

Abstract
Nerve growth factor (NGF) is critical for the differentiation and maintenance of neurons in the peripheral and central nervous system. Sustained autophosphorylation of the TrkA receptor tyrosine kinase and long-lasting activation of downstream kinase cascades are hallmarks of NGF signaling, yet our knowledge of the molecular mechanisms underlying prolonged TrkA activity is incomplete. Protein phosphatase 2A (PP2A) is a heterotrimeric Ser/Thr phosphatase composed of a scaffolding, catalytic, and regulatory subunit (B, B′, and B" gene families). Here, we employ a combination of pharmacological inhibitors, regulatory subunit overexpression, PP2A scaffold subunit exchange, and RNA interference to show that PP2A containing B′ family regulatory subunits participates in sustained NGF signaling in PC12 cells. Specifically, two neuron-enriched regulatory subunits, B′β and B′δ, recruit PP2A into a complex with TrkA to dephosphorylate the NGF receptor on Ser/Thr residues and to potentiate its intrinsic Tyr kinase activity. Acting at the receptor level, PP2A/ B′β and B′δ enhance NGF (but not epidermal growth factor or fibroblast growth factor) signaling through the Akt and Ras-mitogen-activated protein kinase cascades and promote neuritogenesis and differentiation of PC12 cells. Thus, select PP2A heterotrimers oppose desensitization of the TrkA receptor tyrosine kinase, perhaps through dephosphorylation of inhibitory Ser/Thr phosphorylation sites on the receptor itself, to maintain neurotrophin-mediated developmental and survival signaling.
The secreted neurotrophins nerve growth factor (NGF), brain-derived neurotrophic factor, and neurotrophin-3 and -4 interact with type I transmembrane receptors to activate signal transduction cascades that regulate neuronal development, plasticity, and survival in most neuronal populations (23, 47). As the prototypical neurotrophin, NGF acts through two receptors, p75 and tropomyosin-related kinase A (TrkA). A member of the receptor tyrosine kinase family of growth factor receptors, TrkA is required for the development and preservation of cholinergic neurons in the brain as well as sympathetic and sensory neurons in the peripheral nervous system (10, 15, 30). TrkB and TrkC, the closely related receptors for brain-derived neurotrophic factor and neurotrophin-3, respectively, have similarly critical functions in the developing and adult central nervous system (23, 47).
Upon neurotrophin binding, Trk receptors dimerize and cross-autophosphorylate on tyrosine residues in the activation loop of the intracellular kinase domain. Trk autophosphorylation continues on additional Tyr residues, which serve as docking sites for adaptor proteins that feed into the mitogen-activated protein kinase (MAPK; also known as extracellular signal-regulated kinase [ERK]), phosphoinositide 3′ kinase, and phospholipase Cγ (PLCγ) signaling cascades (23, 70). Derived from a peripheral nervous system tumor, PC12 cells express both TrkA and p75 receptors and respond to NGF by differentiating into a sympathetic neuron-like phenotype (19). Prolonged MAP kinase activation is both necessary and sufficient for neuronal differentiation of PC12 cells (13, 19, 46), whereas sustained phosphoinositide 3′ kinase/Akt activation is essential for neurotrophin-dependent survival of PC12 cells and primary neurons (4, 28, 47). While it is well established that maintenance of Trk autophosphorylation is required for long-lasting activation of downstream effectors (48, 71), only a few regulators of TrkA autophosphorylation have been identified thus far (9, 33, 39, 54).
Protein phosphatase 2A (PP2A) is a family of ubiquitous and essential serine/threonine phosphatases that target a wide spectrum of signaling molecules, including kinases and receptors (18, 24, 27). The predominant form of PP2A consists of a core dimer of a scaffolding A subunit and a catalytic C subunit which associates with a large repertoire of regulatory subunits to form heterotrimeric PP2A. There are three gene families that encode regulatory subunits, B (PR55), B′ (B56/PR61), and B" (PR72), which have little sequence and structural similarities. In mammals, each regulatory subunit family contains three to five genes that share 70 to 90% sequence identity (22, 40, 41), and alternative splicing of several genes adds further complexity to PP2A regulation. The B family of regulatory subunits are β-propellers with N-terminal differential targeting sequences (14, 59, 66), whereas B" family subunits contain two calmodulin-like EF hands that confer Ca2+-dependent activity to the PP2A heterotrimer (1, 2, 25). Recent crystal structures depict B′ family subunits as elongated, α-helical repeat-containing proteins poised to present substrates for dephosphorylation by the catalytic subunit (12, 67). Several kinases regulate PP2A activity via phosphorylation of B′ regulatory subunits (1, 31, 61, 68).
Here, we identify two neuron-enriched members of the B′ family of PP2A regulatory subunits, B′β and B′δ, as positive regulators of neurotrophin signaling in PC12 cells. B′β/δ recruit the PP2A holoenzyme to the TrkA signaling complex to enhance TrkA autophosphorylation, perhaps by dephosphorylating inhibitory Ser/Thr phosphorylation sites. PP2A/B′β and Bδ thus foster downstream kinase activation, as well as neuritogenesis and neuronal differentiation of PC12 cells.
MATERIALS AND METHODS
Cell culture and plasmids.
PC12 (6-24) TrkA overexpression cells (a gift from Philip Barker [21]) and PC12 cells (PC6-3 subline [45]) were cultured (37°C, 5% CO2) in RPMI 1640 (Gibco) containing 10% horse and 5% fetal bovine serum (both heat inactivated). PP2A/Aα-RNAi, Aα-exchange cells, and B′β-expressing cells derived from PC6-3 cells were cultured in selection medium as previously described (51, 58). Antibiotics were omitted from cultures seeded for experiments. Plasmids encoding Ras V12 and LCK-mCherry were donated by Philip Stork (Vollum Institute) and Steven Green (University of Iowa), respectively. pFC-MEK1, a plasmid encoding constitutively active MEK1 (S218, 222D), was supplied as part of the PathDetect Elk1 trans-reporting system (Stratagene). The 4×hemagglutinin (HA)-tagged B′ subunits were a gift from David Virshup (42, 43). PP2A/B′ subunit silencing shRNAs were expressed from the human H1 promoter (pSUPER plasmid) (8). The following B′ cDNA sequences were targeted for RNA interference (RNAi): B′α, AATGATCAGTGCTAACATCTT; B′β, AACCCTGAATTTGACCCTGAA; B′γ, AAGACTCACAGTCCAAAAGAA; B′δ, AAGACCATTTTGCATCGCATC; B′ɛ, AATTGGAGGATCTGGAGTTAA.
Antibodies and other reagents.
Polyclonal antibodies against B′β and B′δ were a gift from David Virshup (Duke-NUS, Singapore). Transferrin receptor antibody was a gift from David Sheff (University of Iowa). Antibodies commercially available and their sources were as follows: pan-TrkC-14 and agarose conjugate, TrkA B-3, total ERK, normal rabbit immunoglobulin G (IgG)-agarose, and protein A-agarose (Santa Cruz Biotechnology); HA epitope and agarose conjugate (Sigma); PP2A catalytic subunit (BD Biosciences); phospho-TrkA Y490, phospho-Ser-473 Akt, total Akt, and phospho-ERK1/2 (Cell Signaling); phospho-Tyr (4G10; Upstate Biotechnology).
Rat NGF (2.5 S) and recombinant human fibroblast growth factor 2 (FGF2) were purchased from Upstate Biotechnology, Sigma, and Alomone Labs, respectively. Growth factors were stored at −20°C in lyophilized aliquots and dissolved to 100× in culture medium prior to use. Okadaic acid and microcystin-LR were purchased from Alexis. Microcystin-LR agarose beads were purchased from Upstate Biotechnology. [32P]orthophosphate (10 mCi/ml) was purchased from Perkin-Elmer.
Immunoprecipitations.
Native PC12, Aα-RNAi, or Aα-exchange cells were seeded in 100-mm dishes and treated or not with doxycycline (Dox; 1 μg/ml) for 3 days as indicated. The four-HA-tagged B′ subunits were transiently expressed in PC12 cells for 2 days using Lipofectamine 2000 (BD Biosciences). Cells were serum starved for at least 2 h and pretreated in some cases with or without 300 nM okadaic acid (OA) prior to adding growth factors at staggered times. After washing with phosphate-buffered saline, cells were harvested in radioimmunoprecipitation assay (RIPA) lysis buffer (1% Triton X-100 [TX-100], 0.5% sodium dodecyl sulfate [SDS], 0.5% deoxycholate, 150 mM Tris, pH 8.0, 300 mM NaCl, 1 mM EDTA, and 1 mM EGTA) and supplemented with protease/phosphatase inhibitor cocktail (1 mM benzamidine, 10 μg/ml [20 μM] leupeptin, 1 mM pepstatin, and 250 μM phenylmethylsulfonyl fluoride, 1 mM β-glycerolphosphate, 2.5 mM sodium pyrophosphate, and 0.5 μM microcystin-LR). Soluble protein was quantified using Bradford reagent and lysates equalized for volume and protein concentration were immunoprecipitated using 1.5 μg of TrkA (C-14) antibody and protein A-agarose or agarose conjugates of antibodies directed against TrkA or the HA epitope for 4 to 6 h. Immunoprecipitates were washed four times with Tris-buffered saline containing 0.5% TX-100, extracted in SDS sample buffer, subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a polyvinylidene difluoride (PVDF) membrane (0.45 μm; Millipore) for Western blotting. Primary antibodies were detected either by chemiluminescence using a Kodak Imager 440 or by infrared fluorescence using an Odyssey imaging system (Licor).
Band intensities were quantified with the ImageJ software Gel Analyzer plug-in (National Institutes of Health). Phospho-specific antibody signals were divided by total protein antibody signals to control for loading differences. Signal intensities were normalized to the maximum intensity in each experiment.
Metabolic labeling.
PC12 6-24 cells cultured to 70 to 80% confluence in six-well dishes were washed twice with phosphate-free RPMI (Invitrogen) and serum starved for 30 min with the same medium containing 0.5% dialyzed fetal bovine serum. Cells were labeled with 500 μCi/ml [32P]orthophosphate for 60 min prior to 250 μM OA or dimethyl sulfoxide vehicle treatment. Beginning 30 min post-OA or dimethyl sulfoxide treatment, cells were stimulated with 50 ng/ml NGF for 0 to 60 min, harvested in RIPA buffer, and TrkA was immunoprecipitated as described above. Immunoprecipitates were divided into three aliquots, two of which were subjected to phosphatase treatment while the third aliquot was incubated in wash buffer alone (15 min, 30°C with intermittent mixing). Tyrosine dephosphorylation was carried out with 1 U/μl yersinia outer protein tyrosine phosphatase (YopH; New England Biolabs, Bedford, MA) in 50 mM Tris, pH 7, 100 mM NaCl, 5 mM dithiothreitol, 2 mM EDTA, 0.05% TX-100; total (Ser/Thr/Tyr) dephosphorylation was performed with 1 U/μl lambda phosphatase (New England Biolabs) in 50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM dithiothreitol, 0.1 mM EGTA, 2 mM MnCl2, 0.05% TX-100. Reactions were quenched by addition of SDS sample buffer and EDTA to 25 mM. After SDS-PAGE and transfer to PVDF membranes, samples were first exposed to phosphorimager screens and then subjected to immunoblotting for TrkA as described above. Relative 32P incorporation was quantified by densitometric analysis with the Gel Analyzer plug-in of ImageJ, dividing 32P signals by antibody signals.
Tissue preparation.
Dorsal root ganglion, spinal cord, and brain tissue extracts were harvested immediately following cervical dislocation of male Taconic C57BL/6 mice as described previously (53). TrkA was immunoprecipitated from lysates equalized for protein concentration as described above. Control immunoprecipitations were performed with 5 μg of nonimmune rabbit IgG. Total extract input (5% of the volume used for immunoprecipitation) was loaded in parallel.
Microcystin affinity precipitation.
Mouse brain lysates were prepared as described above in the absence of microcystin-LR. Normalized lysates were divided into thirds. As a control, one sample was incubated for 30 min with 1 μM free microcystin-LR to block microcystin-LR-agarose binding. Samples were incubated with 15 μl of microcystin-LR-agarose or glutathione-S-transferase-agarose as a control for 2 h. Beads were washed three times in 0.5% TX-100-Tris-buffered saline and extracted in SDS sample buffer, subjected to SDS-PAGE, and transferred to PVDF membrane (0.45 μm; Millipore) for Western blotting. Images were gathered using an Odyssey imaging system (Licor). Total extract input (5% of volume used for immunoprecipitation) was loaded in parallel.
MAPK reporter assays.
To quantify ERK activation, the PathDetect Elk1 trans-reporting system (Stratagene) was modified for the dual-luciferase assay (Promega) as previously described (57). Briefly, native PC12 cells or B′β subunit-overexpressing PC12 cells were transfected with reporter plasmid mix, pSUPER-based plasmids expressing shRNAs directed against B′ subunits, or activator plasmid (Ras V12, pFC-MEK1 S218, 222D) or pcDNA3.1 empty vector. After 2 days with or without Dox treatment or 3 days of shRNA expression, 2 h of serum starvation, and 5 to 6 h of stimulation with growth factors as indicated, cultures were lysed in passive lysis buffer (Promega) and subjected to dual-luciferase assays using a Berthold Sirius tube luminometer. Photinus and Renilla luciferase activity ratios were expressed relative to basal conditions without ERK activator plasmids, Dox, or growth factor stimulation.
Neurite outgrowth assay.
B′β-inducing PC12 cells were plated on collagen-coated 12-well plates and transfected with 1 μg/well membrane-targeted LCK-mCherry using Lipofectamine 2000 to visualize neurites. Wells were treated with or without 1 μg/ml Dox for 2 days to induce B′β expression and then stimulated with 2 ng/ml NGF for 18 h. Native PC12 cells were transiently transfected with B′ subunit shRNA and LCK-mCherry (1:3 mass ratio) for 3 days and stimulated with 2 ng/ml NGF for 26 h. Cells were washed with phosphate-buffered saline before and after 5 min of fixation with 4% paraformaldehyde. Fifty to 100 cells per condition were photographed using the 20× lens of an inverted epifluorescence microscope equipped with a digital camera. Total neurite length from each cell was measured using ImageJ. Image capture and analyses were performed blind to the experimental condition.
Cell cycle analysis.
B′β-inducing PC12 cells were plated on collagen-coated 60-mm well plates at 150,000/plate and cultured with or without 1 μg/ml Dox for 2 days to induce B′β expression. Cultures were then treated with or without 10 ng/ml NGF for 2 and 4 days to stimulate differentiation. Cells detached with trypsin were pelleted at 1,000 × g and lysed in propidium iodide (PI) lysis buffer (0.1% TX-100, 0.1% sodium citrate, 0.05 mg/ml PI, and 2 μg/ml RNase A [Roche]) for 30 min at room temperature and placed on ice. DNA content was analyzed by flow cytometry with a Becton Dickinson FACScan. For each sample 1.5 × 104 events were recorded. PI histograms were generated and analyzed with the FlowJo software (Tree Star) to determine percentages of cells in each phase (G0/G1, S, and G2/M).
Surface biotin labeling.
PC12 cells were grown to 80% confluence on 100-mm dishes, serum starved for 2 h, and pretreated with or without 300 nM OA for 1 h prior to 20 ng/ml NGF stimulation for 0 to 60 min. Cells were chilled on ice, washed two times with cold phosphate-buffered saline, and incubated with 500 nM EZ-link Sulfo-NHS-LC-LC-biotin (Pierce) for 30 min on ice. Cells were harvested in RIPA buffer, and normalized lysates were precipitated with NeutrAvidin agarose (Pierce) for 2 h. Beads were washed three times in 0.5% Triton X-100-Tris-buffered saline, extracted in SDS sample buffer, and subjected to SDS-PAGE and Western blotting for total TrkA and transferrin receptor. TrkA surface expression was determined by quantitative Western blotting as the ratio of TrkA and transferrin receptor signals in the same lane.
Statistical analysis.
Two-way analysis of variance with Bonferroni post tests was performed for multiple comparisons, and Student's t tests were used for individual comparisons using Prism 4.0. P values less than 0.05 were considered significant.
RESULTS
PP2A sustains NGF-dependent TrkA autophosphorylation.
We have previously shown that inducible RNAi of the major PP2A scaffolding subunit Aα promotes apoptotic cell death in PC12 cells starting 4 days after induction with Dox (58). Assayed at earlier time points, Aα knockdown also attenuated epidermal growth factor (EGF), FGF2, and NGF-dependent activation of Akt and ERK1/2 but had no effect on autophosphorylation of the EGF receptor on total tyrosines or on Tyr-1045 (62), a critical SH2 domain recruitment site. Because acute inhibition of PP2A by OA resulted in hyperphosphorylation of Akt and ERK1/2, we concluded that chronic PP2A inhibition by RNAi leads to a feedback downregulation of a signaling molecule(s) downstream of the EGF receptor but upstream of Ras (62).
To investigate the mechanism by which PP2A regulates NGF-dependent kinase cascades, we immunoprecipitated TrkA from PC12 cells stimulated for up to 3 h with saturating concentrations of NGF (20 ng/ml) and assessed TrkA activity by immunoblotting for phospho-Tyr (Fig. 1A). In the absence of Dox, TrkA autophosphorylation peaked at 5 min post-NGF addition and was maintained at 50% maximum levels throughout the time course analyzed (Fig. 1B). ERK1/2 activation (pT-E-pY immunoreactivity) followed a similar profile (Fig. 1D), while Akt phosphorylation at Ser-473 declined more slowly (Fig. 1C). Doxycycline treatment for 3 days resulted in a ∼50% reduction of total PP2A/A subunit levels (Fig. 1A), which was accompanied by a 20 to 30% decrease in PP2A-like phosphatase activity (58). Confirming earlier results (62), PP2A downregulation attenuated NGF signaling to both Akt and ERK1/2. Paralleling inhibition of Akt and ERK phosphorylation, Tyr phosphorylation of immunoprecipitated TrkA was significantly blunted by PP2A/Aα RNAi at all but the first NGF treatment time point (Fig. 1A and B). NGF receptor specificity was indicated by control experiments, according to which EGF-stimulated autophosphorylation of the EGF receptor was unaffected by PP2A/Aα knockdown (Fig. 1E and F) (62). We quantified relative TrkA expression levels in these immunoprecipitation experiments and detected a significant ∼40% decrease with time in NGF (presumably reflecting proteolytic degradation of the activated receptor [26, 52]) but no effect of PP2A downregulation on TrkA protein levels (data not shown).
FIG. 1.

PP2A/Aα knockdown attenuates the sustained phase of TrkA autophosphorylation and NGF signaling to Akt and ERK. (A) PC12 cells that express shRNA targeting PP2A/Aα from a Dox-inducible promoter (PP2A/Aα↓ cells) were treated for 3 days in the presence or absence of Dox and stimulated with 20 ng/ml NGF for the indicated times. TrkA was immunoprecipitated and probed for phospho-Tyr (pY; 4G10 antibody), whereas activation levels of Akt (phospho-Ser-473) and ERK1/2 (phospho-Thr/phospho-Tyr) were assessed in lysates. Arrows on right indicate mature, glycosylated TrkA. (B to D) Quantification of phosphoprotein immunoreactivity divided by total protein immunoreactivity normalized to the maximum ratio. (E and F) PP2A/Aα↓ cells incubated for 3 days with or without Dox were stimulated with 50 ng/ml EGF for the indicated times. Lysates were probed for pY (arrow indicates position of the EGFR) and striatin, which was used as a loading control for the quantification shown in panel F. The line graphs show means ± standard errors of four (F) or five (B to D) independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.005 (compared to control).
To confirm that PP2A positively regulates NGF signaling by promoting TrkA autophosphorylation, we inhibited PP2A-like phosphatases pharmacologically by a 2-h pretreatment with 250 nM OA. Acute PP2A inhibition diminished NGF-dependent TrkA Tyr phosphorylation to a similar extent as PP2A/Aα RNAi (Fig. 2A). Autophosphorylation of TrkA at Tyr-490, a docking site for Shc and other signaling adaptors, is essential for NGF signaling to Akt and ERK (56, 69). TrkA autophosphorylation profiles in response to NGF and OA treatment were indistinguishable, whether a general phospho-Tyr (4G10) or a phospho Tyr-490 TrkA-specific antibody was used (Fig. 2B and C). In the latter experiments, we probed for total and phospho-TrkA in rapidly prepared total lysates of the PC12 6-24 subline, which overexpresses TrkA 15- to 20-fold over native PC12 cells (21). Thus, both pharmacological and RNAi-mediated inhibition of the Ser/Thr phosphatase PP2A attenuates the sustained, but not the transient, phase of TrkA activation by NGF in two neuronal cell lines with different receptor levels.
FIG. 2.
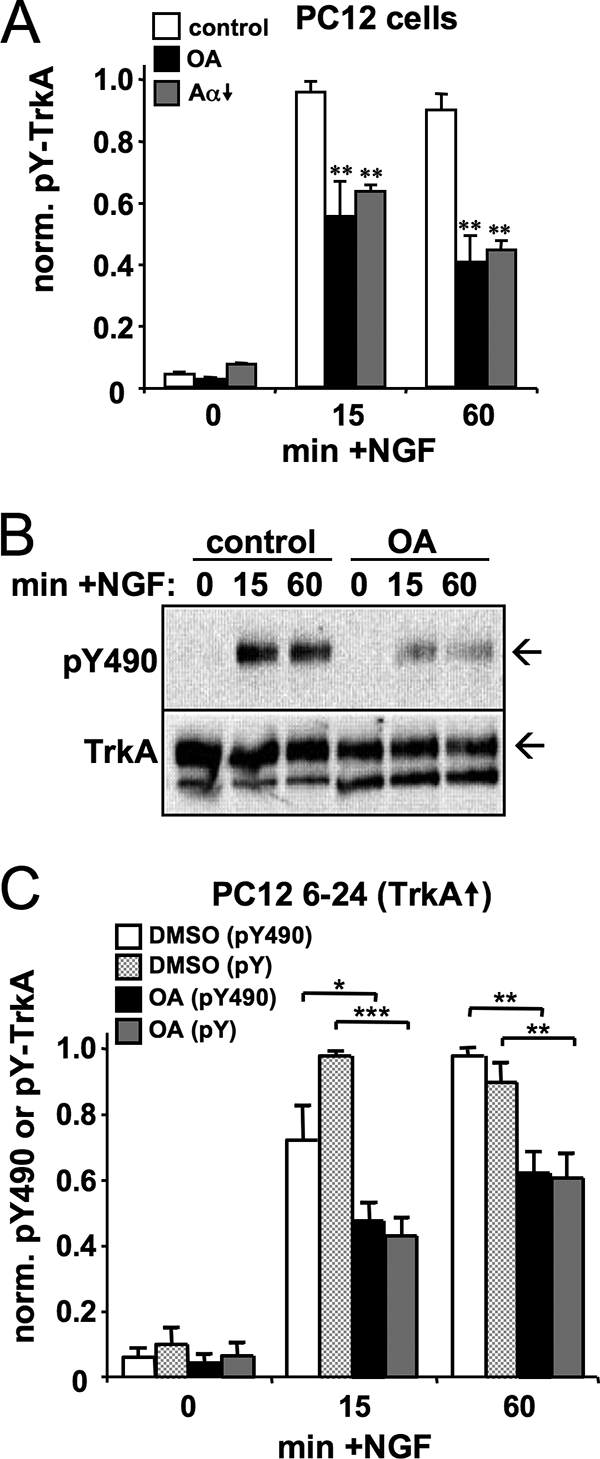
PP2A inhibition blunts Tyr-490 phosphorylation of TrkA in response to NGF. (A) Quantification of total Tyr autophosphorylation of TrkA immunoprecipitated from PP2A/Aα↓ PC12 cells that were treated either for 2 h with 250 nM OA or for 3 days with 1 μg/ml Dox to induce shRNA expression (Aα↓). Cells were stimulated with 20 ng/ml NGF as indicated. (B and C) Representative blot (arrows indicate mature TrkA) and quantification summary of TrkA phosphorylation (total pY and phospho-Tyr-490 [pY490]) in total lysates of PC12 6-24 cells, which overexpress TrkA 15- to 20-fold relative to parental PC12 cells. Graphs show phospho-TrkA divided by total TrkA as the mean ± standard error of four independent experiments. *, P < 0.05; **, P < 0.01.
While PP2A downregulation did not alter total TrkA expression, it remained possible that phosphatase activity is important for surface delivery of the receptor. We therefore carried out biotinylation assays in PC12 cells to analyze cell surface levels of TrkA at different time points following NGF treatment and in the presence or absence of OA. Biotinylated TrkA levels were normalized to the transferrin receptor, which cycles constitutively between surface and endosomal membranes (52). PP2A inhibition had no significant effect on the number of TrkA receptors that were available at the cell surface for ligand stimulation, nor did it affect the rate of receptor internalization after NGF stimulation (Fig. 3). Similar experiments in TrkA-overexpressing PC12 cells (PC12 6-24) supported the same conclusion (n = 5) (data not shown). Taken together, these results suggest that PP2A is important for efficient coupling between NGF binding and autophosphorylation of TrkA.
FIG. 3.
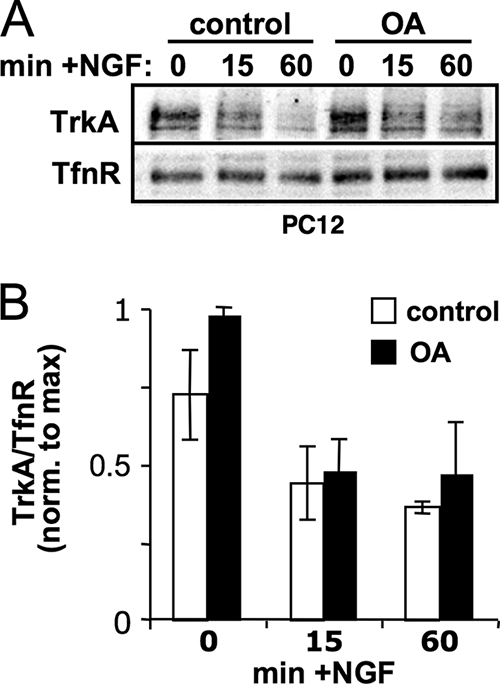
PP2A does not affect TrkA receptor surface expression. (A) PC12 cells pretreated with or without 300 nM OA for 1 h were stimulated with 20 ng/ml NGF for the indicated times. Cell surface proteins were covalently labeled with biotin on ice, recovered with streptavidin-agarose, and probed for TrkA and the transferrin receptor (TfnR) as a loading control. (B) Densitometric analysis of TrkA surface receptors normalized to TfnR (means ± standard errors; n = 3 experiments).
PP2A inhibition causes hyperphosphorylation of TrkA on Ser/Thr residues.
TrkA has been reported to become Ser phosphorylated in response to NGF treatment. Serine phosphorylation of TrkA also occurs following selective activation of the p75 neurotrophin receptor or treatment with ceramide, a p75 signaling intermediate, both of which are associated with decreased responsiveness to NGF (37). We therefore used metabolic radiolabeling to ascertain whether TrkA is a PP2A substrate. PC12 cells overexpressing TrkA in which the cellular ATP pool was labeled by incubation with [32P]orthophosphate were stimulated with NGF for various times in the presence or absence of 250 nM OA, and TrkA phosphorylation was assessed after immunoprecipitation. PP2A inhibition resulted in the expected decrease in TrkA Tyr autophosphorylation but actually increased total 32P incorporation, both in the absence of NGF and 60 min after NGF addition (Fig. 4A). To confirm that PP2A promotes Tyr but attenuates non-Tyr (i.e., Ser/Thr) phosphorylation of TrkA, we subjected metabolically labeled and immunoprecipitated TrkA to in vitro dephosphorylation with yersinia protein tyrosine phosphatase. Tyrosine phosphatase treatment abolished phospho-Tyr antibody reactivity while having minimal effects on total 32P incorporation into TrkA, indicating that Ser/Thr phosphorylation exceeds Tyr phosphorylation of TrkA both with and without NGF stimulation (Ser/Thr phosphorylation values as the percentage of total 32P: no NGF, 83% ± 16%; 15 min with NGF, 54% ± 20%; 60% min with NGF, 81% ± 15%; n = 5). In control reactions with nonspecific lambda phosphatase, phospho-Tyr immunoreactivity and 32P labeling of TrkA were abrogated to the same extent (Fig. 4B). Quantifying tyrosine phosphatase-resistant 32P labeling of TrkA over multiple experiments, we found that PP2A inhibition increased Ser/Thr phosphorylation of TrkA after 0, 15, and 60 min of NGF stimulation (Fig. 4C). Since Ser/Thr phosphorylation of several growth factor receptors has been associated with tyrosine kinase inhibition (5, 7, 11, 16, 34, 35, 44, 55), these results raise the possibility that PP2A promotes TrkA activity by dephosphorylating inhibitory Ser/Thr phosphorylation sites.
FIG. 4.
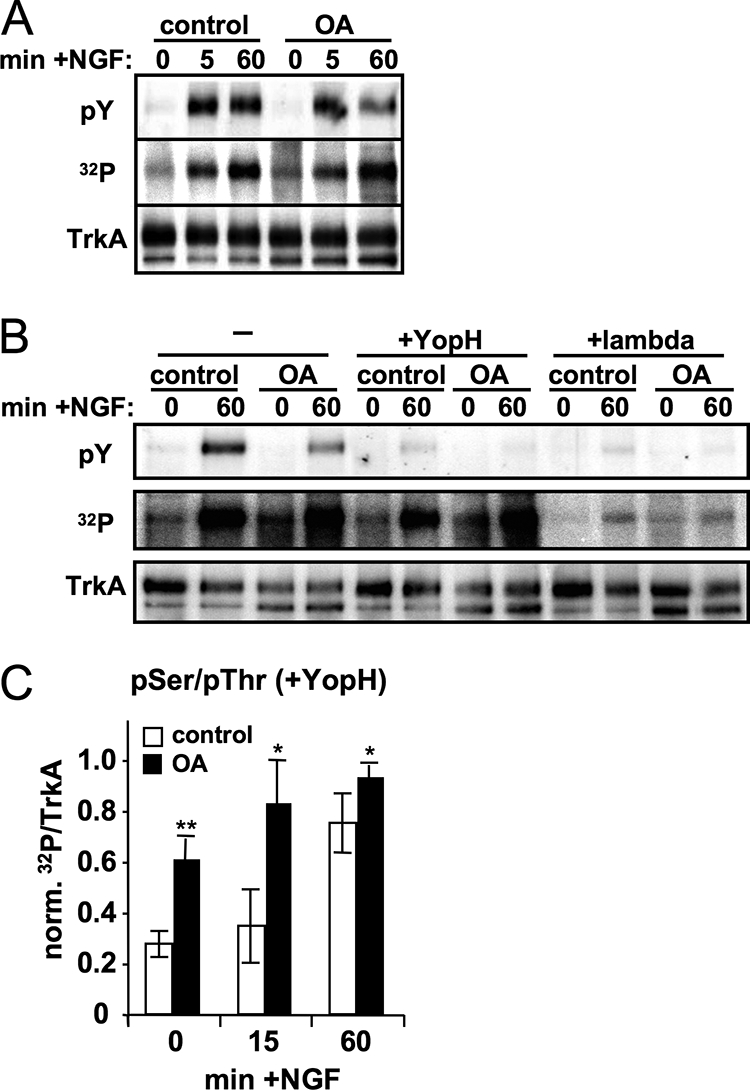
PP2A inhibition increases Ser/Thr phosphorylation of TrkA. PC12 6-24 cells were metabolically labeled prior to the addition of 250 nM OA or vehicle only and then stimulated with 50 ng/ml NGF for 0 to 60 min. Immunoprecipitated TrkA was first exposed to a PhosphorImager screen to quantitate 32P incorporation and then immunoblotted for phospho-Tyr and total TrkA. (A) Representative experiment showing that PP2A inhibition decreases phosphorylation on Tyr but increases total 32P incorporation into TrkA after 60 min in NGF. (B) TrkA immunoprecipitates from PC12 6-24 cells, stimulated in the presence or absence of NGF and OA as indicated, were incubated without phosphatase, with tyrosine phosphatase (YopH), or with nonspecific lambda phosphatase for 15 min at 30°C. Samples were then analyzed for phospho-Tyr and total TrkA immunoreactivity and 32P incorporation. (C) Densitometric quantification of TrkA Ser/Thr phosphorylation (32P remaining after YopH treatment) after immunoprecipitation from cells that had been stimulated as indicated (means ± standard errors of 32P over total TrkA signal from six independent experiments). *, P < 0.05; **, P < 0.01.
The PP2A/B′ family of regulatory subunits fosters TrkA autophosphorylation.
PP2A substrate specificity and subcellular localization are dictated by core dimer association with regulatory subunits from three gene families (B, B′, B"), and any given cell type expresses on the order of a dozen functionally distinct PP2A heterotrimers. To identify the family of holoenzymes that positively regulates TrkA activity, we utilized Aα-exchange PC12 cell lines that inducibly knock down the endogenous scaffolding subunit and concomitantly express a mutant Aα with selective affinity for specific families of regulatory subunits (58, 62) (Fig. 5A). Loss of PP2A dimer association leads to degradation of B and B′, but not B", family regulatory subunits (58) (Fig. 5B and C). Controlling for nonspecific effects of doxycycline, RNAi, or epitope-tagged Aα expression, we found that inducible exchange of endogenous Aα with wild-type, RNAi-resistant Aα had no significant effect on TrkA activation by NGF (Fig. 5D). However, introduction of an Aα mutant that cannot bind any regulatory subunit (EE100RR) (49) resulted in a ∼40% decrease in TrkA activity following 15 and 60 min of NGF stimulation (Fig. 5B and E), an effect similar to pharmacological PP2A inhibition and Aα RNAi (Fig. 1 and 2). Even though a significant fraction of dimeric PP2A has been shown to exist in cells (29), we conclude that this form of the phosphatase is insufficient for maintenance of TrkA activity.
FIG. 5.

The B′ family of PP2A regulatory subunits maintains TrkA activity. (A) Schematic of Aα-subunit exchange. In stable PC12 cell lines, Dox treatment simultaneously induces endogenous Aα silencing and expression of RNAi-resistant mutants defective in select regulatory subunit association. (B to G) The indicated Aα-exchange cell lines were grown for 3 days with or without Dox and stimulated with 20 ng/ml NGF for the indicated times. TrkA immunoprecipitates were probed with pY and total TrkA antibodies. As part of the representative blots shown in panels B and C, lysates were probed for the indicated PP2A subunits, showing specific degradation patterns. (D to G). Summary of densitometric analyses showing pY signals normalized to total TrkA (means ± standard errors of three independent experiments). **, P < 0.01.
Exchange with an Aα mutant that can only associate with B-family regulatory subunits (DWF139AAA) significantly downregulated TrkA receptor activity, as quantified in Fig. 5F. Mutating Aα to selectively destabilize B′-family subunits by preventing their incorporation into the PP2A heterotrimer (DTP177AAA) blunted NGF-induced receptor autophosphorylation to a similar extent (Fig. 5C and G). In supporting experiments, we found that inducible knockdown of Bα, the predominant B-family regulatory subunit in PC12 cells, had no effect on TrkA phospho-Tyr levels (data not shown). Taken together, these data implicate PP2A holoenzymes containing members of the B′ family of regulatory subunits as positive regulators of NGF signaling.
PP2A/B′β expression promotes TrkA receptor activity and downstream signaling.
PP2A/B′β is a neuron-enriched subunit that is abundantly expressed in most neuronal populations in the adult rat brain (17, 42, 51). B′β mRNA levels are also high in embryonic and neonatal rat brain (63), which is when Trk receptors are most highly expressed. We utilized PC12 cell lines that overexpress B′β from a Dox-inducible promoter (B′β↑) (51) to explore the role of this PP2A subunit in NGF signaling. In these cells, Dox increased B′β protein levels by ∼5-fold (Fig. 6A), which was of similar magnitude as B′β mRNA induction during differentiation of a neuroblastoma cell line (42) and as B′β protein upregulation during neuronal differentiation of PC12 cells (57). Dox-induced expression of B′β resulted in a significant potentiation of TrkA Tyr phosphorylation at 15 and 60 min in NGF (Fig. 6A and B). The B′β-mediated enhancement of receptor activity translated into an amplification of Akt and ERK1/2 phosphorylation quantified at 60 and 180 min post-NGF addition (Fig. 6C to F).
FIG. 6.

B′β potentiates TrkA signaling by promoting receptor autophosphorylation. (A to F) PC12 cells that inducibly express B′β were treated for 2 to 3 days with or without Dox and stimulated with 20 ng/ml NGF for the indicated times. TrkA Tyr phosphorylation was assessed in immunoprecipitates, while levels of phospho-Akt and -ERK were determined in cell lysates. Representative blots are shown in panels A, C, and E and quantification summaries of phospho-TrkA over total protein signals are shown in panels B, D, and F (means ± standard errors; n = 3 experiments). *, P < 0.05; **, P < 0.01. (G) B′β-inducing PC12 cells were transiently transfected with empty vector, Ras-V12, or constitutively active (CA)-MEK1 (S218, 222D mutant) and treated for 2 days with or without Dox. After 5 to 6 h of growth factor stimulation (2 ng/ml NGF or 50 ng/ml FGF2, as indicated), MAPK activity was measured in reporter assays based on transactivation of the Elk1 transcription factor. The graph shows dual luciferase activity ratios relative to empty vector-transfected, non-growth factor-treated, and non-Dox-treated samples (means ± standard deviations of triplicate wells) from one experiment representative of four. (H) Interpretation of epistasis experiments.
To obtain further support for the notion that PP2A/B′β boosts NGF signaling at the receptor level, we employed dual luciferase reporter assays, in which ERK-dependent phosphorylation of the transcription factor Elk1 served as a readout for activation of the Ras-MAP kinase cascade (57) (Fig. 6H). We measured luciferase accumulation 5 to 6 h after NGF addition as the integral of MAP kinase phosphorylation and nuclear translocation during this time course. Without B′β induction, subsaturating NGF concentrations (2 ng/ml) resulted in a ∼60-fold increase in Elk1 transcriptional activity. Doxycycline treatment amplified this response by 100 to 200% (200-fold transactivation compared to non-NGF-treated cells in the experiment shown in Fig. 6G). PP2A/B′β displayed striking receptor tyrosine kinase specificity, since B′β overexpression failed to potentiate MAP kinase activation by FGF receptors (Fig. 6G). Notably, FGF2 reproduces many of the biological activities of NGF in PC12 cells, including long-lasting ERK activation and neurite outgrowth (50). Bypassing growth factor receptors, we next stimulated Ras-MAP kinase signaling by cotransfection of constitutively active forms of Ras and MEK1, titrating plasmid concentrations to achieve ∼50-fold Elk1 transcriptional activation over empty vector-transfected cells. Under these conditions, and similar to FGF2-stimulated cells, B′β induction resulted in a slight but significant attenuation of MAP kinase reporter activity (Fig. 6G), which is consistent with the recent identification of PP2A/B′ holoenzymes as MAP kinase phosphatases (31). Nevertheless, TrkA receptor potentiation by the PP2A/B′β heterotrimer evidently overrides the phosphatase's modulatory effect on downstream effector kinases, resulting in an overall amplification of NGF-dependent transcriptional responses (Fig. 6H).
Endogenous B′ subunits associate with TrkA and regulate its activity.
The mammalian B′ subunit gene family gives rise to five gene products (α, β, γ, δ, and ɛ) with a highly conserved tandem α-helical repeat region that interacts with both A and C subunits (12, 67), while the divergent N and C termini may be involved in binding to specific substrates and anchoring proteins (6). To determine if B′ subunits can recruit PP2A to the NGF receptor, we immunoprecipitated endogenous TrkA from PC12 cells that inducibly express B′β. Induction of B′β not only increased the specific association of this subunit with TrkA but also enhanced the recruitment of the endogenous PP2A catalytic subunit to the receptor complex (44% ± 2% increase; n = 3) (Fig. 7A). The robust association of PP2A/C with TrkA in the absence of Dox implies the involvement of endogenous B′ subunits and suggests that B′β induction increases total B′ family subunit levels only moderately.
FIG. 7.

B′β and B′δ recruit PP2A into a TrkA receptor complex. (A) PC12 cells that inducibly express B′β were treated for 3 days with or without Dox and TrkA and normal IgG immunoprecipitates were probed for the indicated proteins. B′β expression increased association of PP2A/C with TrkA by 44% (n = 3). (B) PC12 cells transiently expressing the indicated HA-tagged B′ regulatory subunits or an irrelevant control protein (CTRL) were analyzed for coimmunoprecipitation with TrkA. (C) TrkA and normal IgG immunoprecipitates from PC12 cells and mouse spinal cord and brain were probed for the indicated proteins. (D) Microcystin-LR agarose (MC-ag.) affinity purification of PP1 and PP2A-like phosphatases from mouse brain lysate. TrkA coisolation was blocked by preincubating brain lysates with 5 μM free microcystin-LR (block) or by using agarose only. Approximately 5% of the lysate used for each precipitation was immunoblotted as a reference. (E) TrkA and normal IgG immunoprecipitates from PC12 cells stimulated or not with NGF for 30 min were analyzed for the association of endogenous B′δ. The two B′δ bands in this and in other blots likely correspond to alternative splice products. (F) Densitometric analysis of B′δ coimmunoprecipitation with TrkA (means ± standard errors, n = 5 experiments). ***, P < 0.001.
To investigate selectivity among B′ family members, we isolated endogenous TrkA from PC12 cells that had been transfected with epitope-tagged B′α-ɛ. The two neuron-enriched B′ regulatory subunits, B′β and B′δ, associated robustly with TrkA, while B′α, B′γ, and B′ɛ recovery in the immunoprecipitate was weaker and variable. Additional TrkA coimmunoprecipitation experiments revealed complexes of endogenous B′δ, the PP2A catalytic subunit, and Trk receptors in PC12 cells, in mouse spinal cord, dorsal root ganglia, and forebrain (Fig. 7C and data not shown). We could not show an association of endogenous B′β with TrkA, since B′β comigrates with the IgG heavy chain on SDS-PAGE. Because the immunoprecipitating antibody recognizes epitopes common to all Trk isoforms (K. Deinhardt and M. Chao, personal communication), we cannot rule out the possibility that PP2A interacts with TrkB and TrkC in the forebrain. The PP2A::TrkA complex was also demonstrated in reverse, isolating PP2A-associated proteins from forebrain lysates via immobilized microcystin, a cyclic peptide inhibitor that binds tightly to the catalytic subunits of PP1- and PP2A-like phosphatases. Microcystin-agarose isolated specifically the mature, glycosylated form of TrkA, and the association could be competed with free microcystin (Fig. 7D).
Since PP2A enhances specifically the late phase of NGF signaling (15 min and later [Fig. 1]), we asked whether PP2A is recruited to the activated TrkA signaling complex. Indeed, we found that stimulating PC12 cells for 30 min with NGF resulted in a 70% increase in endogenous PP2A/B′δ coimmunoprecipitation with TrkA (Fig. 7E and F). Given that TrkA is largely internalized at this time point (Fig. 3), these data suggest that PP2A interacts with the NGF receptor at endosomal compartments.
We next sought to expand our data implicating the B′ subunit family in the maintenance of TrkA autophosphorylation (Fig. 5) by examining the role of individual B′ isoforms in NGF signaling. To this end, small hairpin RNAs (shRNAs) were generated against all five B′ subunits and tested for silencing of the cotransfected, epitope-tagged protein in PC12 cells. Knockdown efficiency ranged from 60 to 88% and, in each case, silencing was specific for the targeted subunit (Fig. 8A and data not shown). Silencing of endogenous B′β with the B′β-directed shRNA was previously shown to be ∼50% effective (51). shRNA-expressing plasmids were cotransfected with Elk1 luciferase reporter constructs to measure MAP kinase activation in response to NGF. Silencing of B′β and B′δ, the two B′ isoforms that formed stable complexes with TrkA (Fig. 7), brought about an up to 80% decrease in NGF-dependent gene transcription, while shRNAs targeting the other B′ isoforms had insignificant effects on luciferase activity (Fig. 8B and C). Demonstrating TrkA specificity once more, RNAi of B′β or B′δ was without consequence for MAP kinase reporter activity when cells were stimulated with EGF (Fig. 8C). Inhibition of NGF-dependent MAP kinase activation was dependent on the amount of B′β and B′δ shRNA-expressing plasmid cotransfected with reporter plasmids, with a combination of the two shRNAs plasmids (1:1 ratio) resulting in about same inhibition as either plasmid alone at twice the concentration (Fig. 8B). These data indicate that PC12 cells require both B′β and B′δ for full NGF, but not EGF, responsiveness.
FIG. 8.

Silencing of B′β and B′δ inhibits NGF-dependent MAP kinase activation. (A) PC12 cells were cotransfected with the indicated PP2A subunits with or without the corresponding shRNAs, and the percent knockdown (%KD) was assessed by immunoblotting for the HA tag and for the loading control, Bα/δ (means ± standard errors of four experiments). (B) The indicated amounts of shRNA plasmids targeting B′β and B′δ were expressed for 3 days, and MAP kinase activation in PC12 cells was quantified by dual-luciferase reporter assays. NGF-stimulated MAP kinase activation (2 ng/ml for 5 h) is expressed relative to unstimulated, control (CTRL) shRNA-transfected cells (a representative experiment is shown, with means ± standard deviations of triplicate wells). (C) shRNAs targeting the indicated B′ subunits or the control shRNA were expressed for 3 days, and MAP kinase activation in response to EGF (5 ng/ml for 5 to 6 h) or NGF (2 ng/ml) was quantified by dual-luciferase reporter assays. Activity is expressed relative to NGF/EGF-treated, control shRNA-transfected cells (means ± standard errors of three to six independent experiments). ***, P < 0.001.
B′β and B′δ subunits potentiate NGF-dependent neurite outgrowth and cell cycle inhibition. NGF slows and eventually halts proliferation of PC12 cells and promotes their differentiation into a sympathetic neuron-like cell type with elaborate processes that can propagate action potentials (19, 60). Given that MAP kinase signaling is both necessary and sufficient for process outgrowth of PC12 cells (13, 46) and given that B′β and B′δ augment NGF-induced MAP kinase activation, we first asked whether PP2A regulatory subunits mediate neuritogenesis. We found that inducible expression of B′β led to a 50% increase in total neurite length in PC12 cells treated for 18 h with 2 ng/ml NGF (Fig. 9A). Conversely, silencing of B′β or B′δ resulted in a similarly robust (40 and 60%, respectively) inhibition of neurite outgrowth. Transfection with shRNAs targeting B′α on the other hand, did not affect neurite length compared to scrambled shRNA controls, paralleling the inability of the B′α subunit to bind to TrkA and enhance signaling (Fig. 7B and 8C).
FIG. 9.

PP2A/B′β and B′δ mediate neurite outgrowth in PC12 cells. (A and B) PC12 cells with inducible B′β expression (2 days in the presence of absence of Dox) were stimulated with 2 ng/ml NGF for 18 h, fixed, and analyzed for neurite length. Shown are representative images (A) and summary data (B) (means ± standard errors of three independent experiments with 50 to 100 cells each). (C and D) PC12 cells were transfected with the indicated shRNAs, stimulated after 3 days with NGF (2 ng/ml for 26 h), and scored for neurite outgrowth as above. Representative images in panel C show decreased neuritogenesis after B′β or B′δ, but not B′α, silencing, which is quantified in panel D (means ± standard errors of three independent experiments with 50 to 100 cells each). Membrane-targeted (Lck N terminus) mCherry was used to mark transfected cells and visualize small-caliber neurites. ***, P < 0.001. Bars, 10 μm.
To address whether PP2A/B′β can also enhance NGF-mediated cell cycle exit, B′β-inducible PC12 cells were incubated with 10 ng/ml NGF for 0, 2, and 4 days prior to flow cytometry to analyze DNA content. As shown in the sample histogram of cells analyzed after 2 days in NGF (Fig. 10A), B′β induction increased the proportion of growth arrested cells (G0/G1) at the expense of proliferating cells (G2/S/M). Similar effects were seen after 4 days in the presence of NGF. Significantly, B′β expression had no effect on cell cycle progression of cells growing in the absence of NGF (Fig. 10B), indicating that this PP2A regulatory subunit partakes specifically in neurotrophin-mediated cell cycle control.
FIG. 10.

PP2A/B′β accelerates cell cycle arrest by NGF. (A) PC12 cells were induced to express B′β (2 days with or without Dox) and then stimulated with 10 ng/ml NGF for 0, 2, and 4 days, and cell cycle profiles (representative histograms) were obtained by flow cytometry. (B) Summary graph plotting percentages of dividing cells (G2 + S phase; means ± standard errors of four independent experiments). *, P < 0.05.
DISCUSSION
The present study identifies two isoforms of the ubiquitous and essential Ser/Thr phosphatase PP2A as master regulators of neurotrophin signal transduction. We found that pharmacological and RNAi-mediated inhibition of PP2A inhibits the sustained phase of NGF receptor autophosphorylation (at total tyrosines, as well as Tyr-490, the Shc adaptor site), while EGF and FGF receptor activities were unaffected (62) (Fig. 6G and 8C). The magnitude of the effect (1.5- to 2-fold reduction of TrkA autophosphorylation) is remarkable, considering that we could only partially inhibit PP2A because of its essential roles in cell viability (58). Conversely, forced expression of the neuronal PP2A regulatory subunit B′β enhanced TrkA Tyr phosphorylation and signal transduction to Akt and ERK, as well as neurotrophin-dependent transcriptional activation, neurite outgrowth, and cell cycle exit. Since endogenous B′β is upregulated during neuronal differentiation of PC12 cells (57), this PP2A subunit may be part of a positive feedback mechanism that confers switch-like behavior to NGF responses. Remarkably, inhibition of PP2A with okadaic acid had opposite effects on Tyr and non-Tyr phosphorylation of TrkA, suggesting a mechanism in which the phosphatase targets inhibitory Ser/Thr phosphorylation sites in the intracellular domain of the receptor tyrosine kinase. In aggregate, our data indicate that recruitment of PP2A to a TrkA signaling complex via B′β and the related B′δ regulatory subunit attenuates NGF receptor desensitization and sustains the activity of neurotrophin effector kinases critical for neuronal development, plasticity and survival.
PP2A regulatory subunits impart substrate selectivity, subcellular targeting, and second messenger regulation to the phosphatase. Indeed, several PP2A/B′ holoenzyme-specific substrates and interacting proteins have recently been reported. For instance, tyrosine hydroxylase, the rate-limiting enzyme in catecholamine synthesis, is uniquely dephosphorylated and inactivated by the PP2A/B′β heterotrimer (51). PP2A/B′β also preferentially binds to the kinase Pim-1 to promote its degradation (36). Ankyrin-B, a linker between the actin cytoskeleton and membrane receptors and ion channels, binds to the variable C-terminal tail of the B′α regulatory subunit (6). The same PP2A regulatory subunit also selectively interacts with and downregulates c-myc (3), while PP2A/B′γ interacts with and dephosphorylates the tumor suppressor p53 (32) and PP2A/B′δ preferentially binds to the dual-specificity phosphatase and mitotic activator Cdc25 (38). Available structural information for the PP2A/B′γ heterotrimer (12, 67) will aid in the elucidation of specificity determinants for B′ subunit association with TrkA and other targets.
TrkA-directed PP2A activity is likely regulated at multiple levels. At the level of complex formation, NGF was found to drive the association of endogenous B′δ with TrkA (Fig. 7E and F). We suspect that the PP2A/B′β holoenzyme also undergoes activity-dependent translocation to the NGF receptor, although we were not able to demonstrate this experimentally due to the similar relative mobilities of B′β and the IgG heavy chain. At the level of posttranslational modification, both regulatory subunits can be phosphorylated by ERK at an identified Ser residue, which leads to dissociation of the PP2A holoenzyme (31) and could therefore be a mechanism for TrkA feedback inhibition. Also, protein kinase A has been shown to activate PP2A/B′δ via phosphorylation of a Ser in its unique C terminus (1, 61), allowing for possible integration of cAMP and neurotrophin signals.
As the most commonly used model cell line for the study of neurotrophin signal transduction, PC12 cells undergo cell fate decisions based on the temporal profile of MAP kinase activation by growth factors (reviewed in reference 64). Specifically, transient activation of ERK1/2 (by, for instance, EGF) is mitogenic, whereas sustained signaling through the Ras-MAP kinase module causes cell cycle arrest and neuronal differentiation. One of the key differences between EGF and NGF signaling lies in the regulation by receptor endocytosis: whereas removal of the ligand-bound EGF receptor from the plasma membrane rapidly terminates signal transduction, endocytosis of the TrkA receptor is required for the formation of a signaling endosome, which in peripheral neurons undergoes retrograde transport from the synapse or growth cone to affect gene expression in the soma (48, 71). Proteins critical for translating sustained TrkA autophosphorylation to persistent effector kinase activation include the endosomal small GTPase Rap1, its activators Crk and C3G, and the tetramembrane spanning adaptor protein ARMS/Kidins220 (reviewed in reference 70). To our knowledge, PP2A is the first intracellular signaling molecule that has been shown to enhance NGF signaling by promoting the sustained, but not the initial phase of, receptor autophosphorylation.
There are multiple possible mechanisms by which PP2A/B′β and B′δ could boost TrkA activity. Our cell surface biotinylation experiments argue against a role of PP2A in NGF-dependent internalization of TrkA, but it remains possible that PP2A regulates the partitioning of TrkA into lipid rafts (33), or an interaction with p75 to form high-affinity NGF binding sites (65). However, the finding that PP2A inhibition does not affect TrkA autophosphorylation until 15 min after NGF addition (Fig. 1) suggests that the Ser/Thr phosphatase opposes a delayed, Trk-specific desensitization mechanism. Ser/Thr phosphorylation of several receptor tyrosine kinases has been reported, often as a consequence of feedback phosphorylation by downstream Ser/Thr kinases (5, 7, 11, 16, 34, 35, 44, 55). In the case of the hepatocyte growth factor receptor cMET, PP2A dephosphorylates an inhibitory protein kinase C phosphorylation site in the tyrosine kinase domain, thereby keeping cMET active (20). Indeed, previous work by Barker and colleagues showed that NGF treatment or selective stimulation of the p75 receptor induces TrkA phosphorylation on Ser residues (37). We have shown here that non-Tyr (presumably Ser [37]) phosphorylation accounts for the majority of 32P incorporation into unliganded and ligand-activated TrkA and that PP2A dephosphorylates these sites (Fig. 4). TrkA Ser(/Thr) phosphorylation sites have to be identified before we can ask whether TrkA dephosphorylation is the mechanism by which PP2A/B′β and B′δ potentiate NGF signaling.
Acknowledgments
We are grateful to Tom Cribbs and Ronald Merrill for invaluable technical assistance and helpful discussions. We also thank Gene Hess and Fred Quelle for assistance with flow cytometry.
This work was supported by National Institute of Health grants NS43254 (to S.S.) and a predoctoral fellowship from the Pharmaceutical Research and Manufacturers of America Foundation (to M.J.V.).
Footnotes
Published ahead of print on 24 November 2008.
REFERENCES
- 1.Ahn, J. H., T. McAvoy, S. V. Rakhilin, A. Nishi, P. Greengard, and A. C. Nairn. 2007. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56δ subunit. Proc. Natl. Acad. Sci. USA 1042979-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn, J. H., J. Y. Sung, T. McAvoy, A. Nishi, V. Janssens, J. Goris, P. Greengard, and A. C. Nairn. 2007. The B"/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 1049876-9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold, H. K., and R. C. Sears. 2006. Protein phosphatase 2A regulatory subunit B56α associates with c-myc and negatively regulates c-myc accumulation. Mol. Cell. Biol. 262832-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashcroft, M., R. M. Stephens, B. Hallberg, J. Downward, and D. R. Kaplan. 1999. The selective and inducible activation of endogenous PI 3-kinase in PC12 cells results in efficient NGF-mediated survival but defective neurite outgrowth. Oncogene 184586-4597. [DOI] [PubMed] [Google Scholar]
- 5.Barbier, A. J., H. M. Poppleton, Y. Yigzaw, J. B. Mullenix, G. J. Wiepz, P. J. Bertics, and T. B. Patel. 1999. Transmodulation of epidermal growth factor receptor function by cyclic AMP-dependent protein kinase. J. Biol. Chem. 27414067-14073. [DOI] [PubMed] [Google Scholar]
- 6.Bhasin, N., S. R. Cunha, M. Mudannayake, M. S. Gigena, T. B. Rogers, and P. J. Mohler. 2007. Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293H109-H119. [DOI] [PubMed] [Google Scholar]
- 7.Bioukar, E. B., N. C. Marricco, D. Zuo, and L. Larose. 1999. Serine phosphorylation of the ligand-activated beta-platelet-derived growth factor receptor by casein kinase I-γ2 inhibits the receptor's autophosphorylating activity. J. Biol. Chem. 27421457-21463. [DOI] [PubMed] [Google Scholar]
- 8.Brummelkamp, T. R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296550-553. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarti, K., R. Lin, N. I. Schiller, Y. Wang, D. Koubi, Y. X. Fan, B. B. Rudkin, G. R. Johnson, and M. R. Schiller. 2005. Critical role for kalirin in nerve growth factor signaling through TrkA. Mol. Cell. Biol. 255106-5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, K. S., M. C. Nishimura, M. P. Armanini, C. Crowley, S. D. Spencer, and H. S. Phillips. 1997. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J. Neurosci. 177288-7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheusova, T., M. A. Khan, S. W. Schubert, A. C. Gavin, T. Buchou, G. Jacob, H. Sticht, J. Allende, B. Boldyreff, H. R. Brenner, and S. Hashemolhosseini. 2006. Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev. 201800-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho, U. S., and W. Xu. 2007. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 44553-57. [DOI] [PubMed] [Google Scholar]
- 13.Cowley, S., H. Paterson, P. Kemp, and C. J. Marshall. 1994. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77841-852. [DOI] [PubMed] [Google Scholar]
- 14.Dagda, R. K., C. A. Barwacz, J. T. Cribbs, and S. Strack. 2005. Unfolding-resistant translocase targeting: a novel mechanism for outer mitochondrial membrane localization exemplified by the Bβ2 regulatory subunit of protein phosphatase 2A. J. Biol. Chem. 28027375-27382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fagan, A. M., M. Garber, M. Barbacid, I. Silos-Santiago, and D. M. Holtzman. 1997. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J. Neurosci. 177644-7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freedman, N. J., L. K. Kim, J. P. Murray, S. T. Exum, L. Brian, J. H. Wu, and K. Peppel. 2002. Phosphorylation of the platelet-derived growth factor receptor-beta and epidermal growth factor receptor by G protein-coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J. Biol. Chem. 27748261-48269. [DOI] [PubMed] [Google Scholar]
- 17.Fukunaga, K., D. Muller, M. Ohmitsu, E. Bako, A. A. DePaoli-Roach, and E. Miyamoto. 2000. Decreased protein phosphatase 2A activity in hippocampal long-term potentiation. J. Neurochem. 74807-817. [DOI] [PubMed] [Google Scholar]
- 18.Gallego, M., and D. M. Virshup. 2005. Protein serine/threonine phosphatases: life, death, and sleeping. Curr. Opin. Cell Biol. 17197-202. [DOI] [PubMed] [Google Scholar]
- 19.Greene, L. A., and A. S. Tischler. 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 732424-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashigasako, A., M. Machide, T. Nakamura, K. Matsumoto, and T. Nakamura. 2004. Bi-directional regulation of Ser-985 phosphorylation of c-met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J. Biol. Chem. 27926445-26452. [DOI] [PubMed] [Google Scholar]
- 21.Hempstead, B. L., S. J. Rabin, L. Kaplan, S. Reid, L. F. Parada, and D. R. Kaplan. 1992. Overexpression of the Trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron 9883-896. [DOI] [PubMed] [Google Scholar]
- 22.Hendrix, P., P. Turowski, R. E. Mayer-Jaekel, J. Goris, J. Hofsteenge, W. Merlevede, and B. A. Hemmings. 1993. Analysis of subunit isoforms in protein phosphatase 2A holoenzymes from rabbit and Xenopus. J. Biol. Chem. 2687330-7337. [PubMed] [Google Scholar]
- 23.Huang, E. J., and L. F. Reichardt. 2003. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72609-642. [DOI] [PubMed] [Google Scholar]
- 24.Janssens, V., and J. Goris. 2001. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353417-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssens, V., J. Jordens, I. Stevens, C. Van Hoof, E. Martens, H. De Smedt, Y. Engelborghs, E. Waelkens, and J. Goris. 2003. Identification and functional analysis of two Ca2+-binding EF-hand motifs in the B"/PR72 subunit of protein phosphatase 2A. J. Biol. Chem. 27810697-10706. [DOI] [PubMed] [Google Scholar]
- 26.Jullien, J., V. Guili, L. F. Reichardt, and B. B. Rudkin. 2002. Molecular kinetics of nerve growth factor receptor trafficking and activation. J. Biol. Chem. 27738700-38708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Junttila, M. R., S. P. Li, and J. Westermarck. 2007. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 22954-965. [DOI] [PubMed] [Google Scholar]
- 28.Klesse, L. J., K. A. Meyers, C. J. Marshall, and L. F. Parada. 1999. Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene 182055-2068. [DOI] [PubMed] [Google Scholar]
- 29.Kremmer, E., K. Ohst, J. Kiefer, N. Brewis, and G. Walter. 1997. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in cells. Mol. Cell. Biol. 171692-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuruvilla, R., L. S. Zweifel, N. O. Glebova, B. E. Lonze, G. Valdez, H. Ye, and D. D. Ginty. 2004. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 118243-255. [DOI] [PubMed] [Google Scholar]
- 31.Letourneux, C., G. Rocher, and F. Porteu. 2006. B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J. 25727-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, H. H., X. Cai, G. P. Shouse, L. G. Piluso, and X. Liu. 2007. A specific PP2A regulatory subunit, B56γa, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 26402-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Limpert, A. S., J. C. Karlo, and G. E. Landreth. 2007. Nerve growth factor stimulates the concentration of TrkA within lipid rafts and extracellular signal-regulated kinase activation through c-Cbl-associated protein. Mol. Cell. Biol. 275686-5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu, F., and R. A. Roth. 1994. Identification of serines-1035/1037 in the kinase domain of the insulin receptor as protein kinase C alpha mediated phosphorylation sites. FEBS Lett. 352389-392. [DOI] [PubMed] [Google Scholar]
- 35.Lonic, A., E. F. Barry, C. Quach, B. Kobe, N. Saunders, and M. A. Guthridge. 2008. Fibroblast growth factor receptor 2 phosphorylation on serine 779 couples to 14-3-3 and regulates cell survival and proliferation. Mol. Cell. Biol. 283372-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma, J., H. K. Arnold, M. B. Lilly, R. C. Sears, and A. S. Kraft. 2007. Negative regulation of Pim-1 protein kinase levels by the B56β subunit of PP2A. Oncogene 265145-5153. [DOI] [PubMed] [Google Scholar]
- 37.MacPhee, I. J., and P. A. Barker. 1997. Brain-derived neurotrophic factor binding to the p75 neurotrophin receptor reduces TrkA signaling while increasing serine phosphorylation in the TrkA intracellular domain. J. Biol. Chem. 27223547-23551. [DOI] [PubMed] [Google Scholar]
- 38.Margolis, S. S., J. A. Perry, C. M. Forester, L. K. Nutt, Y. Guo, M. J. Jardim, M. J. Thomenius, C. D. Freel, R. Darbandi, J. H. Ahn, J. D. Arroyo, X. F. Wang, S. Shenolikar, A. C. Nairn, W. G. Dunphy, W. C. Hahn, D. M. Virshup, and S. Kornbluth. 2006. Role for the PP2A/B56δ phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell 127759-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsh, H. N., C. I. Dubreuil, C. Quevedo, A. Lee, M. Majdan, G. S. Walsh, S. Hausdorff, F. A. Said, O. Zoueva, M. Kozlowski, K. Siminovitch, B. G. Neel, F. D. Miller, and D. R. Kaplan. 2003. SHP-1 negatively regulates neuronal survival by functioning as a TrkA phosphatase. J. Cell Biol. 163999-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayer, R. E., P. Hendrix, P. Cron, R. Matthies, S. R. Stone, J. Goris, W. Merlevede, J. Hofsteenge, and B. A. Hemmings. 1991. Structure of the 55-kDa regulatory subunit of protein phosphatase 2A: evidence for a neuronal-specific isoform. Biochemistry 303589-3597. [DOI] [PubMed] [Google Scholar]
- 41.McCright, B., A. R. Brothman, and D. M. Virshup. 1996. Assignment of human protein phosphatase 2A regulatory subunit genes B56α, B56β, B56γ, B56δ, and B56ɛ (PPP2R5A-PPP2R5E), highly expressed in muscle and brain, to chromosome regions 1q41, 11q12, 3p21, 6p21.1, and 7p11.2→p12. Genomics 36168-170. [DOI] [PubMed] [Google Scholar]
- 42.McCright, B., A. M. Rivers, S. Audlin, and D. M. Virshup. 1996. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 27122081-22089. [DOI] [PubMed] [Google Scholar]
- 43.McCright, B., and D. M. Virshup. 1995. Identification of a new family of protein phosphatase 2A regulatory subunits. J. Biol. Chem. 27026123-26128. [DOI] [PubMed] [Google Scholar]
- 44.Parvaresch, S., T. Yesilkaya, K. Baer, H. Al-Hasani, and H. W. Klein. 2002. 14-3-3 binding to the IGF-1 receptor is mediated by serine autophosphorylation. FEBS Lett. 532357-362. [DOI] [PubMed] [Google Scholar]
- 45.Pittman, R. N., S. Wang, A. J. DiBenedetto, and J. C. Mills. 1993. A system for characterizing cellular and molecular events in programmed neuronal cell death. J. Neurosci. 133669-3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qui, M. S., and S. H. Green. 1992. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron 9705-717. [DOI] [PubMed] [Google Scholar]
- 47.Reichardt, L. F. 2006. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B 3611545-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reichardt, L. F., and W. C. Mobley. 2004. Going the distance, or not, with neurotrophin signals. Cell 118141-143. [DOI] [PubMed] [Google Scholar]
- 49.Ruediger, R., H. T. Pham, and G. Walter. 2001. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene 2010-15. [DOI] [PubMed] [Google Scholar]
- 50.Rydel, R. E., and L. A. Greene. 1987. Acidic and basic fibroblast growth factors promote stable neurite outgrowth and neuronal differentiation in cultures of PC12 cells. J. Neurosci. 73639-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saraf, A., D. M. Virshup, and S. Strack. 2007. Differential expression of the B′β regulatory subunit of protein phosphatase 2A modulates tyrosine hydroxylase phosphorylation and catecholamine synthesis. J. Biol. Chem. 282573-580. [DOI] [PubMed] [Google Scholar]
- 52.Saxena, S., C. L. Howe, J. M. Cosgaya, P. Steiner, H. Hirling, J. R. Chan, J. Weis, and A. Kruttgen. 2005. Differential endocytic sorting of p75NTR and TrkA in response to NGF: a role for late endosomes in TrkA trafficking. Mol. Cell. Neurosci. 28571-587. [DOI] [PubMed] [Google Scholar]
- 53.Schnizler, K., L. P. Shutov, M. J. Van Kanegan, M. A. Merrill, B. Nichols, G. S. McKnight, S. Strack, J. W. Hell, and Y. M. Usachev. 2008. Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J. Neurosci. 284904-4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shintani, T., and M. Noda. 2008. Protein tyrosine phosphatase receptor type Z dephosphorylates TrkA receptors and attenuates NGF-dependent neurite outgrowth of PC12 cells. J. Biochem. 144259-266. [DOI] [PubMed] [Google Scholar]
- 55.Sorensen, V., Y. Zhen, M. Zakrzewska, E. M. Haugsten, S. Walchli, T. Nilsen, S. Olsnes, and A. Wiedlocha. 2008. Phosphorylation of fibroblast growth factor (FGF) receptor 1 at Ser777 by p38 mitogen-activated protein kinase regulates translocation of exogenous FGF1 to the cytosol and nucleus. Mol. Cell. Biol. 284129-4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stephens, R. M., D. M. Loeb, T. D. Copeland, T. Pawson, L. A. Greene, and D. R. Kaplan. 1994. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron 12691-705. [DOI] [PubMed] [Google Scholar]
- 57.Strack, S. 2002. Overexpression of the protein phosphatase 2A regulatory subunit Bγ promotes neuronal differentiation by activating the MAP kinase (MAPK) cascade. J. Biol. Chem. 27741525-41532. [DOI] [PubMed] [Google Scholar]
- 58.Strack, S., J. T. Cribbs, and L. Gomez. 2004. Critical role for protein phosphatase 2A heterotrimers in mammalian cell survival. J. Biol. Chem. 27947732-47739. [DOI] [PubMed] [Google Scholar]
- 59.Strack, S., R. Ruediger, G. Walter, R. K. Dagda, C. A. Barwacz, and J. T. Cribbs. 2002. Protein phosphatase 2A holoenzyme assembly: identification of contacts between B-family regulatory and scaffolding A subunits. J. Biol. Chem. 27720750-20755. [DOI] [PubMed] [Google Scholar]
- 60.Toledo-Aral, J. J., P. Brehm, S. Halegoua, and G. Mandel. 1995. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron 14607-611. [DOI] [PubMed] [Google Scholar]
- 61.Usui, H., R. Inoue, O. Tanabe, Y. Nishito, M. Shimizu, H. Hayashi, H. Kagamiyama, and M. Takeda. 1998. Activation of protein phosphatase 2A by cAMP-dependent protein kinase-catalyzed phosphorylation of the 74-kDa B"δ regulatory subunit in vitro and identification of the phosphorylation sites. FEBS Lett. 430312-316. [DOI] [PubMed] [Google Scholar]
- 62.Van Kanegan, M. J., D. G. Adams, B. E. Wadzinski, and S. Strack. 2005. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J. Biol. Chem. 28036029-36036. [DOI] [PubMed] [Google Scholar]
- 63.van Lookeren Campagne, M., K. Okamoto, C. Prives, and R. Gill. 1999. Developmental expression and localization of cyclin G1 and the B′ subunits of protein phosphatase 2a in neurons. Brain Res. Mol. Brain Res. 641-10. [DOI] [PubMed] [Google Scholar]
- 64.Vaudry, D., P. J. Stork, P. Lazarovici, and L. E. Eiden. 2002. Signaling pathways for PC12 cell differentiation: making the right connections. Science 2961648-1649. [DOI] [PubMed] [Google Scholar]
- 65.Wehrman, T., X. He, B. Raab, A. Dukipatti, H. Blau, and K. C. Garcia. 2007. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 5325-38. [DOI] [PubMed] [Google Scholar]
- 66.Xu, Y., Y. Chen, P. Zhang, P. D. Jeffrey, and Y. Shi. 2008. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31873-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu, Y., Y. Xing, Y. Chen, Y. Chao, Z. Lin, E. Fan, J. W. Yu, S. Strack, P. D. Jeffrey, and Y. Shi. 2006. Structure of the protein phosphatase 2A holoenzyme. Cell 1271239-1251. [DOI] [PubMed] [Google Scholar]
- 68.Xu, Z., and B. R. G. Williams. 2000. The B56α regulatory subunit of protein phosphatase 2A is a target for regulation by double-stranded RNA-dependent protein kinase PKR. Mol. Cell. Biol. 205285-5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoon, S. O., S. P. Soltoff, and M. V. Chao. 1997. A dominant role of the juxtamembrane region of the TrkA nerve growth factor receptor during neuronal cell differentiation. J. Biol. Chem. 27223231-23238. [DOI] [PubMed] [Google Scholar]
- 70.Zampieri, N., and M. V. Chao. 2006. Mechanisms of neurotrophin receptor signalling. Biochem. Soc. Trans. 34607-611. [DOI] [PubMed] [Google Scholar]
- 71.Zweifel, L. S., R. Kuruvilla, and D. D. Ginty. 2005. Functions and mechanisms of retrograde neurotrophin signalling. Nat. Rev. Neurosci. 6615-625. [DOI] [PubMed] [Google Scholar]