgi-infected rhesus macaques in absence or presence of treatment with three types of anti-phospholipid effectors. Rhesus monkeys were infected with P. cynomolgi blood-stage parasites, and drug treatment was started at low parasitemia in all monkeys (arrow day 0). Start and end of treatment are indicated by arrows below the x axis. The “T” represents the start of pyrimethamine or chloroquine treatment of the control groups. Parasitemia was determined as described in Fig. 1. In each graph average parasitemia and standard deviation for treated and control groups are shown, except in Fig. C, where for reasons of legibility standard deviations are only shown for the control group. Symbols used are explained in the figures. (A) G25 treatment by twice-daily i.m. doses for eight consecutive days for five experimental and five non-drug-treated control monkeys. (B) TE4gt treatment by once daily oral doses for 8 consecutive days for three experimental monkeys and four non-drug-treated controls. (C) TE3 treatment by once-daily oral doses of three different dosages given for 4 consecutive days, with experimental groups each comprising four monkeys and a non-drug-treated group of four monkeys.
Potent antimalarial activity of injected drug and orally administered prodrugs as revealed by NLME modeling and statistical analysis.
All three drugs were highly effective in clearing P. cynomolgi blood-stage infections in rhesus macaques (Fig. 2). NLME modeling provides statistical evaluation of between-group differences. For G25-treated animals (Fig. 2A) peak parasitemia, expressed as the percent infected RBC (and 95% confidence interval), is significantly lower in treated monkeys: 0.5% (−0.4 to 1.5) (note that negative values in 95% confidence interval for parasitemias and days result from statistical calculations but have no biological significance) versus 6.1% (5.1 to 7.1) (degrees of freedom [df] = 109; t-value = −8.1; P < 0.0001, for group comparison) in control monkeys. Peak parasitemia in treated monkeys is reached at the second dosing (day 0.7 (−0.6 to 2), whereas peak parasitemia in controls was at day 3.5 (3.4 to 3.6) (df = 109; t-value = 63.6, P < 0.0001, for group comparison). Parasitemia in G25-treated animals is below detection level by day 3, after which monkeys remain free from parasites (no recrudescence), indicating a complete cure.
A prodrug approach using TE4gt and TE3 allowed oral administration of the biscationic antiphospholipid effectors (22). Although these drugs have a slightly later onset of activity than G25 (peak parasitemia at days 1.0 and 1.9 for TE4gt and TE3 [see below], respectively, versus day 0.7 for G25) both are fully curative. The delay in activity seems likely to be attributable to the differing route of administration. In TE4gt-treated macaques (Fig. 2B), complete cure was obtained at day 4, and peak parasitemia was significantly lower than in controls (1.5% [−0.6 to 3.7] versus 4.7% [2.8 to 6.6]; df = 73; t-value = 2.2; P = 0.03, for group comparison) and occurred shortly after the second dose (day 1.0 [0.6 to 1.3]), while peak parasitemia in controls occurred at day 2.3 (2.2 to 2.4; df = 73; t-value = −6.5; P < 0.0001, for group comparison). A dose-ranging study was undertaken for TE3 (22), and all doses cured parasitemia by days 4 to 5 (Fig. 2C).
Parasitemia development in the TE3 dose-ranging study, as observed and as modeled by NLME, is presented in Fig. 3. While both primary peak and recrudescent parasitemia are evident in all control monkeys (Fig. 3A to D) the second peak (recrudescence) does not occur in TE3-treated macaques at any dose (Fig. 3E to P). Peak parasitemias were significantly lower in treated monkeys (0.6% [0.1 to 1.1] versus 3.0% [2.2 to 3.9] in controls; df = 310; t-value = −5.1; P < 0.0001 for all dosages compared to controls), and the peak parasitemia occurred significantly earlier in treated monkeys (day 1.8 [1.6 to 2.0] versus day 3.9 [3.8 to 4.0] in controls; df = 310; t-value = −18.8; P < 0.0001 for all dosages compared to controls) (Table 1).
FIG. 3.

Observed and NLME-predicted parasitemias in the absence of treatment or during TE3 treatment at three doses. Parasitemia development in the control group (A to D) can be compared to TE3-treated monkeys. The three groups of four P. cynomolgi-infected monkeys were treated (starting at day 0) with TE3 at 3 mg/kg (E to H), 9 mg/kg (I to L), and 27 mg/kg (M to P), respectively. In each panel the observed parasitemia (dotted lines) and the predicted parasitemia by NLME (solid lines), determined as described in Fig. 1, are indicated.
Statistical evaluation reveals dose-effect relationship for TE3.
Dose effects in TE3-treated animals were analyzed by modeling the first parasitemia peak only, since no recrudescence was evident in treated animals. The dose-effect relationship for TE3 becomes apparent after applying NLME to relate the applied drug dose to reduction of parasitemia and time point after treatment (Table 2). At the two highest doses evaluated, peak parasitemia time was observed after the second dose (day 1.7 [1.5 to 1.8] and day 1.6 [1.5 to 1.8], for 9 and 27 mg/kg, respectively; not significantly different), whereas the peak parasitemia time at the lowest dose occurred significantly later (day 2.0 [1.9 to 2.1]; df = 154 and 154; t-values −2.8 and 3.6; P < 0.005 and P < 0.0006, respectively, for 9 and 27 mg/kg compared to 3 mg/kg). In addition, the peak parasitemia was reduced in all treated groups (Fig. 2C and 3), with a trend for more reduction at higher dosages, although not statistically significant. Complete cure with TE3 was observed at day 4 (two highest doses) or day 5 (lowest dose), and no recrudescence was apparent (Table 1).
DISCUSSION
Malaria parasites have restricted host ranges. For the major parasites of humans, P. falciparum and P. vivax, experimental in vivo models are restricted to parasite strains that have been adapted to growth in splenectomized or intact nonnatural hosts, Aotus or Saimiri monkeys (5, 9). P. vivax is increasingly being recognized as a clinically important parasite (15); however, its biological characteristics (such as limited host range and restriction to reticulocytes) make it a difficult parasite to work with experimentally. Here, we report on studies undertaken in preclinical development of a new class of antimalarial drugs targeting phospholipid biosynthesis, in which we have modeled P. vivax infections by using a natural parasite-host combination (20) of P. cynomolgi, a parasite with zoonotic capacity that is phylogenetically closely related to P. vivax (7), in M. mulatta, the rhesus macaque, a species that is phylogenetically closely related to humans.
We confirm the reproducible characteristics of blood-stage infection in this model (18) and show these qualities to be valuable for monitoring drug activity. After infection with 106 P. cynomolgi blood-stage parasites, peak parasitemia of between 1 and 10% infected RBC is reached between days 9 and 12. Within each control group, peak parasitemias of all animals occurred within a 24-h window. The initial peak parasitemia resolves and is followed by a recrudescence peak 5 to 8 days later. NLME allows for accurate modeling of this consistent parasitemia development due to inclusion of random effects for parasitemia peak time and height.
The biological characteristics of this system make this in vivo nonhuman primate malaria model very well suited to monitoring drug activity in a highly relevant preclinical setting, as demonstrated here for anti-phospholipid effectors. The effects on the first peak parasitemia provide a readout for the general antiparasitic activity of the test compounds (i.e., whether the treatment significantly reduce parasite levels in the host), and activity on the recrudescence in the absence of continued drug treatment is a measure for the curative effect of the compounds. An added benefit of the P. cynomolgi-rhesus model that has not been utilized here but that nevertheless is important in work targeting pre-erythrocytic P. vivax developmental stages is that P. cynomolgi also allows for activity testing of compounds on hypnozoites, the dormant liver stages that are characteristic for sporozoite-induced P. vivax infections (3).
With all three compounds in the phospholipid synthesis inhibitor series tested against blood stages, whether given i.m. or orally, there was a strong antiparasite activity reflected in both the timing and the height of peak parasitemia (Fig. 2 and Table 1). Furthermore, all compounds were completely curative, as illustrated by the absence of any recrudescence following the first peak parasitemia. This finding further underlines the potential of the phospholipid synthesis inhibitor approach, especially with establishment of an effective prodrug approach allowing oral application (22).
The NLME models allowed us to demonstrate that treatment with G25 or G25-like compounds (i) reduces the height of peak parasitemia and, as a result, peak parasitemia is reached at a significantly earlier time point; (ii) prevents recrudescence of parasitemia; and (iii), of considerable significance, the NLME models allowed us to discriminate between the antimalarial pharmacological activity of three doses of one compound that was fully curative at each of these doses. Indeed, in the case of TE3, timing of peak parasitemia was dose dependent (Table 2), and a trend was observed showing that with increasing dose, height of peak parasitemia is reduced. The data presented here demonstrate that the P. cynomolgi-rhesus monkey model for human malaria, analyzed using NLME to model parasitemia, allowing for accurate quantitation of treatment effects, provides a powerful in vivo model to guide compound development for human malaria to clinical trials.
To date, this is the first report we are aware of on the use of NLME models for the analysis of parasitemia in an experimental malaria infection. In vivo infection models for other Plasmodium species may also be modeled using parameterization as described here, depending on the course of parasitemia development. The analysis is applicable to measurements of both in vivo drug efficacy and vaccine efficacy in animal models for human malaria and in clinical studies themselves and offers the possibility to significantly increase the data output in the search for new effective malaria drugs and vaccines. In addition, NLME can be used to analyze simulated experiments and thus calculate the minimal number of animals needed in an experimental setting to reach a predefined significant difference between control and treatment group. The NLME approach therefore offers the prospect of significant gains following the principles of refinement and reduction in experimental animal use.
Supplementary Material
Acknowledgments
This study was supported by the European Community grant QLK2-CT-2000-01166 and is part of the activities of the BioMalPar European Network of Excellence supported by a European grant (LSHP-CT-2004-503578) from the Priority 1 Life Sciences, Genomics, and Biotechnology for Health in the 6th Framework Programme. This study was generated in the context of the AntiMal project, funded under the 6th Framework Programme of the European Community (contract IP-018834).
The authors are solely responsible for the content this study; it does not represent the opinion of the European Community, and the Community is not responsible for any use that might be made of the information contained herein.
We thank S. H. Heisterkamp (Biometrics Department, Schering-Plough, Oss, The Netherlands, and Groningen Bioinformatics Centre, Rijksunversiteit Groningen, Groningen, The Netherlands) for help in defining the parameterization for the NLME models and in performing the NLME analyses.
Footnotes
Published ahead of print on 17 November 2008.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Baird, J. K. 2004. Chloroquine resistance in Plasmodium vivax. Antimicrob. Agents Chemother. 48:4075-4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calas, M., M. L. Ancelin, G. Cordina, P. Portefaix, G. Piquet, V. Vidal-Sailhan, and H. Vial. 2000. Antimalarial activity of compounds interfering with Plasmodium falciparum phospholipid metabolism: comparison between mono- and bisquaternary ammonium salts. J. Med. Chem. 43:505-516. [DOI] [PubMed] [Google Scholar]
- 3.Coatney, G. R., W. E. Collins, W. McWarren, and P. G. Contacos. 1971. The primate malarias. U.S. Government Printing Office, Washington, DC.
- 4.Coatney, G. R., H. A. Elder, P. G. Contacos, M. E. Getz, R. Greenland, R. N. Rossan, and L. H. Schmidt. 1961. Transmission of the M strain of Plasmodium cynomolgi to man. Am. J. Trop. Med. Hyg. 10:673-678. [DOI] [PubMed] [Google Scholar]
- 5.Collins, W. E., G. G. Galland, J. S. Sullivan, and C. L. Morris. 1994. Selection of different strains of Plasmodium falciparum for testing blood-stage vaccines in Aotus nancymai monkeys. Am. J. Trop. Med. Hyg. 51:224-232. [DOI] [PubMed] [Google Scholar]
- 6.Escalante, A. A., D. E. Freeland, W. E. Collins, and A. A. Lal. 1998. The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc. Natl. Acad. Sci. USA 95:8124-8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyles, D. E., G. R. Coatney, and M. E. Getz. 1960. Vivax-type malaria parasite of macaques transmissible to man. Science 131:1812-1813. [DOI] [PubMed] [Google Scholar]
- 8.Guerra, C. A., R. W. Snow, and S. I. Hay. 2006. Mapping the global extent of malaria in 2005. Trends Parasitol. 22:353-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herrera, S., B. L. Perlaza, A. Bonelo, and M. Arevalo-Herrera. 2002. Aotus monkeys: their great value for anti-malaria vaccines and drug testing. Int. J. Parasitol. 32:1625-1635. [DOI] [PubMed] [Google Scholar]
- 10.Mendis, K. 1995. Simian malaria models for research on human disease. Ceylon Med. J. Sci. 38:31-35. [Google Scholar]
- 11.Mendis, K., B. J. Sina, P. Marchesini, and R. Carter. 2001. The neglected burden of Plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 64:97-106. [DOI] [PubMed] [Google Scholar]
- 12.Paterson, S., and J. Lello. 2003. Mixed models: getting the best use of parasitological data. Trends Parasitol. 19:370-375. [DOI] [PubMed] [Google Scholar]
- 13.Perera, K. L., S. M. Handunnetti, I. Holm, S. Longacre, and K. Mendis. 1998. Baculovirus merozoite surface protein 1 C-terminal recombinant antigens are highly protective in a natural primate model for human Plasmodium vivax malaria. Infect. Immun. 66:1500-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinheiro, J. C., and D. M. Bates. 2000. Mixed-effects models in S and S-PLUS, statistics, and computing. Springer-Verlag, Berlin, Germany.
- 15.Price, R. N., E. Tjitra, C. A. Guerra, S. Yeung, N. J. White, and N. M. Anstey. 2007. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 77:79-87. [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe, A. W., E. Eyster, and A. Kellner. 1968. Liquid nitrogen preservation of red blood cells for transfusion; a low glycerol-rapid freeze procedure. Cryobiology 5:119-128. [DOI] [PubMed] [Google Scholar]
- 17.Salom-Roig, X. J., A. Hamze, M. Calas, and H. J. Vial. 2005. Dual molecules as new antimalarials. Comb Chem. High Throughput Screen. 8:49-62. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt, L. H., D. V. Cramer, R. N. Rossan, and J. Harrison. 1977. The characteristics of Plasmodium cynomolgi infections in various Old World primates. Am. J. Trop. Med. Hyg. 26:356-372. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt, L. H., R. Greenland, and C. S. Genther. 1961. The transmission of Plasmodium cynomolgi to man. Am. J. Trop. Med. Hyg. 10:679-688. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt, L. H., R. Greenland, R. Rossan, and C. Genther. 1961. Natural occurrence of malaria in rhesus monkeys. Science 133:753. [DOI] [PubMed] [Google Scholar]
- 21.Snow, R. W., C. A. Guerra, A. M. Noor, H. Y. Myint, and S. I. Hay. 2005. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434:214-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vial, H. J., S. Wein, C. Farenc, C. Kocken, O. Nicolas, M. L. Ancelin, F. Bressolle, A. Thomas, and M. Calas. 2004. Prodrugs of bisthiazolium salts are orally potent antimalarials. Proc. Natl. Acad. Sci. USA 101:15458-15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waters, A. P., D. G. Higgins, and T. F. McCutchan. 1993. Evolutionary relatedness of some primate models of Plasmodium. Mol. Biol. Evol. 10:914-923. [DOI] [PubMed] [Google Scholar]
- 24.Wengelnik, K., V. Vidal, M. L. Ancelin, A. M. Cathiard, J. L. Morgat, C. H. Kocken, M. Calas, S. Herrera, A. W. Thomas, and H. J. Vial. 2002. A class of potent antimalarials and their specific accumulation in infected RBC. Science 295:1311-1314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.