

Abstract
Methods for increasing the proteolytic stability of EFK17 (EFKRIVQRIKDFLRNLV), a new peptide sequence with antimicrobial properties derived from LL-37, were evaluated. EFK17 was modified by four d-enantiomer or tryptophan (W) substitutions at known protease cleavage sites as well as by terminal amidation and acetylation. The peptide variants were studied in terms of proteolytic resistance, antibacterial potency, and cytotoxicity but also in terms their adsorption at model lipid membranes, liposomal leakage generation, and secondary-structure behavior. The W substitutions resulted in a marked reduction in the proteolytic degradation caused by human neutrophil elastase, Staphylococcus aureus aureolysin, and V8 protease but not in the degradation caused by Pseudomonas aeruginosa elastase. For the former two endoproteases, amidation and acetylation of the terminals also reduced proteolytic degradation but only when used in combination with W substitutions. The d-enantiomer substitutions rendered the peptides indigestible by all four proteases; however, those peptides displayed little antimicrobial potency. The W- and end-modified peptides, on the other hand, showed an increased bactericidal potency compared to that of the native peptide sequence, coupled with a moderate cytotoxicity that was largely absent in serum. The bactericidal, cytotoxic, and liposome lytic properties correlated with each other as well as with the amount of peptide adsorbed at the lipid membrane and the extent of helix formation associated with the adsorption. The lytic properties of the W-substituted peptides were less impaired by increased ionic strength, presumably by a combination of W-mediated stabilization of the largely amphiphilic helix conformation and a nonelectrostatic W affinity for the bilayer interface. Overall, W substitutions constitute an interesting means to reduce the proteolytic susceptibility of EFK17 while also improving antimicrobial performance.
The cathelicidins are a major family of antibacterial peptides in mammals (66). The human cathelicidin hCAP-18 is released by several types of epithelial and immune cells, notably neutrophils, in response to inflammation and subsequently cleaved to generate the active C-terminal LL-37 peptide (18, 52). LL-37 is an extensively investigated peptide displaying strong antibacterial effects also at a high ionic strength (57). It also displays a range of other interesting properties, e.g., relating to lipopolysaccharide neutralization, immune cell stimulation and attraction, reepithelization, and wound closure (13). Although LL-37 displays some cytotoxicity against human cells, possibly originating from a hydrophobic interaction with cell membranes (31), this is largely absent in human serum due to interactions with apolipoproteins and other serum factors (61). It was previously found that reducing the number of residues from the N terminus of LL-37 decreases its cytotoxicity while retaining the bactericidal properties, although such truncations may also result in an increased proteolytic sensitivity (8, 31). Also, a larger truncation of the N terminus with simultaneous C-terminal truncations, generating internal peptide segments, has been found to result in reduced cytotoxicity at a largely retained bactericidal potency (28, 47, 48).
A challenge with LL-37 in various therapeutic contexts, e.g., chronic wounds or eye infections, is its susceptibility to proteolytic degradation. Thus, LL-37 is readily degraded by a number of bacterial proteolytic enzymes, which has initiated research aimed at stabilizing LL-37 against degradation using either surfactant formulations (34) or coformulations of LL-37 with inhibitors of such enzymes or with compounds complexing with LL-37, thereby precluding enzyme access to the cleavage sites on the peptide (43). Naturally, such proteolytic susceptibility may also have an effect on the direct bactericidal performance of LL-37. For example, both Pseudomonas aeruginosa and Staphylococcus aureus are normally cleared by LL-37 but are also known to develop countermeasures against the peptide. In both cases, this resistance is at least partly mediated by the secretion of peptidases that degrade LL-37 (43, 47). Both elastase from P. aeruginosa and aureolysin from S. aureus inactivate the peptide and have all their collective nine cleavage sites within 20 residues of the C terminus (43, 47).
It is notable that P. aeruginosa elastase and S. aureus aureolysin belong to the family of M4 peptidases (thermolysin family), comprising numerous bacterial proteases such as gelatinase of Enterococcus faecalis as well as the 50-kDa metalloproteinase of Proteus mirabilis. All these proteases have similar specificities, reflecting the ubiquitous occurrence of proteases among potentially pathogenic bacteria. These proteases preferentially hydrolyze positions involving hydrophobic side chains at the P1′ position (most notably L and F) (10). Since most antimicrobial peptides are rich in alternating sequences comprising cationic and hydrophobic amino acids, the secretion of bacterial proteases may represent a basic innate immune evasion mechanism employed by pathogenic bacteria (23). The findings that various microbes such as Proteus mirabilis (2), Candida albicans (39), and Salmonella enterica (19) degrade and thereby inactivate antimicrobial peptides further support the notion of a global role of bacterial proteases in bacterial resistance to antimicrobial peptides. Bacterial proteases have profound effects on and modulate both adaptive and innate immune responses during infection (22, 56), and the inactivation of antimicrobial peptides could therefore be one factor behind many chronic and acute infective diseases. For example, chronic leg ulcerations are characterized by a continuous infective state where enzymes of P. aeruginosa, Enterococcus faecalis, Proteus mirabilis, and Streptococcus pyogenes may degrade LL-37 (43), leading to low levels of this peptide (20). Clearly, considering the vast spectrum of diseases involving bacteria such as Pseudomonas (e.g., keratitis, chronic otitis, postoperative wound infections, and pneumonia), staphylococci (such as infected eczema, impetigo, conjunctivitis, deeper infections, and abscesses), and streptococci (erysipelas, sepsis, and sore throat) together with increasing resistance to conventional antibiotics (e.g., methicillin-resistant S. aureus), there is a need for novel antibiotics, motivating the development of potent and proteolytically stable antimicrobial peptides.
The aim of the present study was to isolate a short but lytic segment against P. aeruginosa and S. aureus from LL-37 and to alter the sequence in order to decelerate the enzymatic degradation at maintained high levels of antibacterial potency and low levels of cytotoxicity. From previous studies of isolations of bactericidal fragments of LL-37 (8, 28, 31, 47, 48), the positions of the peptidase cleavage sites, and a process of screening, we identified a new 17-amino-acid internal and antimicrobial part of LL-37, named EFK17 (Table 1). The fragment chosen included eight out of the nine protease cleavage sites used by P. aeruginosa elastase and S. aureus aureolysin that lead to the inactivation of LL-37, which emphasizes the importance of the chosen peptide sequence (43, 47). The native EFK17 peptide was compared to variants substituted with either four d-enantiomers (d) or four tryptophan residues (W) at typical protease cleavage sites as well as to variants with acetylated and amidated terminals (a) (Table 1). The amphiphilicity of the generated peptides is visible in an idealized helical wheel projection (Fig. 1).
TABLE 1.
Amino acid sequences of LL-37, EFK17, and the modified EFK17 variants
| Peptide | Sequencea |
|---|---|
| LL-37 | NH2-LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES-COOH |
| EFK17 | NH2-E1FKR2IVQR2IKD3FLRN3L4V-COOH |
| d | NH2-EdFKRdIVQRdIKDdFLRNLV-COOH |
| W | NH2-EWKRWVQRWKDWLRNLV-COOH |
| a | CH3CONH-EFKRIVQRIKDFLRNLV-CONH2 |
| d/a | CH3CONH-EdFKRdIVQRdIKDdFLRNLV-CONH2 |
| W/a | CH3CONH-EWKRWVQRWKDWLRNLV-CONH2 |
In the LL-37 sequence, the internal EFK17 segment is highlighted in boldface type, as are the modification sites on the EFK17 variants. All EFK17 variants have a net charge of +3 at pH 7.4 and a pI of ∼11. The known proteolytic cleavage sites within EFK17 are marked for V8 protease, aureolysin, and Pseudomonas aeruginosa elastase (PE) and are shown as 1 to 4 (43, 47). 1, V8 and PE; 2, aureolysin and PE; 3, PE; 4, aureolysin.
FIG. 1.

Helical wheel projection of EFK17 with enantiomeric (dF or dI) and tryptophan (W) substitutions outlined. Black and white circles represent hydrophilic and hydrophobic residues, respectively.
MATERIALS AND METHODS
Peptides and bacteria.
The peptides analyzed in this study were synthesized by Innovagen AB (Lund, Sweden) and were of >95% purity. Human neutrophil elastase (HNE) and Pseudomonas aeruginosa elastase (PE) were obtained from Calbiochem (La Jolla, CA), while Staphylococcus aureus aureolysin and V8 protease (V8) were both obtained from BioCol GmbH (Potsdam, Germany). The bacterial strains used were P. aeruginosa ATCC 27853 and S. aureus ATCC 29213 from the American Type Culture Collection (Rockville, MD), P. aeruginosa 15159 and S. aureus 2844 from the Department of Bacteriology at Lund University (Lund, Sweden), and S. aureus FDA 486 from the State Serum Institute (Copenhagen, Denmark).
Lipids and buffers.
The phospholipid used was 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (>99%) from Avanti Polar Lipids (Alabaster, AL). Cholesterol (>99%) and n-dodecyl-β-d-maltoside (DDM) (>98%) were both from Sigma-Aldrich (Steinheim, Germany). Except for the cytotoxicity experiments where other media were used, all measurements were performed at 37°C in 10 mM Tris buffer (pH 7.4) containing 5 mM glucose (ionic strength, 0.006) and, when indicated, also with 150 mM NaCl (ionic strength, 0.156). The water used was from a Millipore Milli-Q Plus 185 ultrapure water system. Other chemicals used were of analytical grade.
Protease activity.
Peptides (136 μM) were incubated at a peptide-to-enzyme ratio of 300:1 for 4 h at 37°C in Tris buffer. The samples were supplemented with Laemmli loading buffer (25), and proteolysis was heat terminated by 3 min of boiling. Gel electrophoresis was done using Criterion precast Tris-Tricine 16.5% polyacrylamide gels and Tris-Tricine-sodium dodecyl sulfate running buffer from Bio-Rad (Hercules, CA) at 150 V for 35 min, submerged in ice. The gels were subsequently stained for 1 h with 0.25% Coomassie brilliant blue R-250, destained, and scanned. The gel bands from the resulting images were quantified with ImageJ v1.37 software (U.S. National Institutes of Health, Bethesda, MD [http://rsb.info.nih.gov/ij]), from where the undegraded fraction can be obtained from the sum and grayscale of all pixels in each gel band. The fractions were normalized within the same gel with the background and with bands from identically treated and handled peptides of the same variant but incubated without protease. The results from the proteolysis were reproduced in two separate experiments. The size standard used was SeeBlue prestained standard from Invitrogen (Lidingö, Sweden).
Antimicrobial activity.
The MIC assay was carried out by a microtiter broth dilution method as previously described (64), with slight modifications. For the preparation of refined medium (58), 100 ml Luria-Bertani (LB) broth was prepared in Tris buffer (10 mM) (pH 7.4) and applied onto a column packed with 40 ml DEAE Sephacel (Sigma-Aldrich, Schnelldorf, Germany). The column was previously rinsed with water, 2 M NaCl, 0.1 M NaOH, water, and 70% ethanol, respectively, and finally equilibrated in Tris buffer before use. The medium was passed through the column twice and finally filter sterilized (0.22-μm Millex GP filter unit) and stored at 8°C until use. The peptides were dissolved in Tris buffer at a concentration 10 times higher than the required range by serial dilutions from a stock solution. Ten microliters from each concentration was added to a 96-well microtiter plate (polypropylene; Costar Corp., Cambridge, MA), resulting in concentrations of 0.6, 1.2, 2.5, 5, 10, 20, 40, or 80 μM. Bacteria grown overnight in 3% trypticase soy broth were rinsed with Tris buffer and diluted in refined LB medium to get a concentration of approximately 106 CFU per ml. Ninety microliters from the bacterial solution was subsequently added to each well. The plate was incubated at 37°C for 21 h. Bacterial growth was determined by visual inspection of pellet formation and by determining the optical density at 540 nm (62). Experiments were reproduced in two separate experiments and were found to be consistent within each dilution factor. The MIC was taken as the concentration at which greater than 95% of growth inhibition and no visible pellet were observed.
In the viable-count analysis, bacteria were grown to the mid-logarithmic phase in Todd-Hewitt medium. Bacteria were washed and diluted in Tris buffer containing 5 mM glucose. Following this, bacteria (50 μl with 2 × 106 CFU/ml) were incubated with the peptides at a concentration of 6 or 30 μM in Tris buffer with 150 mM NaCl at 37°C for 2 h. To quantify the bactericidal activity, serial dilutions of the incubation mixtures were plated onto Todd-Hewitt agar, followed by incubation at 37°C overnight, and the number of CFU was determined. One hundred percent survival was defined as the total survival of bacteria in the same buffer and under the same conditions in the absence of peptide.
Cytotoxicity.
A hemolysis assay was performed with EDTA-treated human blood centrifuged at 800 × g for 10 min, after which serum and buffy coat were removed. The erythrocytes were washed three times and resuspended, yielding a 5% suspension in phosphate-buffered saline (PBS) (pH 7.4). For hemolysis in the presence of serum, citrated blood was diluted with an equivalent volume of 5% PBS. The cells were then incubated with end-over-end rotation for 1 h at 37°C in the presence of peptide. The samples were then centrifuged at 800 × g for 10 min. The absorbance of hemoglobin release was measured at 540 nm and compared to hemolysis induced by 2% Triton X-100 (Sigma-Aldrich, Steinheim, Germany) that served as a positive control. Spontaneous leakage was found to be less than 2% and was used as background.
For the lactate dehydrogenase (LDH) assay, HaCaT keratinocytes were grown in 96-well plates (3,000 cells/well) in serum-free keratinocyte medium (SFM) supplemented with bovine pituitary extract and recombinant epidermal growth factor (BPE-rEGF) (Invitrogen) to confluence. The medium was then removed, and 100 μl of 6 or 60 μM peptide in SFM-BPE-rEGF solution was added to the well plate and incubated overnight. The LDH-based TOX-7 kit (Sigma-Aldrich, Schnelldorf, Germany) was used for the quantification of LDH release from the cells. The results are presented as fractions of the positive control where 1% Triton X-100 was added. Spontaneous LDH release was less than 7% and used as background.
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay used sterile-filtered MTT (Sigma-Aldrich, Schnelldorf, Germany) solution (5 mg/ml in PBS), which was stored protected from light at −20°C until use. HaCaT keratinocytes (3,000 cells/well) were seeded into 96-well plates and grown in SFM-BPE-rEGF medium to confluence. The peptides were then added to 6 or 60 μM. After incubation overnight, 20 μl of the MTT solution was added to each well, and the plates were incubated for 1 h at 37°C. The MTT-containing medium was then removed by aspiration. The blue formazan product generated was dissolved by the addition of 100 μl of 100% dimethyl sulfoxide per well. The plates were then gently swirled for 10 min at room temperature to dissolve the precipitate. The absorbance was monitored at 550 nm, and the results are fractions compared to untreated cells. The background MTT signal from 1% Triton X-100-treated keratinocytes was less than 7% and was used as background.
Liposome preparation.
Dry lipid films were formed on flask walls by dissolving DOPC and cholesterol in chloroform, followed by evaporation under nitrogen gas flow and, subsequently, also in a vacuum overnight at room temperature. After preparation of the dried phospholipid film, the leakage marker 5(6)-carboxyfluorescein was added at a self-quenching concentration of 100 mM in Tris buffer, the latter with or without additional 150 mM NaCl, and the lipids were resuspended. The suspensions were then freeze-thawed between liquid nitrogen and 62°C and vortexed for eight cycles. Liposome polydispersity was then reduced by extrusion through polycarbonate filters with 100-nm pores using a LipoFast miniextruder (Avestin, Ottawa, Canada). Nonencapsulated carboxyfluorescein was removed by filtration through two subsequent Sephadex G-25 medium PD-10 columns (Amersham Biosciences, Uppsala, Sweden). The resulting DOPC-cholesterol (3:2 molar ratio) liposomes thus obtained were unilamellar and had a diameter of about 110 nm. Due to the long, symmetric, and unsaturated acyl chains of DOPC, several methodological advantages were reached. In particular, membrane cohesion was good, which facilitated very stable, unilamellar, and largely defect-free liposomes (observed from cryo-transmission electron microscopy) and well-defined supported lipid bilayers (observed by ellipsometry and atomic force microscopy), allowing very detailed values for leakage and adsorption density to be obtained. Although there are differences between zwitterionic, anionic, or cholesterol-containing membranes in their interactions with cationic peptides, it was previously shown that qualitatively similar effects of peptide modifications such as charge, length, hydrophobicity, and topology were obtained for these membranes (35, 36, 38). The large proportion and diversity of nonphospholipid components in biomembranes, as well as the diversity within the phospholipids, prevent close mimicry in most model systems without creating methodological and interpretative predicaments. Although our present model probably has more in common with eukaryotic membranes, we believe that it is sufficient for comparison with data from both human cells and bacteria in the context of being a predominately phospholipid bilayer, which is indicated by the correlation between results found in the present investigation.
Liposome leakage.
Liposome integrity was monitored by examining 5(6)-carboxyfluorescein (Acros Organics, Geel, Belgium) efflux from the liposomes to the external lower-concentration environment, resulting in a loss of self-quenching and thus in an increased fluorescence signal (7). This fluorescence increase was measured with a Spex (Edison, NJ) Fluorolog-2 spectrofluorometer. Measurements were conducted, under constant stirring, at a fixed lipid concentration of 10 μM and with five peptide concentrations between 0.01 and 1 μM in individual experiments. In each experiment, initial signal acquisition for 10 min was performed to reveal any spontaneous leakage. The effect of the addition of peptide to the system was then monitored for 45 min, well after the leakage induction had reached its limiting value. To establish the 100% leakage signal, Triton X-100 was added to a concentration of 0.05%.
Ellipsometry.
Peptide adsorption to supported lipid bilayers was studied by in situ null ellipsometry using an Optrel (Kleinmachnow, Germany) Multiskop ellipsometer equipped with a 100-mW argon laser (532 nm) at an angle of incidence of 67.66°. The adsorption was monitored by monitoring adsorption-induced changes in the amplitude and phase of light reflected at the adsorbing surface. From these data, the refractive index (n) and layer thickness (d) of the adsorbed layer were determined. With these parameters, the adsorbed amount (Γ) was calculated according to methods described previously by De Feijter et al. (9), using a refractive index increment (dn/dc) of 0.154 cm3/g (54, 59). Corrections were routinely made to compensate for any change in the bulk refractive index caused by changes in temperature or excess electrolyte concentrations. The bilayer-supporting surfaces were polished silica slides with an oxide layer thickness of 30 nm (Okmetic, Espoo, Finland). These slides were first cleaned at 80°C for 5 min in water solutions of 3.6% NH4OH and 4.3% H2O2, followed by 4.6% HCl and 4.3% H2O2. The slides were then stored in 99.7% ethanol and were cleaned by plasma discharges for 5 min at 18 W in 0.2-torr residual air just prior to use with a Harrick Plasma (Ithaca, NY) PDC-32G cleaner. Lipid bilayers were deposited as described in detail previously (38, 54). In brief, a mixed micellar solution containing DOPC-cholesterol (3:2 molar ratio) and DDM was prepared by the addition of a 19 mM DDM water solution to DOPC-cholesterol dry lipid films, followed by stirring overnight, yielding a solution containing 97.3 mol% DDM, 1.6 mol% DOPC, and 1.1 mol% cholesterol. The micellar solution was added to the substrates when submerged in 5 ml water in the cuvette at 25°C, and the resulting adsorption was monitored. After the lipid adsorption had stabilized, residual micelles as well as a fraction of DDM adsorbed were removed by rinsing with Milli-Q water (5 ml/min) until desorption stabilized. By repeating this procedure and gradually lowering the concentrations of the micellar solution (730, 150, 76, 38, and 19 μM), stable, densely packed bilayers were formed, with a defect density of less than 10% and with structural characteristics similar to those of bulk lamellar structures of the lipids (54). After lipid bilayer formation and after adequate buffer and temperature had been adjusted, the peptides were added cumulatively to concentrations of 0.01, 0.1, 0.5, and 1 μM, and the adsorption in each case was monitored for 45 min, well after adsorption saturation was reached. Results were obtained from duplicates of individual experiments.
Circular dichroism.
The peptide secondary structure was determined using Tris buffer with or without additional 150 mM NaCl as well as liposomes (100 μM lipid). A J810 spectropolarimeter (Jasco Corporation, Easton, MD) was used in the range of 190 to 260 nm at 37°C in a quartz cuvette at a peptide concentration of 10 μM. The helix content was quantified at 222 nm (17) to 225 nm (49) using reference spectra for samples containing 100% α-helix and 100% random coil. As a reference, samples of 0.133 mM (monomer concentration) poly-l-lysine in 0.1 M NaOH or in 0.1 M HCl, respectively, were used (49). To avoid any instrumental baseline drift between measurements, the background value (detected at 250 to 260 nm, where no peptide signal is present) was subtracted for each individual sample measurement. Signals from nonpeptide components were also corrected for.
Statistics.
Values are reported as means ± standard deviations of the means. To determine significance, analysis of variance (ANOVA) (SigmaStat, SPSS Inc., Chicago, IL) followed by post hoc testing using the Holm-Sidak method or a Student's t test was used as indicated in the figure legends, where “n” denotes the number of independent experiments. Significance was accepted at a P value of <0.05.
RESULTS AND DISCUSSION
Protease degradability.
From protease incubation and subsequent gel electrophoresis of each peptide variant, it is clear that both LL-37 and its native EFK17 fragment showed a low level of proteolytic resistance to the enzymes used, with less than 20% of the peptide remaining undigested at the conditions employed for all enzymes (Fig. 2a and b). In contrast, the EFK17 homologues containing four d-enantiomer substitutions (d and d/a) at known proteolytic cleavage sites (Table 1) did not show any appreciable signs of proteolysis. These results are in line with those reported previously in the literature. For example, terminal d-enantiomers are known to increase the peptide life span in serum (21, 33), and antibiotic peptides that are all d-enantiomeric are virtually indigestible by a number of proteases (3, 21, 60). Also, one should bear in mind that side chains as far as 3 residues away from the cleavage site can have an effect on protease affinity (42). From the d-enantiomer results, we can conclude that all cleavage sites for all proteases used are covered from the four substitution sites.
FIG. 2.

(a) Representative pictures from Tris-Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis of peptides after proteolysis. The proteases used were human neutrophil elastase (HNE), Pseudomonas aeruginosa elastase (PE), aureolysin (Aur), and V8 protease. The proteolysis was performed at a peptide-to-enzyme ratio of 300:1 and at a peptide concentration of 136 μM and with incubation at 37°C for 4 h. For EFK17-d/a, size references for 7 and 4 kDa are included (SeeBlue prestained standard). (b) All proteolysis results summarized as the fraction of undegraded peptide after 4 h of proteolytic exposure. * indicates a statistically significant difference compared to EFK17 (P < 0.05 by t test).
The target affinity of serine proteases such as HNE is very much governed by the residue at the P1′ position of the cleavage site. The preference follows the following order: cationic or aliphatic > aromatic ≫ anionic, d-enantiomer, or proline (41). Thus, I→W modifications are expected to result in an increased level of protease resistance. Indeed, the EFK17 homologues containing four W substitutions (W and W/a) all displayed increased stability against three out of four enzymes. The one protease not affected by the W substitutions was Pseudomonas aeruginosa elastase. This enzyme is known to have a high level of affinity for the other two aromatic amino acids, F and Y, at the P1′ position (32) and, as the present results show, also seems to be able to accommodate W. What can be said of the other two enzymes is that the V8 protease is known to cleave only at sites with a P1 anionic residue and that aureolysin needs a P1′ hydrophobic side chain (4, 12). The effect of a W residue at P1′ is, however, not documented for these proteases. The reduced proteolytic effect by the W substitutions for V8 and aureolysin could originate from the specificity pocket being unsuitable to accommodate the large indole side chain, such as in the case of HNE, or because there is increased oligomerization as a result of the W substitutions. Aromatic residues such as W also mediate helix-helix interactions and oligomerization (40), with the latter being a possible reason behind the slower proteolysis found for LL-37 than that found for a truncated monomeric variant (31).
Terminal modification of EFK17 by acetylation of the N terminus and amidation of the C terminus, on its own, showed little effect on protease degradation. However, when terminal modifications were combined with W substitution (W/a), the protease resistance against aureolysin and HNE was further improved. In this context, it is interesting that N-terminal acetylation, and, to a lower extent, also C-terminal amidation, is known to increase the peptide life span in cell cultures and serum, which has been suggested to be associated with exopeptidase activity (26, 33). Although the enzymes used in the present study are all endopeptidases, two of them are nevertheless restricted by these terminal modifications. In the case of aureolysin, it is possible that the two cleavage sites next to the W substitutions are inactivated in the W variant and that the last cleavage site close to the C terminus is solely responsible for the observed proteolysis. When the terminals are also modified, as in the W/a variant, the end cap created at the C terminus would then inhibit the last aureolysin cleavage site and increase proteolytic stability further. Irrespective of the detailed mechanism involved, the results imply a more general role of terminal modifications in the proteolytic stability of antimicrobial peptides.
Antibacterial and cytotoxic effect.
Although attractive from a cytotoxic perspective (see below), the antimicrobial potency of EFK17 was significantly lower than that of its precursor peptide LL-37, illustrated, e.g., by the MIC of EFK17 being much higher than that of LL-37 for the bacterial stains investigated (Table 2). However, both the W substitution and terminal modifications (W, a, and W/a) resulted in an enhanced antibacterial potency in all three S. aureus and two P. aeruginosa strains, as reflected by their MICs. This difference also prevailed under physiological salt conditions, which was illustrated by data obtained by viable-count analysis of EFK17 and its W/a homologue (Fig. 3). The d-enatiomer substitutions (d and d/a), on the other hand, resulted in a reduced antibacterial potency compared to that of the native EFK17 peptide (Table 2).
TABLE 2.
MIC of each peptide variant for five strains of Pseudomonas aeruginosa and Staphylococcus aureus
| Peptide | MIC (μM)a
|
||||
|---|---|---|---|---|---|
|
P. aeruginosa
|
S. aureus
|
||||
| ATCC 27853 | 15159 | ATCC 29213 | FDA 486 | 2844 | |
| EFK17 | 80 | 80 | 80 | 40 | 20 |
| d | >80 | >80 | >80 | >80 | >80 |
| W | 10 | 20 | 10 | 5 | 5 |
| a | 10 | 10 | 10 | 5 | 5 |
| d/a | >80 | >80 | >80 | >80 | >80 |
| W/a | 10 | 10 | 10 | 5 | 5 |
| LL-37 | 2.5 | 2.5 | 5 | 2.5 | 5 |
The concentration intervals used were 0.6, 1.2, 2.5, 5, 10, 20, 40, and 80 μM.
FIG. 3.
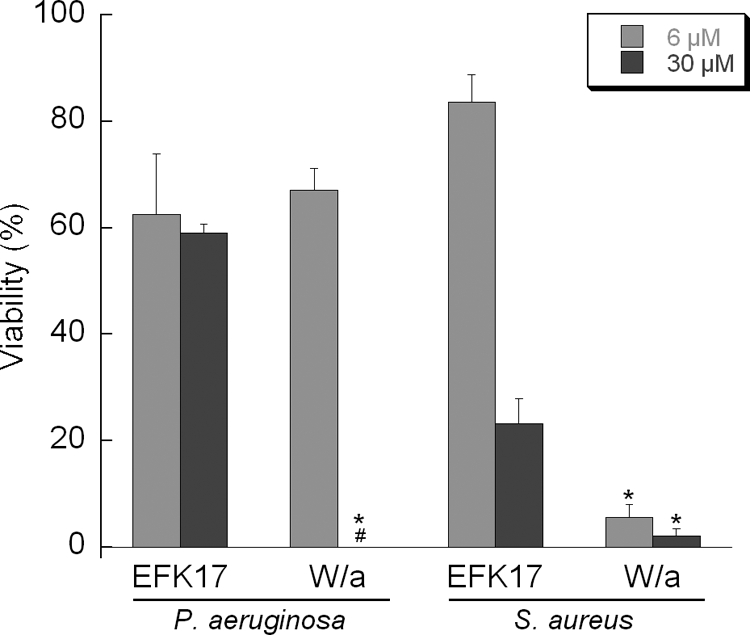
Analysis of viable counts of the native EFK17 peptide and its W/a homologue for Pseudomonas aeruginosa and Staphylococcus aureus in Tris-buffered 150 mM NaCl solution (pH 7.4). # indicates a viability of 0%; * indicates a statistically significant difference compared to EFK17 (P < 0.05 by ANOVA; n = 3).
When looking at cytotoxicity through hemolysis as well as LDH and MTT release, the bactericidal and cytotoxic effects largely coincided (Fig. 4) (the correlations are further demonstrated in Fig. S1a and S1b in the supplemental material). Thus, the W and terminal modifications both resulted in increased cytotoxicity in all three assays. These cytotoxicity levels were comparable to those of LL-37. Similarly to LL-37 (61), the levels of cytotoxicity of all EFK17 variants were found to be drastically reduced in the presence of serum (Fig. 4a).
FIG. 4.

Peptide-induced cytotoxicity monitored by hemolysis (a), LDH (b), and MTT (c) assays. For LDH and MTT assays, the negative controls are less than 7%, while those for hemolysis are less than 2% of the total and are used as background. All measurements were performed in triplicate. # indicates hemolysis of ∼0%, which occurs in serum-containing samples; * indicates a statistically significant difference when modified peptides were compared to EFK17 (P < 0.05 by ANOVA; n = 3).
Interaction with model lipid membranes.
In order to further characterize the origin of the peptide composition effects on bactericidal and cytotoxic properties, peptide-induced liposome leakage was investigated (Fig. 5). The results showed that at a low ionic strength, all terminally modified and W-substituted variants were as potent as the native EFK17 peptide in causing membrane defects and subsequent liposome leakage. The d-enantiomeric variants were significantly less potent. Under physiological salt conditions, the level of liposome leakage was substantially lower overall, illustrating the importance of electrostatic interactions for the action of these highly charged peptides. The W substitutions were less affected by the increased ionic strength, with significantly higher leakage levels than those of their unsubstituted counterparts. In general, the liposome leakage results followed the same order of potency for the peptides as that gained from both the bactericidal experiments and cytotoxicity results from serum-free cell cultures. Hence, in analogy to what has been found in numerous previous investigations (35, 36, 38, 51), we conclude that there is a close analogy between the effects on the lipid model systems on the one hand and bactericidal and cytotoxic effects on the other.
FIG. 5.

Peptide-induced liposome leakage as a function of peptide concentration. In these measurements, the lipid concentration was fixed at 10 mM (DOPC-cholesterol, 3:2 molar ratio), while the peptide concentrations were 0.01, 0.1, 0.5, or 1 mM added in separate experiments. Each leakage level was obtained from kinetic curves monitored during 45 min of peptide incubation at 37°C, after which all samples had well reached their maximum leakage induction levels. The right two graphs have 150 mM NaCl added to the Tris buffer. ¤ indicates that at least the three highest leakage levels of that series are statistically significantly higher than those of its d-enantiomer variant; * indicates that the value is statistically significantly higher than that of its non-W-modified variant (P < 0.05 by ANOVA; n = 3).
By combining the results from liposome leakage and ellipsometry (Fig. 5 and 6), one can see that there is also a close correlation between peptide-induced liposome leakage and the peptide adsorption density at the lipid membrane. Also in the circular dichroism results (Fig. 7), the magnitude of adsorption-induced helicity correlates with the adsorption density so that higher levels of adsorption and higher levels of helix induction are associated with each other. The latter is a reasonable finding given the amphiphilic nature of the idealized helix formed in these systems (Fig. 1). By comparing the system with and without salt, the presence of salt decreased helix induction by 3, 5, and 7% for EFK17, W, and a, respectively. Helix formation is a strong driving force behind amphiphilic peptide adsorption to lipid bilayers (63) and/or is a means to reduce the system free energy for a peptide localized at lipid membranes. EFK17-a has the strongest decline in the level of helix induction due to the addition of salt and, subsequently, also the strongest decline in the level of adsorption. The W-substituted peptide displays only weaker salt-dependent adsorption reduction despite reduced levels of helix induction. Hydrophobicity scales typically have tryptophan as being equal to or less hydrophobic than the substituted phenylalanine and isoleucine (24, 55). If anything, the W substitutions reduce the hydrophobicity of the peptide and would thereby reduce hydrophobically driven adsorption. The reason behind the increased salt tolerance with adsorption for the W-substituted peptide may be explained by the structural attraction to the membrane interface that W residues give peptides. The side chain of W has a preference to interact with the bilayer interface in the vicinity of the glycerol group, which is not driven by classical hydrophobic interactions or by dipolar forces (65). As that attraction is not dictated by electrostatics or helix formation, it provides a higher salt tolerance for adsorption. This seems to more than compensate for any loss of hydrophobicity resulting from the W substitutions. In agreement with this argument, the F→W substitution in amphiphilic peptides was previously found to significantly increase the affinity of peptide for lipid membranes (1).
FIG. 6.

Peptide adsorption monitored by ellipsometry on supported lipid bilayers (DOPC-cholesterol, 3:2 molar ratio). Peptides were cumulatively added to concentrations of 10 μM, 100 μM, 500 μM, and 1 mM. Each adsorption was quantified kinetically for 45 min, well past when the adsorption equilibrium was reached, and the saturation adsorption after this time is shown. (a) Representative kinetic adsorption plots for W and d peptides in NaCl-free buffer. Peptide additions to each concentration are indicated. (b) Adsorption levels for EFK17 and its homologues d, W, and a are summarized for measurements in Tris buffer with and without 150 mM NaCl. # indicates that the adsorption level was not determinable due to bilayer destabilization as a result of peptide-bilayer interactions; * indicates a statistically significant different value compared to that of EFK17 at an identical ionic strength (P < 0.05 by t test).
FIG. 7.
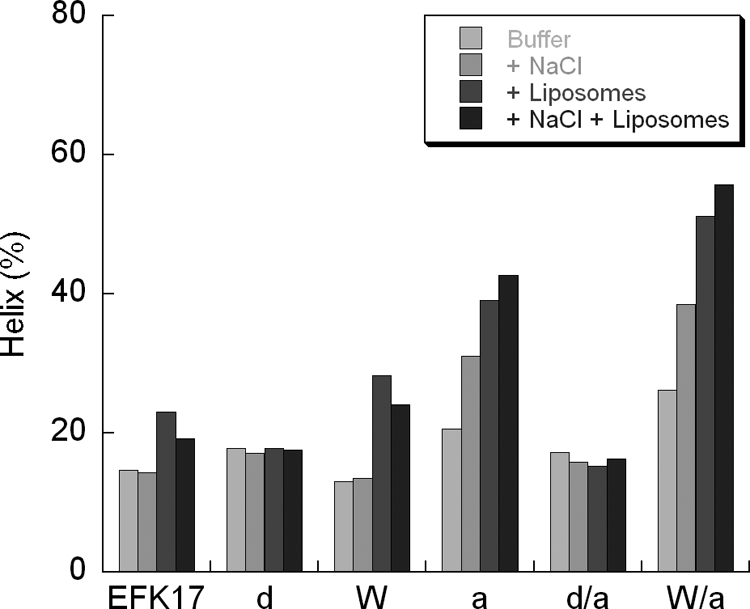
Results from circular dichroism measurements showing α-helix content versus peptide composition and environment. Each peptide was analyzed using Tris buffer with or without additional 150 mM NaCl and/or liposomes (DOPC-cholesterol, 3:2 molar ratio) at a 10 μM peptide concentration and a 100 mM lipid concentration. The error margin in these measurements is estimated to be less than ±3%.
The oppositely charged side chains in our peptides are organized in a manner that would generate helix-stabilizing salt bridges through E1-R4 and the strong cooperative triad R8-N11-R14 (30, 50). The reduced helicity seen when salt is included in liposome solutions is likely a result of the increased ionic strength of the medium, which interferes with these helix-stabilizing salt bridges (50).
Aromatic residues can form π interactions with each other and with cationic residues. These can stabilize helix formation much in the same way as do intermolecular salt bridges if they are interspaced by 2 or 3 residues (5, 46, 50). Consequently, the four W substitutions presently employed in the EFK17-W and -W/a variants give the peptide the opportunity to stabilize the helix, e.g., illustrated by the I→W substitutions at residues 5 and 9, which produces a well-known helix-stabilizing constellation and which may account for the increase in helix seen in those peptides (44). The exchange of the nonaromatic I and the less quadropolar F for the strong quadropole indole group of W can also effectively shield proximal R or K cationic residues from the hydrophobic bilayer (11, 15, 27). This would facilitate the penetration of the these charged side chains into the hydrophobic bilayer and, thus, possibly increase lytic properties (6). It is likely that shielding the cationic residues also interferes with electrostatic arrest at the interface of anionic membranes (in analogy with melittin and phosphatidylinositol membranes) (53), resulting in a more profound effect of W substitutions on bacteria. Furthermore, since the W substitutions may in our case have a reducing effect on peptide hydrophobicity depending on the hydrophobicity scale employed, it is of interest in the present context since it has been found that an increased level of hydrophobicity for helical amphiphilic AMP may specifically increase the lytic property on erythrocytes rather than bacteria (67). The lower hydrophobicity and increased penetrability of negative membranes both indicate that the W-modified variant would have a higher bactericidal-to-cytotoxic ratio. This is also the case in the present study: when the cytotoxicity is increased, the bactericidal gain is proportionally greater (∼20%).
Terminal acetylation and amidation increase helicity by stabilizing the helix by hydrogen bonding to an unsatisfied main-chain NH or CO group, respectively (14). The effect of this is clearly seen in our circular dichroism measurements. C-terminal amidation was previously found to increase helicity in antimicrobial peptides and also to yield increased antibacterial potency, although the latter may in part be a result of an increased net positive charge (45). In our case, the net charge was constant, which emphasizes the helix-inducing ability as the parameter that increases bactericidal effect. If, as in our study, both N-acetylation and C-amidation are implemented, there is no net charge alteration of the peptide, but it reduces the gross charge, which could possibly improve oligomerization and bilayer penetration. Recently, an improved amphiphilic version of a 24-mer segment of LL-37 with acetylated and amidated terminals did not show any toxicity in both topical and systemic administrations in animal testing while having improved antibiotic properties compared with those of LL-37 (28).
Furthermore, the d-enantiomer substitutions resulted in substantially lower levels of adsorption and liposome leakage induction. As can be seen in Fig. 7, there is no sign of helix induction upon adsorption, and thereby, one of the major driving forces behind adsorption is removed. Internal d-enantiomer substitutions and the loss of antibacterial activity were previously coupled with a loss of helicity (21). In analogy, other means for reducing helicity, such as proline substitutions, have also been found to reduce the peptide interaction with phospholipid membranes (1). Decreasing helicity, by inclusion of d-enantiomers or proline, for amphiphilic antimicrobial peptides has been correlated with decreased bactericidal and cytotoxic effects (16), although gram-negative bacteria were still sensitive to the helix-impaired peptides (16). Destabilizing helixes for antimicrobial peptides have therefore been suggested to be a means of reducing cytotoxicity while still having an antibacterial effect on gram-negative bacteria such as Pseudomonas aeruginosa (68). The conclusions from our present results, on the other hand, do not support this conclusion. Thus, we could not observe any inhibitory effect of the helix-impaired peptides on the presently investigated Pseudomonas aeruginosa strains. Instead, the extent of helix induction upon the peptide interaction with the lipid membrane correlates with the adsorption density and the antibacterial potency as well as with cytotoxicity.
From the results for peptide-induced liposome leakage and peptide adsorption at the phospholipid bilayer on the one hand and bactericidal and cytotoxic effects on the other, it is clear that lytic properties in biological systems correlate with liposome leakage induction and that increased liposome leakage correlates with increased peptide adsorption. The latter effect, also found previously for a number of other antimicrobial peptides (35-38, 51), indicates that the formation of membrane defects is due to local packing defects in the immediate vicinity of the adsorbed peptide molecules. Different factors affecting the peptide adsorption density, including peptide length, charge, hydrophobicity, and topology, have all been found to correlate with liposome leakage induction and also with bactericidal potency (35-38, 51). In the present case, peptide adsorption density is at least partly given by the degree of helix induction, as described above, since in a perfect helix projection, the peptide is perfectly amphiphilic (Fig. 1).
Finally, it is worth noting that both S. aureus and P. aeruginosa are known to develop resistance to both conventional antibiotics and antimicrobial peptides (29). One of the key mechanisms in the latter is the proteolytic degradation of AMPs; a possible indication of where the presently investigated W-substituted and proteolytically stabilized peptides may be of interest is atopic dermatitis. This chronic disease is characterized by excessive S. aureus colonization, for which long-term AMP therapy based on peptides of endogenous origin may be of interest. Similarly, bacterial conjunctivitis frequently involves P. aeruginosa and may be another indication candidate for these modified peptides.
Supplementary Material
Acknowledgments
We give our deepest gratitude to Mina Davoudi and Lotta Wahlberg for their expert technical assistance.
This work was financed by the Swedish Research Council (projects 13471 and 2006-4469) and DermaGen AB.
Footnotes
Published ahead of print on 24 November 2008.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Andrushchenko, V. V., H. J. Vogel, and E. J. Prenner. 2007. Interactions of tryptophan-rich cathelicidin antimicrobial peptides with model membranes studied by differential scanning calorimetry. Biochim. Biophys. Acta 1768:2447-2458. [DOI] [PubMed] [Google Scholar]
- 2.Belas, R., J. Manos, and R. Suvanasuthi. 2004. Proteus mirabilis ZapA metalloprotease degrades a broad spectrum of substrates, including antimicrobial peptides. Infect. Immun. 72:5159-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bessalle, R., A. Kapitkovsky, A. Gorea, I. Shalit, and M. Fridkin. 1990. All-D-magainin: chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 274:151-155. [DOI] [PubMed] [Google Scholar]
- 4.Bjoorklind, A., and H. Jornvall. 1974. Substrate specificity of three different extracellular proteolytic enzymes from Staphylococcus aureus. Biochim. Biophys. Acta 370:524-529. [DOI] [PubMed] [Google Scholar]
- 5.Butterfield, S. M., P. R. Patel, and M. L. Waters. 2002. Contribution of aromatic interactions to alpha-helix stability. J. Am. Chem. Soc. 124:9751-9755. [DOI] [PubMed] [Google Scholar]
- 6.Chan, D. I., E. J. Prenner, and H. J. Vogel. 2006. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta 1758:1184-1202. [DOI] [PubMed] [Google Scholar]
- 7.Chen, R. F., and J. R. Knutson. 1988. Mechanism of fluorescence concentration quenching of carboxyfluorescein in liposomes: energy transfer to nonfluorescent dimers. Anal. Biochem. 172:61-77. [DOI] [PubMed] [Google Scholar]
- 8.Ciornei, C. D., T. Sigurdardottir, A. Schmidtchen, and M. Bodelsson. 2005. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL-37. Antimicrob. Agents Chemother. 49:2845-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Feijter, J. A., J. Benjamins, and F. A. Veer. 1978. Ellipsometry as a tool to study the adsorption behavior of synthetic and biopolymers at the air-water interface. Biopolymers 17:1759-1772. [Google Scholar]
- 10.de Kreij, A., G. Venema, and B. van den Burg. 2000. Substrate specificity in the highly heterogeneous M4 peptidase family is determined by a small subset of amino acids. J. Biol. Chem. 275:31115-31120. [DOI] [PubMed] [Google Scholar]
- 11.Dougherty, D. A. 1996. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 271:163-168. [DOI] [PubMed] [Google Scholar]
- 12.Drapeau, G. R., Y. Boily, and J. Houmard. 1972. Purification and properties of an extracellular protease of Staphylococcus aureus. J. Biol. Chem. 247:6720-6726. [PubMed] [Google Scholar]
- 13.Durr, U. H., U. S. Sudheendra, and A. Ramamoorthy. 2006. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 1758:1408-1425. [DOI] [PubMed] [Google Scholar]
- 14.Fairman, R., K. R. Shoemaker, E. J. York, J. M. Stewart, and R. L. Baldwin. 1989. Further studies of the helix dipole model: effects of a free alpha-NH3+ or alpha-COO− group on helix stability. Proteins 5:1-7. [DOI] [PubMed] [Google Scholar]
- 15.Gallivan, J. P., and D. A. Dougherty. 1999. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA 96:9459-9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giangaspero, A., L. Sandri, and A. Tossi. 2001. Amphipathic alpha helical antimicrobial peptides. Eur. J. Biochem. 268:5589-5600. [DOI] [PubMed] [Google Scholar]
- 17.Greenfield, N., and G. D. Fasman. 1969. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8:4108-4116. [DOI] [PubMed] [Google Scholar]
- 18.Gudmundsson, G. H., B. Agerberth, J. Odeberg, T. Bergman, B. Olsson, and R. Salcedo. 1996. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur. J. Biochem. 238:325-332. [DOI] [PubMed] [Google Scholar]
- 19.Guina, T., E. C. Yi, H. Wang, M. Hackett, and S. I. Miller. 2000. A PhoP-regulated outer membrane protease of Salmonella enterica serovar Typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 182:4077-4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heilborn, J. D., M. F. Nilsson, G. Kratz, G. Weber, O. Sorensen, N. Borregaard, and M. Stahle-Backdahl. 2003. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J. Investig. Dermatol. 120:379-389. [DOI] [PubMed] [Google Scholar]
- 21.Hong, S. Y., J. E. Oh, and K. H. Lee. 1999. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 58:1775-1780. [DOI] [PubMed] [Google Scholar]
- 22.Hornef, M. W., S. Normark, B. Henriques-Normark, and M. Rhen. 2005. Bacterial evasion of innate defense at epithelial linings. Chem. Immunol. Allergy 86:72-98. [DOI] [PubMed] [Google Scholar]
- 23.Kraus, D., and A. Peschel. 2006. Molecular mechanisms of bacterial resistance to antimicrobial peptides. Curr. Top. Microbiol. Immunol. 306:231-250. [DOI] [PubMed] [Google Scholar]
- 24.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 26.Maillere, B., G. Mourier, M. Herve, and A. Menez. 1995. Fine chemical modifications at N- and C-termini enhance peptide presentation to T cells by increasing the lifespan of both free and MHC-complexed peptides. Mol. Immunol. 32:1377-1385. [DOI] [PubMed] [Google Scholar]
- 27.Mecozzi, S., A. P. West, Jr., and D. A. Dougherty. 1996. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc. Natl. Acad. Sci. USA 93:10566-10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nell, M. J., G. S. Tjabringa, A. R. Wafelman, R. Verrijk, P. S. Hiemstra, J. W. Drijfhout, and J. J. Grote. 2006. Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides 27:649-660. [DOI] [PubMed] [Google Scholar]
- 29.Nizet, V. 2006. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 8:11-26. [PubMed] [Google Scholar]
- 30.Olson, C. A., E. J. Spek, Z. Shi, A. Vologodskii, and N. R. Kallenbach. 2001. Cooperative helix stabilization by complex Arg-Glu salt bridges. Proteins 44:123-132. [DOI] [PubMed] [Google Scholar]
- 31.Oren, Z., J. C. Lerman, G. H. Gudmundsson, B. Agerberth, and Y. Shai. 1999. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 341(3):501-513. [PMC free article] [PubMed] [Google Scholar]
- 32.Pauchon, V., C. Besson, J. Saulnier, and J. Wallach. 1993. Peptide synthesis catalysed by Pseudomonas aeruginosa elastase. Biotechnol. Appl. Biochem. 17(2):217-221. [PubMed] [Google Scholar]
- 33.Powell, M. F., T. Stewart, L. Otvos, Jr., L. Urge, F. C. Gaeta, A. Sette, T. Arrhenius, D. Thomson, K. Soda, and S. M. Colon. 1993. Peptide stability in drug development. II. Effect of single amino acid substitution and glycosylation on peptide reactivity in human serum. Pharm. Res. 10:1268-1273. [DOI] [PubMed] [Google Scholar]
- 34.Reijmar, K., A. Schmidtchen, and M. Malmsten. 2007. Bactericidal and hemolytic properties of mixed LL-37/surfactant systems. J. Drug Del. Sci. Tech. 17:293-297. [Google Scholar]
- 35.Ringstad, L., E. A. Nordahl, A. Schmidtchen, and M. Malmsten. 2007. Composition effect on peptide interaction with lipids and bacteria: variants of C3a peptide CNY21. Biophys. J. 92:87-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ringstad, L., L. Kacprzyk, A. Schmidtchen, and M. Malmsten. 2007. Effects of topology, length, and charge on the activity of a kininogen-derived peptide on lipid membranes and bacteria. Biochim. Biophys. Acta 1768:715-727. [DOI] [PubMed] [Google Scholar]
- 37.Ringstad, L., E. Protopapa, B. Lindholm-Sethson, A. Schmidtchen, A. Nelson, and M. Malmsten. 2008. An electrochemical study into the interaction between complement-derived peptides and DOPC mono- and bilayers. Langmuir 24:208-216. [DOI] [PubMed] [Google Scholar]
- 38.Ringstad, L., A. Schmidtchen, and M. Malmsten. 2006. Effect of peptide length on the interaction between consensus peptides and DOPC/DOPA bilayers. Langmuir 22:5042-5050. [DOI] [PubMed] [Google Scholar]
- 39.Ruissen, A. L., J. Groenink, P. Krijtenberg, E. Walgreen-Weterings, W. van't Hof, E. C. Veerman, and A. V. N. Amerongen. 2003. Internalisation and degradation of histatin 5 by Candida albicans. Biol. Chem. 384:183-190. [DOI] [PubMed] [Google Scholar]
- 40.Sal-Man, N., D. Gerber, I. Bloch, and Y. Shai. 2007. Specificity in transmembrane helix-helix interactions mediated by aromatic residues. J. Biol. Chem. 282:19753-19761. [DOI] [PubMed] [Google Scholar]
- 41.Schellenberger, V., C. W. Turck, L. Hedstrom, and W. J. Rutter. 1993. Mapping the S′ subsites of serine proteases using acyl transfer to mixtures of peptide nucleophiles. Biochemistry 32:4349-4353. [DOI] [PubMed] [Google Scholar]
- 42.Schellenberger, V., C. W. Turck, and W. J. Rutter. 1994. Role of the S′ subsites in serine protease catalysis. Active-site mapping of rat chymotrypsin, rat trypsin, alpha-lytic protease, and cercarial protease from Schistosoma mansoni. Biochemistry 33:4251-4257. [DOI] [PubMed] [Google Scholar]
- 43.Schmidtchen, A., I. M. Frick, E. Andersson, H. Tapper, and L. Bjorck. 2002. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46:157-168. [DOI] [PubMed] [Google Scholar]
- 44.Serrano, L., M. Bycroft, and A. R. Fersht. 1991. Aromatic-aromatic interactions and protein stability. Investigation by double-mutant cycles. J. Mol. Biol. 218:465-475. [DOI] [PubMed] [Google Scholar]
- 45.Shalev, D. E., A. Mor, and I. Kustanovich. 2002. Structural consequences of carboxyamidation of dermaseptin S3. Biochemistry 41:7312-7317. [DOI] [PubMed] [Google Scholar]
- 46.Shi, Z., C. A. Olson, and N. R. Kallenbach. 2002. Cation-pi interaction in model alpha-helical peptides. J. Am. Chem. Soc. 124:3284-3291. [DOI] [PubMed] [Google Scholar]
- 47.Sieprawska-Lupa, M., P. Mydel, K. Krawczyk, K. Wojcik, M. Puklo, B. Lupa, P. Suder, J. Silberring, M. Reed, J. Pohl, W. Shafer, F. McAleese, T. Foster, J. Travis, and J. Potempa. 2004. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 48:4673-4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sigurdardottir, T., P. Andersson, M. Davoudi, M. Malmsten, A. Schmidtchen, and M. Bodelsson. 2006. In silico identification and biological evaluation of antimicrobial peptides based on human cathelicidin LL-37. Antimicrob. Agents Chemother. 50:2983-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sjogren, H., and S. Ulvenlund. 2005. Comparison of the helix-coil transition of a titrating polypeptide in aqueous solutions and at the air-water interface. Biophys. Chem. 116:11-21. [DOI] [PubMed] [Google Scholar]
- 50.Smith, J. S., and J. M. Scholtz. 1998. Energetics of polar side-chain interactions in helical peptides: salt effects on ion pairs and hydrogen bonds. Biochemistry 37:33-40. [DOI] [PubMed] [Google Scholar]
- 51.Sonesson, A., L. Ringstad, E. A. Nordahl, M. Malmsten, M. Morgelin, and A. Schmidtchen. 2007. Antifungal activity of C3a and C3a-derived peptides against Candida. Biochim. Biophys. Acta 1768:346-353. [DOI] [PubMed] [Google Scholar]
- 52.Sorensen, O., K. Arnljots, J. B. Cowland, D. F. Bainton, and N. Borregaard. 1997. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 90:2796-2803. [PubMed] [Google Scholar]
- 53.Stromstedt, A. A., P. Wessman, L. Ringstad, K. Edwards, and M. Malmsten. 2007. Effect of lipid headgroup composition on the interaction between melittin and lipid bilayers. J. Colloid Interface Sci. 311:59-69. [DOI] [PubMed] [Google Scholar]
- 54.Tiberg, F., I. Harwigsson, and M. Malmsten. 2000. Formation of model lipid bilayers at the silica-water interface by co-adsorption with non-ionic dodecyl maltoside surfactant. Eur. Biophys. J. 29:196-203. [DOI] [PubMed] [Google Scholar]
- 55.Tossi, A., L. Sandri, and A. Giangaspero. 2002. New consensus hydropathy scale extended to non-proteinogenic amino acids, p. 416-417. In Peptides 2002: Proceedings of the Twenty-Seventh European Peptide Symposium. Edizioni Ziino, Naples, Italy.
- 56.Travis, J., J. Potempa, and H. Maeda. 1995. Are bacterial proteinases pathogenic factors? Trends Microbiol. 3:405-407. [DOI] [PubMed] [Google Scholar]
- 57.Travis, S. M., N. N. Anderson, W. R. Forsyth, C. Espiritu, B. D. Conway, E. P. Greenberg, P. B. McCray, Jr., R. I. Lehrer, M. J. Welsh, and B. F. Tack. 2000. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect. Immun. 68:2748-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner, J., Y. Cho, N.-N. Dinh, A. J. Waring, and R. I. Lehrer. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 42:2206-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vacklin, H. P., F. Tiberg, and R. K. Thomas. 2005. Formation of supported phospholipid bilayers via co-adsorption with beta-D-dodecyl maltoside. Biochim. Biophys. Acta 1668:17-24. [DOI] [PubMed] [Google Scholar]
- 60.Wade, D., A. Boman, B. Wahlin, C. M. Drain, D. Andreu, H. G. Boman, and R. B. Merrifield. 1990. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. USA 87:4761-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang, Y., B. Agerberth, A. Lothgren, A. Almstedt, and J. Johansson. 1998. Apolipoprotein A-I binds and inhibits the human antibacterial/cytotoxic peptide LL-37. J. Biol. Chem. 273:33115-33118. [DOI] [PubMed] [Google Scholar]
- 62.Wiegand, I., K. Hilpert, and R. E. Hancock. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163-175. [DOI] [PubMed] [Google Scholar]
- 63.Wieprecht, T., O. Apostolov, M. Beyermann, and J. Seelig. 1999. Thermodynamics of the alpha-helix-coil transition of amphipathic peptides in a membrane environment: implications for the peptide-membrane binding equilibrium. J. Mol. Biol. 294:785-794. [DOI] [PubMed] [Google Scholar]
- 64.Wu, M., and R. E. Hancock. 1999. Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 274:29-35. [DOI] [PubMed] [Google Scholar]
- 65.Yau, W. M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry 37:14713-14718. [DOI] [PubMed] [Google Scholar]
- 66.Zanetti, M., R. Gennaro, and D. Romeo. 1995. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 374:1-5. [DOI] [PubMed] [Google Scholar]
- 67.Zelezetsky, I., S. Pacor, U. Pag, N. Papo, Y. Shai, H. G. Sahl, and A. Tossi. 2005. Controlled alteration of the shape and conformational stability of alpha-helical cell-lytic peptides: effect on mode of action and cell specificity. Biochem. J. 390:177-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zelezetsky, I., and A. Tossi. 2006. Alpha-helical antimicrobial peptides—using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 1758:1436-1449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.