

Abstract
Several protein synthesis inhibitors are known to inhibit ribosome assembly. This may be a consequence of direct binding of the antibiotic to ribosome precursor particles, or it could result indirectly from loss of coordination in the production of ribosomal components due to the inhibition of protein synthesis. Here we demonstrate that erythromycin and chloramphenicol, inhibitors of the large ribosomal subunit, affect the assembly of both the large and small subunits. Expression of a small erythromycin resistance peptide acting in cis on mature ribosomes relieves the erythromycin-mediated assembly defect for both subunits. Erythromycin treatment of bacteria expressing a mixture of erythromycin-sensitive and -resistant ribosomes produced comparable effects on subunit assembly. These results argue in favor of the view that erythromycin and chloramphenicol affect the assembly of the large ribosomal subunit indirectly.
The ribosome is a target for many medically important antibiotics (42, 56, 63). These drugs bind to either the small or the large subunit and inhibit essential ribosome functions: decoding, peptidyl transfer, transit of the nascent peptide chain, or activation of ribosome-associated GTPases. In addition, antibiotics cause many physiological changes; for example, they induce stress response pathways and alter gene expression patterns in other ways (23, 25, 45, 60).
One of the known ”side effects” of ribosome-targeting antibiotics is inhibition of ribosome assembly. This effect was originally described for chloramphenicol: defective particles sedimenting more slowly than mature ribosomal subunits were observed in cells treated with this drug (19, 29). Such particles contain precursor forms of rRNA and an incomplete set of ribosomal proteins (1, 53). During chloramphenicol treatment, ribosomal proteins are produced in nonstoichiometric amounts (20). In addition, the equilibrium between the production of rRNA and ribosomal proteins is upset, and rRNA is expressed in excess (30, 37, 49). It has been proposed that this unbalanced synthesis of components is responsible for the chloramphenicol-induced defects in ribosomal assembly (21).
More recently, Champney and colleagues suggested that several other inhibitors of protein synthesis stall the assembly of ribosomal subunits by binding to the respective precursor particles. The drugs proposed to have such an effect included macrolides and ketolides (10, 11, 15, 35, 59), streptogramin B (16), lincosamides (16), aminoglycosides (36), evernimicin (14), linezolid (12), and pleuromutilins (13). However, the indirect mechanism of assembly inhibition cannot be excluded for any of these drugs.
To investigate the mechanism by which ribosome assembly is inhibited, we chose two inhibitors of the 50S subunit, erythromycin and chloramphenicol, and carried out experiments designed to differentiate between the two alternative hypotheses: (i) direct inhibition due to the binding of antibiotics to the precursor particles and (ii) indirect inhibition, when an imbalance in the production of components disturbs ribosome assembly. Our results appear to be more compatible with the latter model.
MATERIALS AND METHODS
Strains.
Strain MG1655 (6) was used in all experiments involving Escherichia coli. Clinical Staphylococcus aureus strains carrying mutations in chromosomal copies of rRNA operons (43) were generously provided by Roland Leclercq. The strains were UCN14 (carrying an A2058T mutation in four out of five rrl alleles), UCN17 (with A2058G in four out of six rrl alleles), and UCN18 (with A2059G in three out of five rrl alleles). The MIC for erythromycin in all three strains was 512 to 1,024 μg/ml, as determined in 96-well plates by the microbroth dilution procedure (3).
Analysis of E. coli ribosomes.
Cells were grown at either 25 or 37°C in 200 ml 2× YT medium (46) until the A600 reached 0.2. At this point, either erythromycin (final concentration, 100 μg/ml) or chloramphenicol (final concentration, 7 μg/ml) was added; no antibiotic was added to the control culture. The cultures were then incubated for a further 2 h or 1 h at 25 or 37°C, respectively. The cells were collected by centrifugation in a Sorvall GS-3 rotor at 4,000 rpm and 4°C for 10 min and were resuspended in 1 ml lysis buffer (60 mM KCl, 60 mM NH4Cl, 50 mM Tris-HCl [pH 8], 6 mM MgCl2, 6 mM β-mercaptoethanol, 16% sucrose); lysozyme and DNase I (Amresco, GE Healthcare) were added to final concentrations of 1 mg/ml and 20 U/ml, respectively. The cells were incubated for 15 min at −70°C and then thawed in ice-cold water for 30 min. The freeze-thaw cycle was repeated twice, followed by centrifugation at 13,000 × g and 4°C for 20 min. The supernatant was diluted twofold with buffer D (60 mM KCl, 60 mM NH4Cl, 10 mM Tris-HCl [pH 8], 12 mM MgCl2, 6 mM β-mercaptoethanol). Lysate (2 ml) was first loaded onto a 30-ml, 10 to 25% (wt/wt) sucrose gradient prepared in buffer D and then centrifuged at 23,000 rpm in an SW28 rotor (Beckman) at 4°C for 13.5 h.
Analysis of E. coli rRNA.
RNA was precipitated from the sucrose gradient fractions with 0.7 volume of isopropanol. The precipitate was dissolved in 200 μl buffer D, and 1 ml 5 M guanidinium thiocyanate-4% Triton X-100 was added, followed by vortexing for 20 min at room temperature. An SiO2 suspension (25 μl, 50%) in distilled water (7) was added, followed by shaking for 10 min at room temperature. The silica was precipitated by centrifugation at 3,000 × g for 1 min. The precipitate was washed with 1 ml 5 M guanidinium thiocyanate, followed by a wash with 1 ml 50% ethanol. The RNA was eluted by incubating the silica in water for 5 min at 55°C. Centrifugation was used to precipitate the silica during the washing and elution steps. Samples were stored at −20°C.
Reverse transcription from primers GTTTGGGGTACGATTTGATG and GCCAGCGTTCAATCTGAG was used to map the 5′ ends of 23S and 16S rRNA, respectively (31, 32).
For dot blot analysis, purified RNA was denatured by addition of formamide (20 μl), formaldehyde (6 μl), and 4 μl of buffer DB (0.2 M morpholinepropanesulfonic acid [MOPS; pH 7], 20 mM sodium acetate, 10 mM EDTA [pH 8]), followed by incubation at 65°C for 5 min. The samples were cooled on ice, and an equal volume of 20× SSC (3 M NaCl plus 0.3 M sodium acetate) was added. For every gradient profile, 0.2 pmol of 23S RNA from the 50S fraction was used per sample. Equal volumes from the other gradient fractions were used to prepare the RNA for dot blotting. Samples were applied to a Hybond N+ (Amersham) membrane using a vacuum blotter, followed by UV cross-linking. The oligonucleotides used for primer extension (see above) were 5′ end-labeled with 32P and used for hybridization. Prehybridization was performed for 4 h at 50°C in 6× SSC, 5× Denhardt's solution, 0.5% (wt/vol) sodium dodecyl sulfate (SDS), and 100 μg/ml salmon sperm DNA (46). Labeled probes were added, followed by hybridization for 12 h at 50°C. The filters were washed at 50°C for 5 min in 2× SSC-0.1% SDS (wt/vol) and for 10 min in 1× SSC-0.1% SDS, followed by autoradiography.
Peptide-mediated erythromycin resistance.
E. coli cells containing plasmids coding for an erythromycin resistance peptide (sequence MRLFV) (57) or a control peptide (MNAIK) (61) were grown at 37°C in 500 ml 2× YT medium (46) until the A600 was 0.1. At this point, synthesis of the peptide was induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. When the A600 reached 0.2, the cultures were divided into two 70-ml aliquots. One sample received erythromycin to a final concentration of 50 μg/ml, and the second was grown under the same conditions but without erythromycin. One hour after the addition of erythromycin, the cells were collected and lysed, and the ribosomes were analyzed as described above. No IPTG was added in the otherwise similar control experiments.
Pulse-labeling.
E. coli cells were grown at 37°C in 400 ml of tryptone (10 g/liter)-yeast extract (1 g/liter)-NaCl (10 g/liter) until the A600 reached 0.2. Erythromycin (final concentration, 100 μg/ml) or chloramphenicol (final concentration, 7 μg/ml) was added, and the cultures were divided into 50-ml aliquots. After 5, 10, or 60 min of incubation at 37°C, 2 μCi of [3H]uridine (38 Ci/mmol; GE Healthcare) per 50 ml of culture was added, followed by incubation for 5 min at 37°C. In the experiment for which results are shown in Fig. 4a, incorporation of the label was stopped by adding ice to the culture. The other samples were treated by adding rifampin (rifampicin) (final concentration, 500 μg/ml) for 5 min at 37°C, followed by ice. Cells were collected by centrifugation and lysed, and ribosomes were analyzed by sucrose gradient centrifugation as described above. Each sucrose gradient was fractionated into 40 fractions. High-molecular-weight material was precipitated by adding an equal volume of 20% trichloroacetic acid (TCA). The precipitates were collected on glass fiber filters, and the radioactivity incorporated was measured by scintillation counting.
FIG. 4.

Effect of erythromycin on the assembly of sensitive and resistant ribosomes in a mixed population. S. aureus strain UCN18, carrying an A2059G mutation in three out of five rrl alleles, was grown without (left panels) or with (right panels) erythromycin. The bacteria were lysed, and ribosomal subunits were analyzed in sucrose gradients. (a) Flowthrough optical profiles of the sucrose gradient fractions. (b) Optical densities of individual gradient fractions corresponding to the 30S-to-50S gradient area used in subsequent experiments. (c) Primer extension analysis of wild-type and mutant 23S rRNA in individual gradient fractions. (d) Quantification of the gels in panels c. (e) Design of the primer extension assay.
Preparation and analysis of S. aureus ribosomal subunits and precursors.
Cultures of S. aureus strains UCN14, UCN17, and UCN18 (43) were grown in 2 ml of tryptone soy broth (TSB) medium (4). They were diluted 100-fold into two 50-ml cultures and grown at 37°C until the A600 reached 0.1. Erythromycin was added to one of the flasks to a final concentration of 128 μg/ml, and growth was continued until the A600 reached ca. 0.6. Cells were pelleted in a Beckman JA-25 rotor (7000 rpm, 10 min, 4°C), and the pellets were washed once with 10 ml cold buffer S (10 mM Tris-HCl [pH 7.8], 1 mM magnesium diacetate, 50 mM NH4Cl, 2 mM β-mercaptoethanol (10). After repelleting, the cells were resuspended in 400 μl buffer S. Lysostaphin (Sigma) and an RNase inhibitor (Roche) were added to final concentrations of 50 μg/ml and 100 U/ml, respectively. The cell suspension was incubated for 20 min at room temperature. Two units of RQ DNase (Promega) were added to each sample, and after 5 min of incubation at room temperature, the samples were spun for 10 min at 21,000 × g in a refrigerated tabletop centrifuge.
The cell lysate (A260, 10 to 20) was loaded onto a 10 to 40% sucrose gradient prepared in buffer S. Gradients were centrifuged in a Beckman SW41 rotor at 24,500 rpm for 18 h at 4°C and were then fractionated through the UV monitor; 400-μl fractions were collected. The optical density (A260) of the material in individual fractions was determined using a NanoDrop spectrophotometer, and ribosomal material was precipitated by adding sodium acetate to 0.3 M, followed by 2 vol ethanol, and incubating at −20°C overnight. The ribosomal precipitates were collected by centrifugation for 10 min at 21,000 × g in a refrigerated tabletop centrifuge, resuspended in 150 μl of buffer R (0.3 M sodium acetate, 5 mM EDTA, 0.5% SDS), and extracted with phenol, phenol-chloroform, and chloroform. Sodium acetate was added to 0.3 M. The RNA was ethanol precipitated and resuspended in 15 μl water.
Primer extension analysis was carried out as described previously (5) using primer SaL2060 (GTAAAGCTCCACGGGGTC) and either 1.5 μl (from the 50S peak fractions) or 2.5 μl (from the 30S peak fractions) of RNA solution. The reaction products were resuspended in 8 μl formamide with loading dyes, and 1-μl samples were run on a 12% denaturing polyacrylamide gel. The gel was dried and exposed to a PhosphorImager screen (Molecular Dynamics). Images on the screen were scanned and analyzed using ImageQuant software.
RESULTS
Inhibition of ribosome assembly by erythromycin and chloramphenicol.
As a starting point, we tested the previously described effects of chloramphenicol and erythromycin by analyzing the ribosomes obtained from antibiotic-treated cells by sucrose gradient centrifugation. The drugs were used at concentrations causing similar levels of growth inhibition (after 8 h of growth, the optical density of the culture fell to 70% of that of the untreated control). Particles with sedimentation characteristics different from those of mature subunits accumulated in response to treatment with either of the antibiotics (Fig. 1). These more slowly sedimenting particles indicate ribosomal assembly defects. We reproducibly observed that the defective particles accumulating in response to erythromycin sedimented notably differently from those accumulating in response to chloramphenicol (compare Fig. 1c and e). Ribosomal assembly defects are usually more pronounced at lower growth temperatures (18, 26, 44), so we tested the effects of chloramphenicol and erythromycin at 25°C as well as at 37°C. Antibiotic-induced accumulation of several different assembly-defective particles occurred at both growth temperatures; as expected, the more slowly sedimenting particles accumulated in greater amounts at the lower temperature (Fig. 1).
FIG. 1.

Sucrose gradient profiles of ribosomes isolated from untreated (a and b), erythromycin-treated (c and d), or chloramphenicol-treated (e and f) cells. The bacterial cultures were grown either at 37°C (a, c, and e) or at 25°C (b, d, and f). In panels c through f, solid lines represent profiles from antibiotic-treated cells, and profiles from untreated cells (dotted lines) are added for better comparison. Dot blot analyses of 16S and 23S rRNAs isolated from the sucrose gradient fractions are shown at the bottom of each panel.
With both antibiotics, we observed an accumulation of particles sedimenting more slowly than 30S. To verify the nature of these particles, we isolated RNA from the sucrose gradient fractions and hybridized it with 16S rRNA- or 23S rRNA-specific probes. As expected, 16S RNA was found in the 30S and 70S ribosomes, and 23S RNA was found in the 50S and 70S fractions, in untreated cells (Fig. 1). For cells treated with either of the antibiotics, 23S rRNA was found in the 70S and 50S fractions and also in particles sedimenting at 40S (Fig. 1), consistent with the idea that large ribosomal subunit assembly is inhibited. Particles sedimenting more slowly than 30S after treatment with both erythromycin and chloramphenicol contained 16S RNA (Fig. 1). This shows that in addition to the inhibition of large subunit assembly, both drugs cause defects in the biosynthesis of small subunits. This observation revealed that antibiotics that target the large ribosomal subunit exclusively in fact impede the assembly of both the 50S and 30S subunits.
Effects of the antibiotics on rRNA maturation.
During the assembly of ribosomal subunits, pre-rRNA associates with proteins and undergoes several cleavage events and nucleotide modifications (28, 51, 62). rRNA processing depends on ribosome assembly (51). It is therefore expected that general inhibition of assembly will cause defects in rRNA processing. We characterized the state of 5′ end processing of 16S and 23S RNA in the antibiotic-induced assembly-defective particles.
During the assembly of the large subunit, RNase III cleaves the precursor at positions −3 and −7 (2, 8, 50) relative to the 5′ end of mature 23S rRNA, while the final maturation occurs in functional ribosomes. At 37°C, the ribosomes from untreated cells contain fully processed 23S RNA (marked in Fig. 2b as +1). Only traces of longer molecules were observed in the 50S fraction. At 25°C, processing intermediates that had been cleaved at positions −3 and −7 accumulated, indicating that processing is slower at lower growth temperatures.
FIG. 2.

Primer extension analyses of the 5′ ends of 16S (a) and 23S (b) RNAs isolated from the sucrose gradient fractions. The material was isolated either from untreated cells (none) or from erythromycin (Ery)- or chloramphenicol (Cam)-treated cultures grown at either 25 or 37°C. The sequencing lanes are shown on the left.
Partially processed 23S RNA accumulated in the 30S, 40S, and 50S fractions in response to antibiotic treatment. The 5′ end of 23S rRNA was either at position −7 or position −3, as in the 50S fraction from untreated cells grown at 25°C. The fact that neither erythromycin nor chloramphenicol treatment creates new processing intermediates suggests that assembly does not follow alternative pathways creating dead-end products; rather, the normal pathway is simply slowed down. On the other hand, quantification of the intermediates shows that the main intermediate in the untreated cells results from RNase cleavage at position −3, whereas the intermediate cleaved at position −7 accumulates predominantly in response to antibiotic treatment. These differences might have arisen because the different protein contents of the particles influence the specificity of the RNase. This interpretation is supported by the previous observation that RNase III cleaves protein-free pre-RNA at position −7 and RNA assembled into ribosomal subunits at position −3 (2).
Similar effects were observed for 16S RNA processing. During normal 16S processing, the first cleavage is created by RNase III at position −115 (28, 51, 52). This is followed by maturation steps creating heterogeneous 5′ ends. We observed that the processing intermediate cleaved at position −115 is present in the 30S fraction isolated from untreated cells grown at 25°C and, to a lesser extent, in cells grown at 37°C (Fig. 2). The same intermediate accumulated in the 20S and 30S fractions from the antibiotic-treated cells, indicating that neither erythromycin nor chloramphenicol treatment created 16S RNA processing patterns different from those in untreated cells but that they retarded 16S rRNA processing overall.
Expression of erythromycin resistance peptides rescues drug-induced assembly defects.
The peptide-mediated erythromycin resistance mechanism acts in cis only upon actively translating mature ribosomes and does not influence the availability of erythromycin in the cell (33, 54). Antibiotic resistance results from the synthesis of specific small peptides (E-peptides) that promote the dissociation of erythromycin from the ribosomes synthesizing those peptides (33). This resistance mechanism should allow one to distinguish between inhibition of assembly by direct trapping of precursor particles and inhibition mediated through the general effect of the drug on protein synthesis. If E-peptide expression reverses the erythromycin-induced assembly defects, that would argue in favor of the indirect mechanism of inhibition.
To test the effects of the small peptides, we expressed one E-peptide (MRLFV) and a control peptide (MNAIK) in E. coli, and we examined ribosome assembly after erythromycin treatment. Without erythromycin, E-peptide synthesis causes some accumulation of precursor particles (Fig. 3). This might indicate an inhibitory effect on the translational capacity of the cell because of massive translation of the E-peptide minigene present on a multicopy plasmid (33), possibly leading to reduced production of ribosomal proteins. When erythromycin was added but the E-peptide was not expressed, incompletely assembled subunits accumulated (Fig. 3). This inhibitory effect of erythromycin on assembly was relieved by E-peptide expression (Fig. 3) but not by the control peptide (data not shown). The observation that the resistance peptide can relieve assembly inhibition suggests that erythromycin inhibits ribosome synthesis through its general effect on translation rather than via binding to assembly intermediates.
FIG. 3.
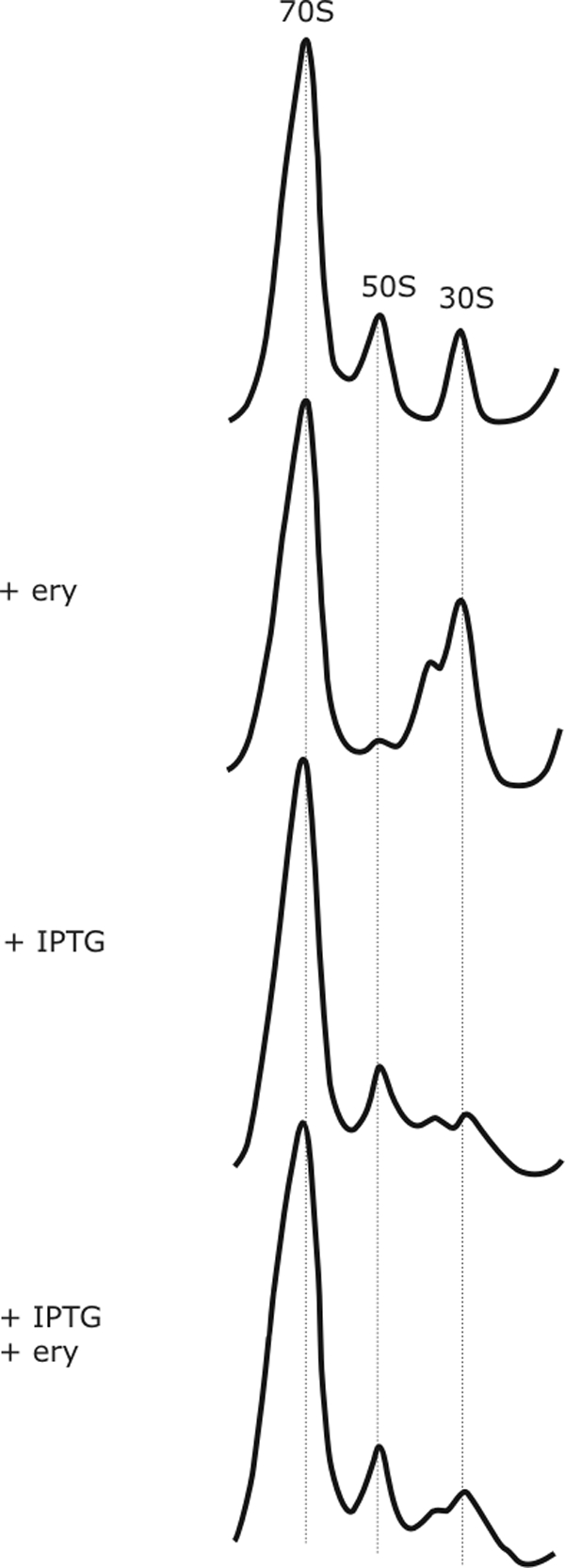
Effects of resistance E-peptide expression on erythromycin-induced assembly defects. Sucrose gradient profiles of ribosomes isolated from cells containing a plasmid for E-peptide expression are shown. Addition of erythromycin (ery) or of IPTG, the inducer of E-peptide expression, is indicated.
As described above, we have observed that erythromycin and chloramphenicol cause rRNA processing defects. If E-peptide expression rescues the erythromycin-induced assembly defect, it is also expected that the inhibition of processing will be alleviated. We have studied the 5′ ends of 23S rRNA isolated from 50S fractions (indicated in Fig. 3). In the 50S fraction isolated from untreated cells, about 85% of the 23S rRNA was fully processed at position +1; if the E-peptide was expressed, 60 to 70% of the RNA was fully processed (consistent with the small assembly defect in response to E-peptide expression); erythromycin treatment reduced the level of mature RNA to 30 to 40%; erythromycin treatment together with E-peptide expression led to 60 to 70% of mature 23S (data not shown). With chloramphenicol treatment, there was about 50% mature 23S rRNA in the 50S fraction, and this percentage did not change in response to E-peptide expression (data not shown). These results show that E-peptide expression specifically alleviates the erythromycin-induced processing defect.
Effect of erythromycin on the assembly of erythromycin-sensitive and erythromycin-resistant ribosomes in a mixed population.
If erythromycin inhibits ribosome assembly via binding to intermediates, then the assembly of the subunits carrying 23S rRNA with erythromycin resistance mutations should not be affected. If cells express a mixed population of wild-type and mutant ribosomes, direct binding of erythromycin to the intermediates is expected to slow down the assembly of wild-type but not mutant ribosomes. On the other hand, if inhibition of ribosome assembly results from the general effect of the drug on protein synthesis, the assembly of both types of ribosomes would be similarly affected. To discriminate between these scenarios, we used clinical strains of S. aureus carrying erythromycin resistance mutations in several copies of the RNA operons (43). We examined ribosome assembly in three such strains: UCN14 (carrying an A2058T mutation in four out of five rrl alleles), UCN17 (with A2058G in four out of six rrl alleles), and UCN18 (with A2059G in three out of five rrl alleles).
S. aureus cells were grown in the absence or presence of erythromycin and then lysed, and ribosomal subunits were analyzed in sucrose gradients (Fig. 4a). The ratio between wild-type and mutant 23S RNA in various fractions was determined by primer extension (Fig. 4e). The results of analysis of UCN18 (Fig. 4) reflect the general pattern observed for all three strains tested. Since these strains are highly resistant to erythromycin (MICs, 512 to 1,024 μg/ml), there was no significant accumulation of assembly-defective particles. Yet the 23S rRNA and 5S rRNA were observed in the 30S peak from cells grown with 128 μg/ml erythromycin (data not shown), indicating that some inhibition of ribosome assembly takes place. The experimental data plots (Fig. 4d) show no significant preferential accumulation of wild-type 23S rRNA in the fraction sedimenting more slowly than 50S. The assembly of 50S subunits carrying wild-type 23S rRNA and that of 50S subunits carrying mutant 23S rRNA were inhibited to comparable extents. This observation suggests that erythromycin does not inhibit assembly by binding to its site in the precursor particles.
Time course of antibiotic-induced inhibition of assembly.
A study of the kinetics of inhibition might provide additional information about the mechanism involved. If the inhibition of ribosome assembly is due to the lack of ribosomal proteins, exposure to the drug would lead to the accumulation over time of particles with gradually decreasing masses and therefore gradually changing sedimentation coefficients. Alternatively, if the drug blocks one particular step in the assembly pathway, defined intermediate particles would accumulate.
To analyze the time course of inhibition of ribosome assembly by chloramphenicol and erythromycin, we profiled ribosomal particles from cells grown at 25°C for different times in the presence or absence of the drugs. The RNA was labeled with [3H]uridine for 5 min, the cells were lysed, and the ribosomal particles were analyzed by sucrose gradient centrifugation (Fig. 5a). The rate-limiting step of ribosomal subunit assembly is the final maturation step, which takes about 1 to 2 min at 37°C (32a). Completion of subunit assembly is manifested by incorporation of the newly assembled subunits into the 70S pool. Consistent with this previous report, we observed that the assembly of ribosomal subunits at 25°C in the absence of the drugs was completed during the 5-min incubation with [3H]uridine: during this short labeling period, a significant fraction of the assembled subunits was already incorporated into 70S ribosomes. Subunit assembly was completed during a further 5-min incubation (Fig. 5b). This additional incubation was performed in the presence of rifampin, which inhibits the initiation of transcription, ensuring that no new rRNA precursors are generated after the initial labeling period.
FIG. 5.

Pulse-labeling of rRNA. Schematic representations of the experiments are shown to the left of the experimental data. Solid lines, optical density patterns of the sucrose gradients; shaded histograms, radioactivity profiles of the TCA-precipitated material. Ery, erythromycin; Cam, chloramphenicol; Rif, rifampin. (a) Experiment performed without rifampin addition; (b) experiments in which pulse-labeling was followed by incubation with rifampin.
In the presence of either chloramphenicol or erythromycin, assembly was significantly retarded even when the antibiotics were added only 5 min before the [3H]uridine (Fig. 5a). With longer antibiotic treatment, broad peaks of assembly intermediates accumulated between the 50S and 20S regions of the gradient. This heterogeneity in the sizes of the defective particles agrees better with the indirect inhibition model (the drugs inhibit ribosome assembly via inhibition of ribosomal protein production and an imbalance in rRNA and ribosomal protein synthesis) than with the model that presumes drug binding to a specific assembly precursor, which would be expected to result in the accumulation of very specific assembly intermediates. Although both erythromycin and chloramphenicol caused the accumulation of assembly-defective particles, there was a slight difference between particles that accumulated after prolonged treatment with the drugs (Fig. 5, compare 10-min and 60-min time points): those that accumulated in the presence of chloramphenicol sedimented more slowly than those that accumulated in the presence of erythromycin. This difference may be attributable to differences in the inhibition of production of specific ribosomal proteins.
DISCUSSION
It has been shown previously that treatment of cells with protein synthesis inhibitors can lead to defects in ribosome assembly. Two alternative explanations for this phenomenon have been proposed. One model suggests that ribosomal subunit assembly is prevented because the production of ribosomal proteins is inhibited, resulting in an imbalance in the amounts of individual proteins synthesized and a lack of stoichiometry with the newly made rRNA (20, 21). The other model suggests that assembly is inhibited by antibiotic binding to precursor particles (59). Sucrose gradient centrifugation showed that radioactively labeled erythromycin cosedimented with the precursor particles (59).
In this study, we carried out experiments designed to differentiate between these two scenarios. We used erythromycin and chloramphenicol as model compounds. These drugs have been used in many previous studies of antibiotic-mediated inhibition of assembly. Both antibiotics bind to the 50S subunit (47, 58). Chloramphenicol inhibits peptide bond formation by preventing the aminoacyl-tRNA 3′ end from binding in the peptidyl transferase center (47), whereas erythromycin hampers the progression of the nascent peptide chain through the ribosomal exit tunnel (34, 55, 58).
Analysis of the ribosomal particles that accumulated in the drug-treated cells revealed precursors that sedimented more slowly than either the 30S or the 50S subunit (Fig. 1). In antibiotic-treated cells, 23S RNA was present not only in 50S subunits but also in fractions sedimenting more slowly than 50S. Similarly, 16S rRNA was also present in fractions sedimenting more slowly than 30S. This result shows that chloramphenicol and erythromycin inhibit the assembly of both the 50S and 30S subunits. Because these drugs do not bind to the small ribosomal subunit, the finding supports the hypothesis that the inhibition of assembly is mediated through general interference with protein synthesis and is not caused by antibiotic binding to precursor particles.
Most conventional antibiotic resistance determinants could not be used to ascertain whether the assembly defect is caused by the inhibition of precursor particles or mature ribosomes: drug efflux pumps and antibiotic modification systems reduce the concentration of the active compound and would therefore have an effect in either case; modification of the binding site might influence drug binding to both precursors and mature particles. On the other hand, E-peptide-mediated erythromycin resistance (33, 54) involves only actively translating mature ribosomes and does not influence the availability of erythromycin in the cell. Therefore, if assembly is inhibited by direct stalling of the process caused by erythromycin binding to the precursor particles, E-peptide-mediated resistance would not relieve the drug-related assembly defects. However, our experiments showed that expression of the resistance E-peptide alleviates the inhibition of assembly (Fig. 3) and defects in 23S rRNA processing (data not shown), arguing that the assembly defects are not mediated through drug binding to intermediates.
In the mixed population of sensitive and mutant ribosomes, stalling of assembly because of drug binding to the precursor particles is expected to influence the assembly of wild-type ribosomes much more significantly than that of mutants that do not bind erythromycin. Yet our data show that in erythromycin-treated cells, the assembly of both sensitive and resistant ribosomes is affected to approximately the same extent, arguing that drug binding to precursors does not stall assembly.
Finally, if assembly were inhibited because the drug binds to precursor particles and prevents subsequent assembly steps, defined assembly intermediates should accumulate. Instead, our kinetics experiments (Fig. 5) showed the appearance of a broad assembly-intermediate peak in drug-treated cells, a result more readily compatible with the hypothesis that assembly defects result from a general inhibition of protein synthesis.
How can the inhibition of protein synthesis lead to defects in ribosome assembly? It has been reported that several inhibitors of protein synthesis, including chloramphenicol and erythromycin, strongly increase the production of rRNA (17, 22, 24, 30, 37, 41, 48, 49). This effect is likely to be caused by the decrease in protein synthesis capacity, activating feedback mechanisms for rRNA transcription (40). On the other hand, as protein synthesis is inhibited, there is less production of ribosomal proteins. As a result, the molar ratio between rRNA and ribosomal proteins will be skewed (20, 53). The excess of rRNA over ribosomal proteins would lead to an accumulation of heterogeneous particles characterized by diverse incomplete sets of ribosomal proteins (21, 38), a scenario fully compatible with our observations (Fig. 5).
In response to treatment with erythromycin or chloramphenicol, partially processed rRNA accumulates and can be detected in several sucrose gradient fractions (Fig. 2). The erythromycin- and chloramphenicol-treated samples contain the same 5′ processing intermediates as those observed in the untreated control; no new cleavage sites are observed. This indicates that antibiotic treatment does not induce completely novel assembly pathways leading to dead-end particles, in agreement with previous reports that upon removal of the drug, the precursor rRNA from the “chloramphenicol particles” is converted into normal 30S and 50S ribosomes without prior degradation (1, 24, 39).
Starting from the middle of the 1990s, Champney and coworkers have reinvestigated the antibiotic-induced ribosomal assembly defects (9, 10). They have described the assembly defects in response to many inhibitors of protein synthesis, including erythromycin. Our observations fully support the previous reports that describe erythromycin-induced assembly inhibition, although our conclusions about the mechanism differ. Usary and Champney have observed binding of radiolabeled erythromycin to precursors of the 50S subunit (59). This was interpreted in the framework of direct inhibition of assembly by the drug. However, mere binding of the drug to the precursor need not necessarily lead to the stalling of assembly. One can easily imagine that precursor particles with bound antibiotic can progress though the remaining steps of the assembly pathway. Furthermore, in assembly experiments using a cell-free system, erythromycin and other macrolide inhibitors of protein synthesis notably stimulated the assembly of the 50S subunit (27).
With both chloramphenicol and erythromycin treatment, precursors of both the large and small ribosomal subunits accumulated (Fig. 1). In contrast, in most of their experiments with several protein synthesis inhibitors acting upon the large ribosomal subunit, Champney et al. observed the accumulation only of 50S subunit precursors. However, it is noteworthy that 30S subunit assembly is inhibited by some macrolides but not others (11, 15, 35). One possible explanation is that the sucrose gradient centrifugation conditions used in those studies do not allow 30S assembly intermediates to be separated reliably from the mature subunits. Another possibility is that the 30S assembly intermediates that accumulate in the presence of some antibiotics are preferentially degraded, leading to seemingly subunit-specific assembly defects.
Acknowledgments
We thank Roland Leclercq for providing the S. aureus mutant strains and Dorota Klepacki for help with some experiments. We thank Ülo Maiväli and Niilo Kaldalu for valuable comments on the manuscript. The English language was corrected by BioMedES, United Kingdom.
This work was supported by The Wellcome Trust International Senior Fellowship (070210/Z/03/Z) (to T.T.), Estonian Science Foundation Grant 7509 (to J. R.), and NIH grant AI072445 from the National Institute of Allergy and Infectious Diseases, NIH (to A.M.).
Footnotes
Published ahead of print on 24 November 2008.
REFERENCES
- 1.Adesnik, M., and C. Levinthal. 1969. Synthesis and maturation of ribosomal RNA in Escherichia coli. J. Mol. Biol. 46:281-303. [DOI] [PubMed] [Google Scholar]
- 2.Allas, U., A. Liiv, and J. Remme. 2003. Functional interaction between RNase III and the Escherichia coli ribosome. BMC Mol. Biol. 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews, J. M. 2001. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 48(Suppl. 1):5-16. [DOI] [PubMed] [Google Scholar]
- 4.Atlas, A. M. 2006. Handbook of microbiological media for the examination of food, 2nd ed. CRC Press, Boca Raton, FL.
- 5.Bailey, M., T. Chettiath, and A. S. Mankin. 2008. Induction of erm(C) expression by noninducing antibiotics. Antimicrob. Agents Chemother. 52:866-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 7.Boom, R., C. J. Sol, M. M. Salimans, C. L. Jansen, P. M. Wertheim-van Dillen, and J. van der Noordaa. 1990. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28:495-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bram, R. J., R. A. Young, and J. A. Steitz. 1980. The ribonuclease III site flanking 23S sequences in the 30S ribosomal precursor RNA of E. coli. Cell 19:393-401. [DOI] [PubMed] [Google Scholar]
- 9.Champney, W. S., and R. Burdine. 1996. 50S ribosomal subunit synthesis and translation are equivalent targets for erythromycin inhibition in Staphylococcus aureus. Antimicrob. Agents Chemother. 40:1301-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Champney, W. S., and R. Burdine. 1995. Macrolide antibiotics inhibit 50S ribosomal subunit assembly in Bacillus subtilis and Staphylococcus aureus. Antimicrob. Agents Chemother. 39:2141-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Champney, W. S., and M. Miller. 2002. Inhibition of 50S ribosomal subunit assembly in Haemophilus influenzae cells by azithromycin and erythromycin. Curr. Microbiol. 44:418-424. [DOI] [PubMed] [Google Scholar]
- 12.Champney, W. S., and M. Miller. 2002. Linezolid is a specific inhibitor of 50S ribosomal subunit formation in Staphylococcus aureus cells. Curr. Microbiol. 44:350-356. [DOI] [PubMed] [Google Scholar]
- 13.Champney, W. S., and W. K. Rodgers. 2007. Retapamulin inhibition of translation and 50S ribosomal subunit formation in Staphylococcus aureus cells. Antimicrob. Agents Chemother. 51:3385-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Champney, W. S., and C. L. Tober. 2000. Evernimicin (SCH27899) inhibits both translation and 50S ribosomal subunit formation in Staphylococcus aureus cells. Antimicrob. Agents Chemother. 44:1413-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Champney, W. S., and C. L. Tober. 2003. Preferential inhibition of protein synthesis by ketolide antibiotics in Haemophilus influenzae cells. Curr. Microbiol. 46:103-108. [DOI] [PubMed] [Google Scholar]
- 16.Champney, W. S., and C. L. Tober. 2000. Specific inhibition of 50S ribosomal subunit formation in Staphylococcus aureus cells by 16-membered macrolide, lincosamide, and streptogramin B antibiotics. Curr. Microbiol. 41:126-135. [DOI] [PubMed] [Google Scholar]
- 17.Cortay, J. C., and A. J. Cozzone. 1983. Effects of aminoglycoside antibiotics on the coupling of protein and RNA syntheses in Escherichia coli. Biochem. Biophys. Res. Commun. 112:801-808. [DOI] [PubMed] [Google Scholar]
- 18.Dabbs, E. R. 1991. Mutants lacking individual ribosomal proteins as a tool to investigate ribosomal properties. Biochimie 73:639-645. [DOI] [PubMed] [Google Scholar]
- 19.Dagley, S., and J. Sykes. 1959. Effect of drugs upon components of bacterial cytoplasm. Nature 183:1608-1609. [DOI] [PubMed] [Google Scholar]
- 20.Dennis, P. P. 1976. Effects of chloramphenicol on the transcriptional activities of ribosomal RNA and ribosomal protein genes in Escherichia coli. J. Mol. Biol. 108:535-546. [DOI] [PubMed] [Google Scholar]
- 21.Dodd, J., J. M. Kolb, and M. Nomura. 1991. Lack of complete cooperativity of ribosome assembly in vitro and its possible relevance to in vivo ribosome assembly and the regulation of ribosomal gene expression. Biochimie 73:757-767. [DOI] [PubMed] [Google Scholar]
- 22.Drobyshev, V. I., and R. S. Shakulov. 1976. Effect of streptomycin on RNA synthesis in E. coli cells. Biokhimiia 41:1193-1199. (In Russian.) [PubMed] [Google Scholar]
- 23.Goh, E. B., G. Yim, W. Tsui, J. McClure, M. G. Surette, and J. Davies. 2002. Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proc. Natl. Acad. Sci. USA 99:17025-17030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosokawa, K., and M. Nomura. 1965. Incomplete ribosomes produced in chloramphenicol- and puromycin-inhibited Escherichia coli. J. Mol. Biol. 12:225-241. [DOI] [PubMed] [Google Scholar]
- 25.Hutter, B., C. Schaab, S. Albrecht, M. Borgmann, N. A. Brunner, C. Freiberg, K. Ziegelbauer, C. O. Rock, I. Ivanov, and H. Loferer. 2004. Prediction of mechanisms of action of antibacterial compounds by gene expression profiling. Antimicrob. Agents Chemother. 48:2838-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iost, I., and M. Dreyfus. 2006. DEAD-box RNA helicases in Escherichia coli. Nucleic Acids Res. 34:4189-4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khaitovich, P., and A. S. Mankin. 1999. Effect of antibiotics on large ribosomal subunit assembly reveals possible function of 5S rRNA. J. Mol. Biol. 291:1025-1034. [DOI] [PubMed] [Google Scholar]
- 28.King, T. C., R. Sirdeskmukh, and D. Schlessinger. 1986. Nucleolytic processing of ribonucleic acid transcripts in procaryotes. Microbiol. Rev. 50:428-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurland, C. G., M. Nomura, and J. D. Watson. 1962. The physical properties of the chloromycetin particles. J. Mol. Biol. 4:388-394. [DOI] [PubMed] [Google Scholar]
- 30.Lazzarini, R. A., and E. Santangelo. 1968. Effect of chloramphenicol on the synthesis and stability of ribonucleic acid in Bacillus subtilis. J. Bacteriol. 95:1212-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liiv, A., and J. Remme. 1998. Base-pairing of 23S rRNA ends is essential for ribosomal large subunit assembly. J. Mol. Biol. 276:537-545. [DOI] [PubMed] [Google Scholar]
- 32.Liiv, A., T. Tenson, T. Margus, and J. Remme. 1998. Multiple functions of the transcribed spacers in ribosomal RNA operons. Biol. Chem. 379:783-793. [DOI] [PubMed] [Google Scholar]
- 32a.Lindahl, L. 1975. Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J. Mol. Biol. 92:15-37. [DOI] [PubMed] [Google Scholar]
- 33.Lovmar, M., K. Nilsson, V. Vimberg, T. Tenson, M. Nervall, and M. Ehrenberg. 2006. The molecular mechanism of peptide-mediated erythromycin resistance. J. Biol. Chem. 281:6742-6750. [DOI] [PubMed] [Google Scholar]
- 34.Lovmar, M., T. Tenson, and M. Ehrenberg. 2004. Kinetics of macrolide action: the josamycin and erythromycin cases. J. Biol. Chem. 279:53506-53515. [DOI] [PubMed] [Google Scholar]
- 35.Mabe, S., J. Eller, and W. S. Champney. 2004. Structure-activity relationships for three macrolide antibiotics in Haemophilus influenzae. Curr. Microbiol. 49:248-254. [DOI] [PubMed] [Google Scholar]
- 36.Mehta, R., and W. S. Champney. 2002. 30S ribosomal subunit assembly is a target for inhibition by aminoglycosides in Escherichia coli. Antimicrob. Agents Chemother. 46:1546-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Midgley, J. E., and W. J. Gray. 1971. The control of ribonucleic acid synthesis in bacteria. The synthesis and stability of ribonucleic acid in chloramphenicol-inhibited cultures of Escherichia coli. Biochem. J. 122:149-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nierhaus, K. H. 1991. The assembly of prokaryotic ribosomes. Biochimie 73:739-755. [DOI] [PubMed] [Google Scholar]
- 39.Nomura, M., and K. Hosokawa. 1965. Biosynthesis of ribosomes: fate of chloramphenicol particles and of pulse-labeled RNA in Escherichia coli. J. Mol. Biol. 12:242-265. [DOI] [PubMed] [Google Scholar]
- 40.Paul, B. J., W. Ross, T. Gaal, and R. L. Gourse. 2004. rRNA transcription in Escherichia coli. Annu. Rev. Genet. 38:749-770. [DOI] [PubMed] [Google Scholar]
- 41.Perel'man, B. V., L. G. Kolibaba, B. R. Belitskii, and R. S. Shakulov. 1979. Effects of different concentrations of chloramphenicol on RNA synthesis in E. coli. Biokhimiia 44:1968-1971. (In Russian.) [PubMed] [Google Scholar]
- 42.Poehlsgaard, J., and S. Douthwaite. 2005. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 3:870-881. [DOI] [PubMed] [Google Scholar]
- 43.Prunier, A. L., B. Malbruny, D. Tande, B. Picard, and R. Leclercq. 2002. Clinical isolates of Staphylococcus aureus with ribosomal mutations conferring resistance to macrolides. Antimicrob. Agents Chemother. 46:3054-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy-Chaudhuri, B., N. Kirthi, T. Kelley, and G. M. Culver. 2008. Suppression of a cold-sensitive mutation in ribosomal protein S5 reveals a role for RimJ in ribosome biogenesis. Mol. Microbiol. 68:1547-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabina, J., N. Dover, L. J. Templeton, D. R. Smulski, D. Soll, and R. A. LaRossa. 2003. Interfering with different steps of protein synthesis explored by transcriptional profiling of Escherichia coli K-12. J. Bacteriol. 185:6158-6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 47.Schlünzen, F., R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R. Albrecht, A. Yonath, and F. Franceschi. 2001. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 413:814-821. [DOI] [PubMed] [Google Scholar]
- 48.Sells, B. H. 1964. RNA synthesis and ribosome production in puromycin-treated cells. Biochim. Biophys. Acta 80:230-241. [DOI] [PubMed] [Google Scholar]
- 49.Shen, V., and H. Bremer. 1977. Chloramphenicol-induced changes in the synthesis of ribosomal, transfer, and messenger ribonucleic acids in Escherichia coli B/r. J. Bacteriol. 130:1098-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sirdeshmukh, R., and D. Schlessinger. 1985. Ordered processing of Escherichia coli 23S rRNA in vitro. Nucleic Acids Res. 13:5041-5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Srivastava, A. K., and D. Schlessinger. 1990. Mechanism and regulation of bacterial ribosomal RNA processing. Annu. Rev. Microbiol. 44:105-129. [DOI] [PubMed] [Google Scholar]
- 52.Srivastava, A. K., and D. Schlessinger. 1989. Processing pathway of Escherichia coli 16S precursor rRNA. Nucleic Acids Res. 17:1649-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sykes, J., E. Metcalf, and J. D. Pickering. 1977. The nature of the proteins in ‘chloramphenicol particles’ from Escherichia coli A19 (Hfr rel met rns). J. Gen. Microbiol. 98:1-16. [DOI] [PubMed] [Google Scholar]
- 54.Tenson, T., A. DeBlasio, and A. Mankin. 1996. A functional peptide encoded in the Escherichia coli 23S rRNA. Proc. Natl. Acad. Sci. USA 93:5641-5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tenson, T., M. Lovmar, and M. Ehrenberg. 2003. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 330:1005-1014. [DOI] [PubMed] [Google Scholar]
- 56.Tenson, T., and A. Mankin. 2006. Antibiotics and the ribosome. Mol. Microbiol. 59:1664-1677. [DOI] [PubMed] [Google Scholar]
- 57.Tenson, T., L. Xiong, P. Kloss, and A. S. Mankin. 1997. Erythromycin resistance peptides selected from random peptide libraries. J. Biol. Chem. 272:17425-17430. [DOI] [PubMed] [Google Scholar]
- 58.Tu, D., G. Blaha, P. B. Moore, and T. A. Steitz. 2005. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 121:257-270. [DOI] [PubMed] [Google Scholar]
- 59.Usary, J., and W. S. Champney. 2001. Erythromycin inhibition of 50S ribosomal subunit formation in Escherichia coli cells. Mol. Microbiol. 40:951-962. [DOI] [PubMed] [Google Scholar]
- 60.VanBogelen, R. A., and F. C. Neidhardt. 1990. Ribosomes as sensors of heat and cold shock in Escherichia coli. Proc. Natl. Acad. Sci. USA 87:5589-5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vimberg, V., L. Xiong, M. Bailey, T. Tenson, and A. Mankin. 2004. Peptide-mediated macrolide resistance reveals possible specific interactions in the nascent peptide exit tunnel. Mol. Microbiol. 54:376-385. [DOI] [PubMed] [Google Scholar]
- 62.Wilson, D. N., and K. H. Nierhaus. 2007. The weird and wonderful world of bacterial ribosome regulation. Crit. Rev. Biochem. Mol. Biol. 42:187-219. [DOI] [PubMed] [Google Scholar]
- 63.Yonath, A. 2005. Antibiotics targeting ribosomes: resistance, selectivity, synergism and cellular regulation. Annu. Rev. Biochem. 74:649-679. [DOI] [PubMed] [Google Scholar]