

Abstract
The development and growth of the skull is controlled by cranial sutures, which serve as growth centers for osteogenesis by providing a pool of osteoprogenitors. These osteoprogenitors undergo intramembranous ossification by direct differentiation into osteoblasts, which synthesize the components of the extracellular bone matrix. A dysregulation of osteoblast differentiation can lead to premature fusion of sutures, resulting in an abnormal skull shape, a disease called craniosynostosis. Although several genes could be linked to craniosynostosis, the mechanisms regulating cranial suture development remain largely elusive. We have established transgenic mice conditionally expressing an autoactivated platelet-derived growth factor receptor α (PDGFRα) in neural crest cells (NCCs) and their derivatives. In these mice, premature fusion of NCC-derived sutures occurred at early postnatal stages. In vivo and in vitro experiments demonstrated enhanced proliferation of osteoprogenitors and accelerated ossification of osteoblasts. Furthermore, in osteoblasts expressing the autoactivated receptor, we detected an upregulation of the phospholipase C-γ (PLC-γ) pathway. Treatment of differentiating osteoblasts with a PLC-γ-specific inhibitor prevented the mineralization of synthesized bone matrix. Thus, we show for the first time that PDGFRα signaling stimulates osteogenesis of NCC-derived osteoblasts by activating the PLC-γ pathway, suggesting an involvement of this pathway in the etiology of human craniosynostosis.
Neural crest cells (NCCs) are ectomesenchymal cells that arise at the dorsolateral edge of the closing neural fold, a region commonly referred to as the neural plate border. The NC can be subdivided into at least four distinct axial populations: cranial, cardiac, vagal, and trunk. The cells of the cranial region migrate ventrally to create the viscerocranium, the anterior skull base, the frontal bones of the skull vault, and the frontal suture (9, 26). However, the parietal and interparietal bones of the skull vault, as well as the sagittal suture between the parietal bones, are of mesodermal origin. The coronal suture is thereby formed between two bones of different origins, the neural-crest-derived frontal bones and the mesodermal parietal bones (26).
During skull development, calvarial growth is regulated by the cranial sutures, which serve as growth centers for osteogenesis. In this process, skeletogenic mesenchyme undergoes intramembranous ossification by direct differentiation into osteoblasts that synthesize the components of the extracellular bone matrix (18). In humans the metopic suture (homologous to the frontal suture in mice) fuses around 18 months after birth, whereas all other sutures do not fuse until an advanced age. In contrast, in the mouse skull the frontal suture fuses within the first 45 days of life, whereas all other sutures remain patent (37, 59). Suture fusion is associated with osteoblast differentiation, which is precisely controlled by several factors expressed either by osteoblasts themselves or by surrounding tissues, such as the dura mater (7, 8, 44, 58). Dysregulation of osteoblast differentiation can lead to premature fusion of one or several sutures and results in the development of an abnormal skull shape, a disease termed craniosynostosis.
Craniosynostosis is one of the most-common human congenital craniofacial deformities, affecting one in 2,500 individuals (11). While nonsyndromic in the majority of cases, it also occurs as a syndromic form associated with more than 150 genetic syndromes (12). Dominant mutations in the receptor tyrosine kinases fibroblast growth factor receptor types 1 to 3 (FGFR1-3) or in the transcription factor TWIST account for 20% of all cases of craniosynostosis (61). Although several genes have been linked to this disorder, the precise mechanisms regulating cranial suture development still remain elusive. Therefore, the identification of genes or of signaling pathways influencing intramembranous ossification and suture development is critical to understand the defective molecular mechanisms leading to craniosynostosis.
Platelet-derived growth factor receptor α (PDGFRα) belongs to the protein family of the receptor tyrosine kinases type III, which are characterized by five immunoglobulin-like domains in the extracellular-ligand-binding domain, a single membrane-spanning motif, and a split intracellular tyrosine kinase domain. Ligand binding induces the dimerization of two receptors and autophosphorylation of specific tyrosine residues in their cytoplasmic domains. These phosphotyrosine residues serve as docking sites for adaptor proteins that initiate signal transduction. PDGFRα can activate three major signal transduction pathways: the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, the phosphatidylinositol 3-kinase/Akt pathway, and the phospholipase C-γ (PLC-γ) pathway (4, 14, 27, 56, 63). The role of PDGFRα during mouse embryogenesis has been intensively analyzed using the naturally occurring patch (Ph) mutant that lacks the Pdgfra gene due to an extensive deletion of chromosome 5 (50, 53). Homozygous Ph/Ph embryos display spina bifida and defects in the development of the lung, the cardiovascular system, and the craniofacial tissue (36, 39, 41, 47, 54). PDGFRα knockout mice display a phenotype similar to that of Patch mutants, particularly in the case of the deformities in the craniofacial region (52). So PDGFRα plays an essential role in the embryonic development of cranial mesenchyme. Conditional ablation of Pdgfra in the NC results in cleft palate formation and incomplete ossification of NC-derived facial bones (55), highlighting the importance of this signaling pathway for the proper development of craniofacial structures. However, the embryonic lethality of Patch mutants and PDGFRα knockout mice exacerbates the elucidation of cell-autonomous functions of the receptor in cranial NCCs.
In this study, we generated transgenic mice conditionally expressing an autoactivated PDGFRα. Conditional expression in NCCs and their derivatives resulted in craniosynostosis at the postnatal stage affecting the NC-derived sutures. Expression of the autoactivated PDGFRα induced hyperproliferation of osteoprogenitors and enhancement of osteoblast differentiation in primary osteoblasts derived from these transgenic mice. Western blot analyses revealed activation of the PLC-γ pathway in the mutant osteoblasts. Moreover, a PLC-γ-specific inhibitor prevented differentiation of osteoblasts. Thus, our study for the first time reveals that PDGFRα influences NC-derived osteogenesis by stimulating the PLC-γ pathway.
MATERIALS AND METHODS
Generation of mice.
The targeting construct used to generate R26hPDGFRaPM embryonic stem (ES) cells is a modification of the plasmid ROSAMER (25). Briefly, a splice acceptor sequence followed by a bovine growth hormone polyadenylation sequence [poly(A)] was inserted into the single XbaI site of pROSA26-1 containing 5.3 kb of genomic ROSA26 DNA followed by a diphtheria toxin A gene (51). A cassette consisting of a loxP-flanked puromycin-N-acetyl-transferase gene terminated with three simian virus 40 polyadenylation sequences was placed behind the splice acceptor sequence to yield plasmid ROSApuro. A PacI site was inserted into the singular KpnI site to allow for linearization of the plasmid prior to electroporation into ES cells. The resulting plasmid was termed RPP and contained a single SalI site in front of the bovine growth hormone poly(A), allowing for the insertion of the mutant PDGFRa cDNA to generate R26hPDGFRaPM.
A PDGFRα D842V mutation was introduced by site-directed mutagenesis of a human wild-type PDGFRa cDNA inserted into a pcDNA3 plasmid (gift of Rainer Heuchel, Department of Genetics and Pathology, Uppsala, Sweden). For insertion of the mutated PDGFRa cDNA into RPP, an additional SalI site was cloned into the multiple cloning site of the pcDNA3 plasmid.
A 25-μg amount of linearized R26hPDGFRaPM was electroporated into Knut1 murine ES cells (42) as previously described (60). After selection with puromycin (1.4 μg/ml), four out of 50 clones were identified as correctly targeted by Southern analyses of EcoRV-restricted genomic DNA using a 32P-labeled DNA probe as described by Soriano (51).
To generate chimeric mice, C57BL/6J blastocysts injected with R26PDGFRaPM ES cells were transferred into pseudopregnant foster mice. Offspring of germ line-transmitting chimeric mice were screened for the presence of the targeted ROSA26 allele by PCR. Transgenic mice were crossed back into the 129/sv genetic background.
Mouse genotyping.
Genotyping of mice was performed by PCR using genomic DNA that was extracted from tail biopsies as described previously (25). Three primers were used in a single PCR to identify the wild-type and the targeted ROSA26 locus. The primers and their sequences which detected the wild-type ROSA26 locus were wt1s (5′-CTC CCA AAG TCG CTC TGA GTT GTT A-3′) and wt1as (5′-CCC ATT TTC CTT ATT TGC CCC TAT T-3′). An additional primer was designed for identifying the targeted ROSA26 locus: SA1as (5′-GAG ATC ATC AAG GAA ACC CTG GAC T-3′). Three-hundred-seventy-base-pair and 250-bp fragments are amplified from the wild-type ROSA26 and the targeted ROSA26 locus, respectively. The primers for the Wnt1Cre transgene, which amplified a 350-bp fragment, were Cre-s (5′-ATT TGC CTG CAT TAC CGG TC-3′) and Cre-as (5′-ATC AAC GTT TTG TTT TCG GA-3′).
Skeletal preparation.
For skeletal preparation, the mice were decapitated and the skulls were skinned and fixed in 95% alcohol. After fixation, the skulls were stained with alcian blue for 1 day following destaining in 95% alcohol for another day. Then, the samples were stained in alizarin red supplemented with 1 to 2% potassium hydroxide, depending on the size of the specimen. Finally, the skulls were cleared in 1 to 2% potassium hydroxide and transferred to solutions containing increasing glycerol concentrations.
Histological analyses and proliferation assay.
The calvaria of mice were prepared (see below), and sections of the posterior frontal suture and coronal suture were fixed in 4% formalin at 4°C overnight. After fixation, the skull bones were decalcified with 0.5 M EDTA, pH 8.0, for 30 to 60 min at room temperature prior to dehydration and paraffin embedding. For histological analyses, sections of the paraffin blocks (<5 μm) were deparaffinized and stained with hematoxylin and eosin (H&E). To investigate the in vivo proliferation of osteoprogenitors, pregnant mice were intraperitoneally injected with bromodeoxyuridine (BrdU; Sigma-Aldrich, Munich, Germany) diluted in phosphate-buffered saline (PBS) (25 mg/ml). Two hours later, the mice were sacrificed and the skulls of the embryos were treated as described above. For detection of BrdU incorporation, sections of the posterior frontal suture were incubated with a monoclonal anti-BrdU antibody and visualized using a streptavidin-biotin staining system (BrdU immunohistochemistry system; Merck, Darmstadt, Germany).
Preparation of primary osteoblasts and cell culture.
Primary osteoblast precursors were isolated from the nasal and frontal bones of postnatal day 5 (P5) mouse calvaria by sequential collagenase digestion. Briefly, after surgical isolation from the skull and adherent tissues, the nasal and frontal bones of the calvaria were treated twice for 7 min at 37°C with PBS containing 4 mM EDTA. After being washed in PBS, the calvaria were treated with 1 ml of 0.1% collagenase for 7 min at 37°C. After the supernatant of the first digestion was discarded, the calvaria were treated three more times with 1% collagenase (20 min at 37°C). The supernatants were pooled and centrifuged, and the cells were resuspended in Dulbecco's modified Eagle's medium with 10% fetal calf serum (growth medium) and placed in 10-cm dishes. Confluence was reached after 5 days, at which time the cells were subcultured. Only the first-passage cells were used for experiments. For the long-term differentiation assay, primary osteoblasts (5 × 104) were seeded in six-well plates for 48 h. Then, maturation was induced with differentiation medium containing 50 μg/ml ascorbic acid and 10 mM β-glycerophosphate. For the in vitro proliferation studies, primary osteoblasts (2 × 104) were grown for 12 h on a two-well chamber slide and labeled with 100 nM BrdU for 6 h in a humidified environment. After the BrdU incubation, cells were fixed in 70% ethanol and stained as described above. In all histological and immunohistological analyses, littermates were compared. To inhibit the PLC-γ pathway, osteoblasts were treated with 5 μM U73122 (Sigma-Aldrich, Munich, Germany) or 80 pmol PLC-γ1-specific small interfering RNA (siRNA) (Santa Cruz, Heidelberg, Germany). PDGFRα-signaling was stimulated by adding 10 ng/ml PDGF-AA to the culture medium (Biosource, Solingen, Germany).
Alkaline phosphatase and mineralized bone matrix formation assay.
Histochemical alkaline phosphatase staining was performed according to the manufacturer's instructions (blue alkaline phosphatase substrate kit III; Vector, Burlingame, CA). To detect mineralized nodules, cultures were fixed in 4% paraformaldehyde and stained with the standard von Kossa method which replaces calcified matrix Ca2+ ions with silver ions, revealing the calcium minerals as black spots.
In situ hybridization.
The Bsp2 probe was digested with either BamHI or NotI to generate sense or antisense riboprobes, respectively. These were generated by standard T7 and sp6 polymerase reactions. In situ hybridization was carried out according to a modified protocol (43).
Preparation of total RNA and semiquantitative RT-PCR.
RNA isolation was performed by using an RNeasy minikit according to the manufacturer's protocol (Qiagen, Hilden, Germany). Reverse transcription (RT) was carried out using 1 μg of total RNA, 0.2 μg oligo(dT) primer, deoxynucleoside triphosphate mix (10 mM), and 100 U Moloney murine leukemia virus-reverse transcriptase (USB, Staufen, Germany) in a volume of 20 μl. RNA and primer were preincubated at 65°C for 5 min and then rapidly cooled on ice before the addition of other reagents. The RT mixture was incubated at 42°C for 1 h. One microliter of cDNA was amplified by PCR in a 50-μl reaction mixture volume containing 2.5 mM deoxynucleoside triphosphate mix, 10 mM specific primers, and 2.5 U Taq DNA polymerase. After an initial denaturation at 94°C for 2 min, 25 to 30 amplifications were performed at 94°C for 30 s, 52 to 63°C for 45 s, and 72°C for 20 to 40 s. As a control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels were estimated by RT-PCR at 25 cycles.
The oligomer sets used for ALP2, Bsp2, Runx2, Msx1, Msx2, osteocalcin, human PDGFRα, murine PDGFRα, FGFR1 to -3, and GAPDH are depicted in Table 1.
TABLE 1.
Primer sequences for RT-PCR
| Gene | Primer pair |
|---|---|
| ALP2 | For 5′-ACT GCC ACT GCC TAC TTG TGT G-3′ |
| Rev 5′-CCT CTG GTG GCA TCT CGT TAT C-3′ | |
| Bsp2 | For 5′-CGG CCA CGC TAC TTT CTT TA-3′ |
| Rev 5′-GCC GTC TCC ATT TTC TTC TG-3′ | |
| Msx1 | For 5′-CTC ATG GCC GAT CAC AGG AA-3′ |
| Rev 5′-GTA CTG CTT CTG GCG GAA CT-3′ | |
| Msx2 | For 5′-CCT CGG TCA AGT CGG AAA AT-3′ |
| Rev 5′-TCT TTT CGC CTT AGC CCT TC-3′ | |
| Osteocalcin | For 5′-CTC TGT CTC TCT GAC CTC ACA G-3′ |
| Rev 5′-CAG GTC CTA AAT AGT GTA ACC G-3′ | |
| Runx2 | For 5′-GAA CCA AGA AGG CAC AGA CA-3′ |
| Rev 5′-AAC TGC CTG GGG TCT GAA AA-3′ | |
| hPDGFRα | For 5′-TGT GCC AGA CCC AGA TGT AG-3′ |
| Rev 5′-TGC CTT TGC CTT TCA CTT CT-3′ | |
| mPDGFRα | For 5′-GAG CAC AAG AAG TTA TGT GAT TTT G-3′ |
| Rev 5′-CCA TGA TCT CAT AGA CTT CAC TGG T-3′ | |
| FGFR1 | For 5′-CTG GAC ATC CCC AGA GAA AA-3′ |
| Rev 5′-GAA GGC ACC ACA GAA TCC AT-3′ | |
| FGFR2 | For 5′-AAG GTA CGA AAC CAG CAC TGG AG-3′ |
| Rev 5′-CAG AGT GAA AGG ATA TCC CGA TAG-3′ | |
| FGFR3 | For 5′-GAG CTA CTT CCG AGC CTC CT-3′ |
| Rev 5′-CCA CCA GAC CTG TAC CAT CC-3′ | |
| GAPDH | For 5′-GAA GCT TGT CAT CAA CGG CAA GCC C-3′ |
| Rev 5′-GCA TCG AAG GTG GAA GAG TGG CAG T-3′ |
Western blot analyses.
Cells were lysed in radioimmunoprecipitation assay buffer (10 mM Tris-HCl, pH 7.2, 150 mM NaCl, 5 mM EDTA, 0.1% sodium dodecyl sulfate, 1% Na deoxycholate, 1% Triton X-100) containing protease inhibitors (complete mini; Roche, Penzberg, Germany). The protein concentration of cell lysates was determined with a bicinchoninic acid protein assay kit according to the manufacturer's protocol (Pierce, Rockford, IL). Equal amounts of protein (15 μg) were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. Nonspecific binding sites were blocked, and membranes were incubated with antibodies to detect β-actin (1:5,000; Abcam, Cambridge, United Kingdom), PDGFRα (1:200; Spring Bioscience, Fremont, CA), phospho-PDGFRα (Tyr 754) (1:150; Santa Cruz, Heidelberg, Germany), Akt (1:1,000; Cell Signaling, Frankfurt a.M., Germany), phospho-Akt (1:1,000; Cell Signaling, Frankfurt a.M., Germany), Erk1/2 (p42/44) (1:1,000; Cell Signaling, Frankfurt a.M., Germany), phospho-Erk1/2 (phospho-p42/44) (1:1,000; Cell Signaling, Frankfurt a.M., Germany), PLC-γ (1:1,000; Cell Signaling, Frankfurt a.M., Germany), and phospho-PLC-γ (1:1,000; Cell Signaling, Frankfurt a.M., Germany), followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoreactivity was detected by using an enhanced chemiluminescence kit (Pierce, Rockford, IL).
RESULTS
Generation of transgenic mice.
To study the role of PDGFRα during intramembranous osteogenesis, we took advantage of the PDGFRα D842V mutant implicated in the etiology of gastrointestinal stroma tumors (GISTs; Online Mendelian Inheritance in Man no. 606764). This mutation leads to autoactivation of the receptor in the absence of ligand and thus to a sustained PDGFRα signaling (22, 23, 30, 34, 57). The respective point mutation was introduced into the human PDGFRa cDNA by site-directed mutagenesis. The mutant cDNA was then inserted into a targeting vector for the ubiquitously expressed ROSA26 (R26) gene locus in such a manner that transcription would be prevented by a loxP-flanked transcriptional stop sequence in the resulting modified allele. Upon Cre-mediated removal of the stop sequence, expression and PDGFRα-mediated signaling will occur (Fig. 1A). Following electroporation into ES cells, clones carrying the targeted ROSA26 locus were identified by Southern blot analysis (Fig. 1B). The chimeras generated transmitted the targeted allele to progeny, and the resulting transgenic mouse line was named R26hPDGFRaPM. In order to activate transgene expression in the anterior skull base, the frontal bones of the skull vault, and the frontal suture, transgenic mice were mated with Wnt1Cre mice expressing the Cre recombinase in NCCs and their derivatives (9, 13). R26hPDGFRaPM; Wnt1Cre doubly transgenic mice were identified by PCR using genomic DNA (Fig. 1C) and were obtained at the predicted Mendelian ratio (25%) after birth, indicating that NC-specific expression of the autoactivated PDGFRα is compatible with embryonic development.
FIG. 1.

Generation of R26hPDGFRaPM mice. (A) Wild-type (wt) ROSA26 locus, with location of the probe indicated. The targeting vector contains a loxP-flanked (black triangles) cassette with a puromycin-selectable marker and a transcriptional stop sequence (PAC-tpA), human PDGFRα cDNA containing the point mutation D846V (hPDGFRαPM) with a polyadenylation sequence, and a PGK-DTA gene for negative selection in ES cells (DTA). Open boxes with numbers show ROSA26 exons, and arrows show primers. SA, splice acceptor; ki, knock-in. (B) Representative Southern blot analysis of ES cell clones. DNA was digested with EcoRV and hybridized with the probe indicated in panel A. The 11-kb band is from the wild-type allele. The 5-kb band is from the targeted allele. (C) PCR genotyping of R26hPDGFRaPM; Wnt1Cre mice. Primers shown in panel A amplify wild-type (370 bp) and targeted alleles (250 bp). Cre-specific primers yield a 350-bp band to identify mice carrying the Wnt1-Cre transgene. The results in lane 1 identify a mouse carrying a Cre-excised allele as depicted in panel A. Lane 2, mouse carrying the PDGFRα transgene but not the Cre allele; lane 3, mouse with neither PDGFRα nor the Cre transgene; lane 4, mouse with the Cre allele but negative for the PDGFRα transgene.
Expression of autoactivated PDGFRα leads to craniosynostosis.
All doubly transgenic mice were viable and could initially not be distinguished visibly from their wild-type littermates. However, during the first week after birth they developed a shortened craniofacial area clearly visible in older animals (Fig. 2A and B). As conditional ablation of Pdgfra in the NC results in cleft palate formation, we also examined the development of the palate. No aberrant development of the palate or of other NCC-derived tissues, like the adrenal gland, the heart, and the melanocytes, could be observed (not shown).
FIG. 2.
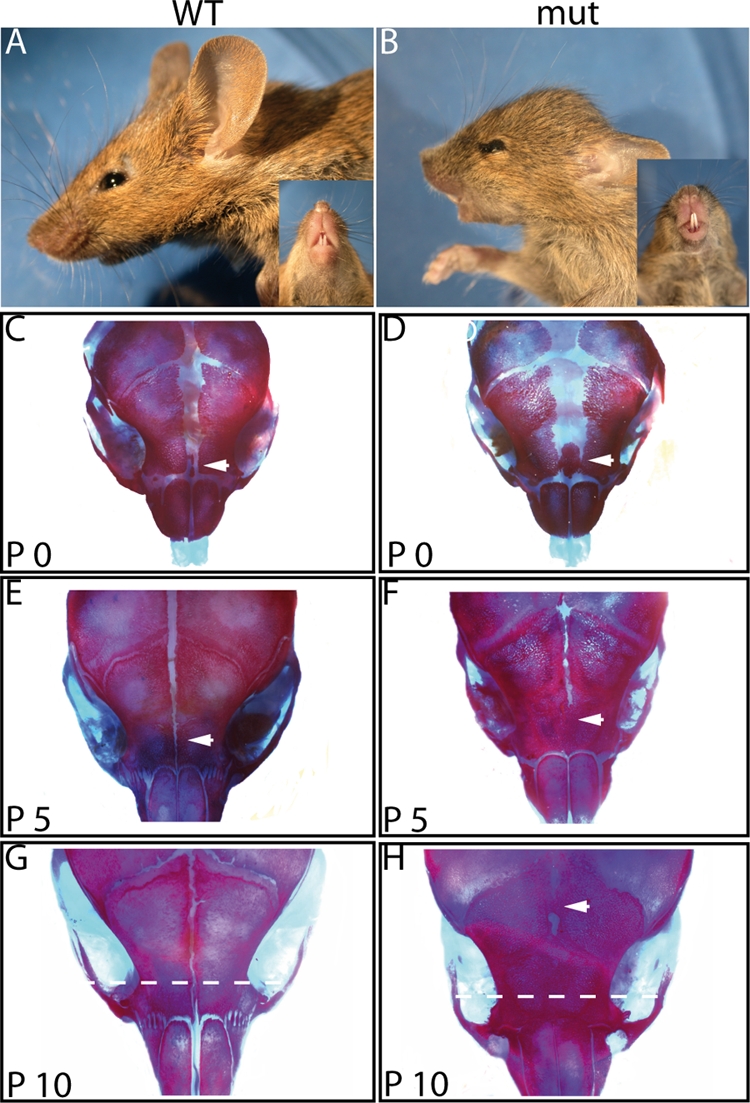
Cranial defects in R26hPDGFRaPM; Wnt1Cre mice. (A, B) Lateral and ventral views of 4-week-old wild-type control (WT) and mutant (mut) mice show abnormal skull growth resulting in a shortened craniofacial area in the mutant mice. (C to H) Alizarin red/alcian blue staining of wild-type and mutant mouse skulls was carried out at postnatal days indicated. No suture fusion was observed in the wild-type controls. The frontal suture of the R26hPDGFRaPM; Wnt1Cre mice did not show fusion at birth, but the interfrontal bone was expanded compared to this bone in the wild-type controls (compare panel D to panel C; arrows). However, fusion of the anterior frontal suture of the mutant mice occurred at P5 (F; arrow). At P10, fusion of the posterior frontal suture could be detected (H; arrow). Original magnification of panels C to H, ×5.8. Dotted lines in panels G and H indicate plane of section for which results of analysis are shown in Fig. 3.
To further analyze the craniofacial phenotype, we performed alizarin red/alcian blue staining of mutant and wild-type skulls at different time points after birth. R26PDGFRaPM;Wnt1Cre newborns (P0) display an overgrowth of the interfrontal bone in the anterior frontal calvaria (Fig. 2D; compare to panel C). At P5, there is a premature fusion of the posterior frontal suture (Fig. 2F), and at P10, fusion of the anterior frontal suture becomes evident (Fig. 2H), in contrast to the frontal sutures of wild-type controls, which remain open at any time point analyzed (Fig. 2C, E, and G). Histological analyses of the anterior frontal suture confirmed the premature suture fusion after birth. During embryogenesis, changes in osteogenesis were not detectable until embryonic day 16.5 (E16.5) in transgenic mice. Beginning from this time point, the interfrontal bone is expanded in R26hPDGFRaPM; Wnt1Cre mice compared to its size in wild-type animals (Fig. 3A and B), and the expansion becomes more prominent with time (Fig. 3C to F). However, in contrast to the remaining frontal suture seen in wild-type animals (Fig. 3G), fusion of the frontal suture occurred from P5 on in R26hPDGFRaPM; Wnt1Cre mice (Fig. 3H).
FIG. 3.

Autoactivation of PDGFRα induces craniosynostosis. Frontal sutures were sectioned as depicted in Fig. 2G and H. (A to H) H&E-stained sections of frontal suture of wild-type controls (WT) and mutant mice (mut) were histologically analyzed at days indicated. Frontal bones are marked by yellow lines. At day E16.5, an expanded interfrontal bone was observed in R26hPDGFRaPM; Wnt1Cre mice (B; arrowheads mark ends of interfrontal bones), which became more expanded during development (D, F) than the wild-type interfrontal bone (A, C, E; arrowheads). Fusion of the frontal suture could not be detected before day P5 in R26hPDGFRaPM; Wnt1Cre mice (H). At this time point, the frontal suture of the wild-type control remains patent (G; arrows). (I to N) H&E-stained sections of coronal sutures of wild-type controls and mutant mice at days indicated. (L) Site of initial fusion of the coronal suture of mutant mice (red arrow). (N) Complete fusion of the coronal suture in mutant mice 20 days after birth. (I, K, M) No fusion of the coronal suture could be detected in wild-type littermates. Scale bars: panels A to F, 500 μm; panels G and H, 100 μm; panels I to N, 200 μm. f, frontal bone; p, parietal bone.
Further histological studies of the coronal sutures of R26hPDGFRaPM; Wnt1Cre mice and of wild-type littermates were carried out. At P10, abnormal development of the frontal bones could be detected in R26hPDGFRaPM; Wnt1Cre mice, suggestive of hyperossification (Fig. 3J; compare to panel I). As expected, no aberrations of parietal bones were detected (Fig. 3J, L, and N), as these bones do not originate from NCCs but from paraxial mesoderm (26). Further analyses revealed a fusion of the coronal suture beginning at P15 (Fig. 3L) and completed by P20 (Fig. 3N). In contrast, no fusion of the coronal suture could be detected in wild-type littermates (Fig. 3I, K, and M).
Giemsa staining of cross-sections within the frontal bones of controls and R26hPDGFRaPM; Wnt1Cre mice at P5 was performed to investigate the structure of these bones in more detail (see Fig. S1 in the supplemental material). The frontal bones of the controls displayed a more planar morphology, while the frontal bones of mutant mice were structured more trabecularly and had an increased portion of calcified bone matrix. These data suggest an altered intramembranous ossification of the suture mesenchyme in R26hPDGFRaPM; Wnt1Cre mice, leading to premature suture fusion and aberrant bone morphology. As sutures serve as growth centers for osteogenesis and regulate proper skull development, the abnormal skull shape is likely caused by the premature fusion of the NC-derived sutures. Furthermore, the established mouse model reflects the phenotype of craniosynostosis in humans.
PDGFRα transgene expression results in hyperproliferation of NC-derived osteoprogenitors.
Previous studies showed that PDGFRα plays an essential role during morphogenesis of the craniofacial region (36, 52, 54, 55). In addition, PDGFs are known to drive the proliferation of undifferentiated mesenchyme and of certain progenitor cell populations during development (6). To elucidate whether the expression of the transgenic PDGFRα influences the proliferation of osteoprogenitors, a BrdU incorporation assay of cross-sections of the frontal suture of wild-type and mutant mice was carried out that revealed an increase in proliferating cells in the R26hPDGFRaPM; Wnt1Cre mice compared to the number in controls (Fig. 4A and B). Further analysis showed that at E16.5 and E19.5, 48% and 32%, respectively, of the mutant cells were proliferating, whereas in the control mice only 36% (E16.5) or 11% (E19.5) were positive for BrdU incorporation (Fig. 4C). Whereas the data from E16.5 show a trend, the results from E19.5 are significant (P < 0.01). These data clearly demonstrate that PDGFRα signaling can induce osteoblast proliferation.
FIG. 4.

PDGFRα transgene stimulates proliferation of osteoprogenitors. Proliferating osteoprogenitors were identified by immunohistological analyses of BrdU incorporation at E16.5 and E19.5. (A, B) Representative sections of wild-type (wt) control and transgenic (mut) mice at E16.5 are shown. Brown, osteoprogenitors positive for BrdU; blue, osteoprogenitors negative for BrdU. Scale bars, 100 μm. (C) A total of 800 cells from the same developmental stage of three different mice were analyzed to obtain the average percentages of BrdU-positive (BrdU+) cells at E16.5 and E19.5. Error bars show standard deviations.
Autoactivated PDGFRα enhances osteoblast differentiation in vitro.
We next isolated primary osteoblasts of calvaria tissue (Fig. 5A and B; see Fig. S2 in the supplemental material) to investigate the functions of PDGFRα in intramembranous ossification in more detail. The results of proliferation assays confirmed the in vivo results, as ∼1.4-fold more cells isolated from R26hPDGFRaPM; Wnt1Cre mice incorporated BrdU than did cells of wild-type mice (not shown).
FIG. 5.

Effect of osteoblast differentiation caused by the autoactivated PDGFRα. NC-derived osteoblasts were isolated from nasal/frontal bones and maintained in growth medium for 5 days before culture in differentiation medium for 15 days. The first day cells were maintained in differentiation medium was set as day 0. (A) Nasal/frontal bones are highlighted by a red box. c, coronal suture; f, frontal suture; F, frontal bone; IP, interparietal bone; l, lambdoid suture; N, nasal bone; P, parietal bone; s, sagittal suture. (B) At day 0 of differentiation, cells showed a spindle-shaped morphology. Scale bar, 200 μm. (C to E) To investigate the effect of the autoactivated PDGFRα on osteoblast differentiation, long-term mineralizing cultures were stained for alkaline phosphatase enzymatic activity and for mineralization by the von Kossa method at indicated time points (d, day) during differentiation. wt, wild type; mut, mutant. (C) Blue cells are positive for ALP expression. (D) Total wild-type and mutant cells from three different fields of vision at day 3 of differentiation were analyzed to determine the average percentages of ALP2-positive (ALP2+) cells. Error bars show standard deviations. (E) Black areas indicate mineralized nodule formation. The results shown in panels C and E are representative of three independent experiments.
Moreover, long-term differentiation assays were performed to study the consequences of transgenic PDGFRα expression during osteoblast differentiation in vitro. Osteoblast differentiation and maturation was analyzed by using osteoblast differentiation markers at various time points. Analysis of alkaline phosphatase (ALP2), an early marker for mature osteoblasts, showed that at day 3 of differentiation, there is a dramatic difference in osteoblast differentiation between wild-type and mutant cells (Fig. 5C). To verify this result, cells were counterstained with hematoxylin and the total cells from three different fields of vision were analyzed to determine the average percentage of ALP2-positive cells. Almost 100% of the mutant cells exhibited ALP2 activity, compared to ∼50% of the control cells, indicating premature differentiation of osteoblasts expressing the autoactivated PDGFRα (Fig. 5D).
Long-term differentiation of osteoblasts in vitro induces the production of extracellular matrix and, subsequently, mineralization of the matrix. To investigate the effect of the autoactivated PDGFRα on mineralization, primary calvarial osteoblasts were stained by the von Kossa method to visualize mineralized nodule formation (Fig. 5E). The staining confirmed a markedly enhanced maturation of the mutant osteoblasts that was clearly visible at day 15. This result suggests that mutant osteoblasts are prone to premature osteoblast differentiation.
Next, RT-PCR analyses were carried out to assess the expression profiles of various bone markers (Fig. 6A). There was similar expression of ALP2 in wild-type and mutant osteoblasts at early stages of differentiation (day 0 to 4). However, in mutant cells, the maximal ALP2 expression was already reached at day 6 of differentiation, whereas in wild-type cells, a comparable expression level was not detectable before day 10. Bone sialoprotein-2 (Bsp2) transcripts, encoding a calcium-binding glycoprotein present in mineralized bone, were only weakly expressed from day 6 on in wild-type osteoblasts and increased up to day 10 of differentiation. In mutant osteoblasts, however, Bsp2 expression was already detectable at day 4 and the expression maximum was reached at day 6 of differentiation. Similar results were obtained for the expression of osteocalcin, a calcium-binding protein expressed in mature osteoblasts undergoing mineralization, confirming the premature differentiation of the mutant osteoblasts.
FIG. 6.

(A) RT-PCR analyses of wild-type and mutant osteoblasts. Long-term differentiation cultures of wild-type and mutant osteoblasts were performed, and total RNA was isolated at different time points (d, day) as indicated. The expression levels of different bone markers (osteocalcin, BSP2, ALP2, Runx2, and Msx1/2), murine PDGFRα (mPDGFRα), human PDGFRα (hPDGFRα), and FGFR1, -2, and -3 were compared. GAPDH was used as an internal control. PC, cDNA of total skull bones. (B to E) Altered expression of Bsp2 during calvarial bone development in mice expressing autoactivated PDGFRα. Calvaria of wild-type controls (WT) and mutant mice (mut) were analyzed by in situ hybridization at days indicated. No differences in the expression pattern were detectable at day E16.5 (B, C). At day E19.5, the Bsp2 mRNA level was strongly upregulated in the frontal bones of mutant mice (E) compared to the level in wild-type controls (D). f, frontal bone; p, parietal bone. Original magnification: panels B and C, ×7; panels D and E, ×5.8.
Upregulation of osteocalcin, Bsp2, and ALP2 is regulated by Runx2, an osteoblast-specific transcription factor (3, 17, 20, 40, 45, 65) which itself is antagonized by the homeobox protein Msx2 (3, 38, 48, 49). Since the Runx2 expression level remained comparable in wild-type and mutant osteoblasts during long-term differentiation, we investigated whether a premature downregulation of Msx2 in mutant osteoblasts could be responsible for the early expression of bone markers. RT-PCR analyses showed a downregulation of Msx2 expression, but not before day 10. Therefore, the upregulation of osteocalcin, Bsp2, and ALP2 may include but is not limited to the loss of the inhibitory effect of Msx2 on Runx2 transcriptional activity.
In the mutant osteoblasts, no upregulation of the genes encoding FGFR1-3, which are mutated in the majority of known genetic causes of craniosynostosis, was detectable. However, transgene-specific RT-PCR revealed mutant PDGFRα expression only in primary osteoblasts isolated from R26hPDGFRaPM; Wnt1Cre mice, whereas endogenous PDGFRα was expressed at equal levels in wild-type and mutant osteoblasts during the course of osteoblast differentiation, indicating the requirement of the receptor throughout proper osteoblast development.
To determine aberrant bone marker expression in vivo, in situ hybridization of calvarial whole-mount preparations was performed at E16.5 and E19.5 using a Bsp2 probe (Fig. 6B to D). At E16.5, an identical expression pattern could be observed in wild-type controls and mutant mice (Fig. 6B and C), with strong expression throughout the calvarial bones and no expression in the suture mesenchyme. In contrast, at E19.5 there was a strong upregulation of Bsp2 expression in the frontal bones of R26hPDGFRaPM; Wnt1Cre mice (Fig. 6E) compared to its expression in wild-type littermates (Fig. 6D). Equal mRNA levels of Bsp2 could be observed in parietal bones at this time point, clearly demonstrating a PDGFRα-dependent effect on BSP2 expression only in bones derived from NCCs. Since Bsp2 is a marker for mature osteoblasts, the upregulation of Bsp2 in the calvaria of R26hPDGFRaPM; Wnt1Cre mice demonstrates a stimulatory role of PDGFRα in osteoblast maturation in vivo.
PDGFRα phosphorylation and activation of the PLC-γ pathway.
Protein lysates of mutant osteoblasts and wild-type controls were prepared at days 0 and 10 during long-term differentiation and analyzed by Western blotting. Using a PDGFRα-specific antibody, we detected a twofold increase in the total PDGFRα level in mutant osteoblasts compared to the level in wild-type controls (Fig. 7A). Moreover, a phospho-PDGFRα-specific antibody revealed an increased amount of activated PDGFRα in the mutant osteoblasts, consistent with autoactivation (Fig. 7B).
FIG. 7.

(A to C) Western blot analyses of wild-type and mutant osteoblasts. Long-term differentiation cultures of wild-type (wt) and mutant (mut) osteoblasts were performed, and total protein was isolated at different time points (d, day) as indicated. Results of Western blotting detecting PDGFRα, phospho-PDGFRα (pPDGFRα), PLC-γ, pPLC-γ, Akt, pAKT, ERK1/2, and pERK1/2 are shown. (D) Wild-type osteoblasts were treated with (+) 10 ng/ml PDGF for 3 days, and PLC-γ protein was detected at different time points as indicated by Western blotting. d 0, protein of wild-type osteoblasts cultivated with growth medium; PC, protein of mutant osteoblasts. (E to G) Inhibition of PLC-γ pathway prevents differentiation of osteoblasts. Primary osteoblasts from nasal/frontal bones of wild-type and mutant mice were maintained in differentiation medium for 15 days with (+) or without (−) 5 μM U73122 after 5 days (E) and 10 days (F). von Kossa staining at day 15 demonstrated a time-dependent inhibition of matrix mineralization in the presence of U73122 in both wild-type and mutant osteoblasts. No change of mineralization was observed when primary osteoblasts were cultured in differentiation medium supplemented with DMSO (dimethyl sulfoxide; diluent of U73122) after 5 days (G). (H) Mutant osteoblasts were transfected with PLC-γ-specific siRNA, and the amount of PLC-γ protein was detected by Western blotting at different time points as indicated. NC1, mutant osteoblasts transfected with scrambled RNA; NC2, mutant osteoblasts transfected only with transfection agent. (I) RT-PCR analyses, at different time points as indicated, of mutant osteoblasts treated (+) or not treated (−) with PLC-γ siRNA. PC, differentiated mutant osteoblasts (day 4).
PDGFRα can activate the MAPK/ERK, phosphatidylinositol 3-kinase/Akt, and PLC-γ pathways, all known to be involved in osteoblast biology (1, 16, 28, 67). We therefore determined the levels of activation of these pathways in wild-type and mutant osteoblasts. Protein lysates were isolated at three different time points during long-term differentiation (day 0, day 5, and day 10) that reflect phases of proliferation, matrix maturation, and mineralization of osteoblast development, respectively, and the levels of expression of total protein and phosphorylated protein were analyzed (Fig. 7C). In both wild-type and mutant osteoblasts, total Akt, ERK1/2, and PLC-γ could be detected at equal amounts. When analyzing the phosphorylation status of Akt and ERK1/2, there were no obvious differences between wild-type and mutant osteoblasts. While the levels of phosphorylated Akt remained constant at all three time points, a reduction of phosphorylated ERK1/2 could be detected toward differentiation, which is consistent with an inhibitory effect of the MAPK pathway on osteoblast differentiation. However, in mutant osteoblasts, the phosphorylation of PLC-γ was increased compared to the phosphorylation of the wild-type controls. Using ImageJ software, the average increase of phospho-PLC-γ in mutants was calculated to be 1.2-fold at day 0, 1.4-fold at day 5, and 1.8-fold at day 10 above the level of control cells. To verify if this effect is dependent on PDGFRα activation, wild-type osteoblasts were incubated with 10 ng/ml PDGF for 3 days. Proteins were isolated after 24, 48, and 72 h and analyzed with a PLC-γ-specific antibody (Fig. 7D). During PDGF treatment, the amount of PLC-γ was clearly upregulated.
In summary, the Western blot data demonstrate a PDGF/PDGFRα-specific upregulation of the PLC-γ pathway in osteoblasts, indicating a general role of this pathway in osteoblast proliferation and differentiation.
Inhibition of the PLC-γ pathway prevents differentiation of osteoblasts.
It is reported that PDGF-mediated PLC-γ activation enhances the synthesis of inorganic phosphate (Pi) carriers and their insertion into the plasma membrane (67). Pi transport is functionally involved in the calcification of bone matrix. To test whether the PLC-γ pathway is required for calcification, we analyzed the ability of the PLC-γ inhibitor U73122 to block matrix mineralization in our cell culture system. Long-term differentiation of primary mutant and wild-type osteoblasts was carried out in the presence of U73122 in the culture medium before matrix mineralization (day 5) or after advanced matrix mineralization (day 10). Matrix mineralization was dramatically reduced when U73122 was added at day 5, as judged by von Kossa staining at day 15 (Fig. 7E). In contrast, matrix mineralization was only slightly reduced when U73122 was added at day 10 (Fig. 7F). Furthermore, treatment with U73122 did not influence the synthesis of bone matrix (not shown), indicating a specific role of the PLC-γ pathway in matrix mineralization. This result was obtained with both wild-type and mutant osteoblasts, demonstrating a general function of PLC-γ in stimulating osteoblast differentiation.
As U73122 might inhibit other molecules than PLC-γ, we next treated mutant osteoblasts with a PLC-γ-specific siRNA. Mutant cells were plated in growth medium and transfected with 80 pmol siRNA. After 6 h, the medium was replaced with differentiation medium. Proteins were isolated after 24, 48, and 72 h, and Western blot analyses were performed using a PLC-γ-specific antibody. Here, PLC-γ protein levels were significantly suppressed after 48 h and slowly recovered after 72 h (Fig. 7H). To monitor the effect of PLC-γ suppression on osteoblast differentiation, the expression of Bsp2 in cells treated or untreated with siRNA was examined by RT-PCR (Fig. 7I). In osteoblasts transfected with siRNA, Bsp2 expression was not detectable before 72 h. In contrast, Bsp2 expression was already visible after 48 h in untreated osteoblasts. Thus, PDGFRα regulates the differentiation of osteoprogenitors into bone-forming cells by activating the PLC-γ pathway.
Sequencing analyses of PDGFRα in craniosynostosis patients.
As the phenotype of R26hPDGFRaPM; Wnt1Cre mice resembles human craniosynostosis, we were interested in whether autoactivating mutations in PDGFRα are present in craniosynostosis patients harboring neither FGFR nor TWIST mutations. Therefore, 15 unrelated craniosynostosis patients were chosen based upon morphological (exhibiting pansynostosis affecting at least the frontal or the coronal suture) and genetic (no mutations of FGFR1 to -3 and TWIST) criteria. Exons 10 (fifth immunoglobulin-like domain), 12 (juxtamembrane domain), 14 (first tyrosine kinase domain), and 18 (second tyrosine kinase domain) of the PDGFRα gene were amplified by PCR using primers within flanking intron regions. These exons are homologous to exons 9, 11, 13, and 17 of c-KIT and display hot-spot regions for mutations leading to an autoactivation of this receptor in GISTs. In one sample analyzed, we observed a common nonsynonymous short nucleotide polymorphism in exon 10 (P478S) which is likely to not be critical for the development of craniosynostosis, as healthy family members harbor the identical short nucleotide polymorphism. Another sample displayed a deletion of 11 bp in intron 17 and an additional polymorphism (V824V) within exon 18. This alteration is found in ∼20% of all c-KIT-negative GISTs but cannot be connected with a phenotype (unpublished data). Thus, the sequencing analysis of the PDGFRα gene in 15 craniosynostosis patients so far does not reveal a role for PDGFRα in human craniosynostosis.
DISCUSSION
We have established and analyzed transgenic mice conditionally expressing an autoactivated PDGFRα in NCCs and their derivatives. These mice allowed the investigation of PDGFRα in NCC after birth and developed craniosynostosis affecting the frontal and coronal sutures at early postnatal stages. The results of in vivo and in vitro experiments demonstrated enhanced proliferation of osteoprogenitors and accelerated ossification of osteoblasts expressing the autoactivated receptor. The results of Western blot analyses revealed an activation of the PLC-γ pathway in the mutant osteoblasts. Moreover, a PLC-γ-specific inhibitor prevented differentiation of osteoblasts. Thus, we have shown for the first time that PDGFRα signaling plays a role in NC-derived osteogenesis by stimulating the PLC-γ pathway.
A role of PDGFRα signaling in the development of cranial NCC derivatives first became clear through analysis of the naturally occurring Ph mutant mouse. Ph is an extensive deletion of chromosome 5 encompassing the Pdgfra gene. By disrupting the Pdgfra gene specifically in NCCs, Tallquist and Soriano were able to verify the role of the receptor in cranial NC (55). These mutant mice exhibit cleft palates and incomplete ossification of NCC-derived bones without defects in migration of NCCs, suggesting a requirement for PDGFRα for postmigratory NC functions. Our experiments provide further evidence for a role for PDGFRα in controlling postmigratory differentiation of cranial NC, as our gain-of-function mutation results in hyperossification of the tissues affected. In their study, Tallquist and Soriano (55) surmised that PDGFRα signaling might promote the differentiation of cranial NCCs. We identified PDGFRα signaling as controlling both proliferation and differentiation of cranial-NC-derived osteoblasts.
Consistent with our results, previous in vivo studies have shown that PDGF can stimulate new bone formation. Mitlak et al. (35) reported that systemically administered PDGF in rodents significantly increased bone density and strength and caused premature closure of the growth plate. In addition, Lynch et al. (32) observed bone formation in fractures of long bones of adult animals under treatment with a combination of insulin-like growth factor I and PDGF. Finally, Horner et al. (24) analyzed the distribution of PDGF-A and PDGFRα in rapidly forming human bone and found that the receptor is localized at sites of bone formation. On the other hand, other investigators suggested that PDGF inhibits osteoinductive processes. Several studies, using either cell cultures of fetal rat calvarial osteoblastic cells (64) or cultures of human bone marrow stromal cells (10, 29), identified PDGF as a potent inductor of osteoblast proliferation but not differentiation and mineralization. However, these latter studies used PDGF-BB to stimulate bone formation, which can activate all kinds of PDGFRα/β homo- and heterodimers. In addition, PDGF-BB is mainly present in platelets and serum (19, 21), whereas the specifically PDGFRα-activating PDGF-AA is the isoform secreted by unstimulated normal bone cells (46, 66). Thus, osteoblastic cells probably are not exposed to PDGF-BB under physiological conditions.
Our transgenic mouse model has the advantage that we were able to investigate bone formation dependent on the specific activation of PDGFRα. As a result, our data support the idea that PDGFRα stimulates bone development in a biphasic manner. First, PDGFRα stimulates the proliferation of osteoprogenitors, which is in agreement with previous studies that describe a role of PDGFRα in tumor proliferation (33, 62). Hyperproliferation of osteoprogenitors is likely to be responsible for the premature fusion of the sutures, as there is an increase in the pool of bone-forming cells. Second, PDGFRα stimulates bone development by regulating the differentiation of osteoblasts. A previous study analyzed the expression pattern of PDGFRα during the differentiation of osteoblasts isolated from rat calvaria (31). The receptor was found to be expressed continuously throughout the development of osteoblasts, and its expression was significantly increased in mature osteoblasts. These data indicate a role of PDGFRα not only during proliferation but also in the stage of differentiation. Our data are consistent with this idea, as osteoblasts expressing the autoactivated PDGFRα display enhanced differentiation, in addition to hyperproliferation. The specific upregulation of the PLC-γ pathway in the mutant osteoblasts suggests this pathway as a possible molecular mechanism controlling the differentiation of osteoblasts.
This idea is corroborated by a previous study that demonstrates a role of the PLC-γ pathway in regulating Pi transport into osteoblastic cells in response to PDGF (67). In addition to its role in cell metabolism, Pi transport is crucially involved in the calcification of bone matrix. During the initial phase of matrix calcification, osteoblasts release matrix vesicles (MVs) that serve as the initial site of bone mineral formation (2). MV-mediated mineralization occurs in two phases. Phase 1 involves the formation of the first bone mineral by uptake of Ca2+ and Pi within MVs. MV-associated phosphatases, including ALP2, thereby degrade β-glycerophosphate to Pi (5, 15). In phase 2, the newly formed mineral penetrates the vesicular membrane, and the rate of mineral proliferation is governed by the extracellular Ca2+ and Pi concentrations in an autocatalytic process. The essential role of PLC-γ-mediated Pi uptake in osteoblast differentiation became apparent after the treatment of osteoblasts with a PLC-γ inhibitor at different time points during long-term differentiation. When treatment was initiated before matrix calcification occurred, the development of nodules and bone matrix but no calcification of matrix could be observed. In contrast, there was less influence on matrix calcification when osteoblasts were treated with the PLC-γ inhibitor after matrix mineralization had started. This time-dependent effect of PLC-γ inhibition is consistent with biphasic bone mineral formation. At an early time point during long-term differentiation there are only phase 1 MVs, and PLC-γ inhibition prevents initial mineral formation. When osteoblasts were treated with the PLC-γ inhibitor at later time points, mineral proliferation was independent of PLC-γ-mediated Pi uptake in MVs because bone minerals were already exposed to the extracellular environment. As phase 1 and phase 2 occur concomitantly, we concluded that PLC-γ inhibition at a later time point had less influence on differentiation, because only the formation of new bone minerals, but not the extension of minerals formed earlier, was suppressed. In addition, the upregulation of ALP2 activity in the mutant osteoblasts might enhance differentiation by increasing the amounts of Pi transported into the MVs. Thus, the osteoinductive processes in the mutant osteoblasts demonstrate a role of PDGFRα not only in proliferation but also in differentiation of osteoprogenitors into bone-forming cells.
Because the phenotype of our transgenic mice resembles human craniosynostosis, we aimed to detect autoactivating mutations in the PDGFRα gene in craniosynostosis patients. A sequencing analysis of the PDGFRα gene in 15 patients did not reveal PDGFRα mutations in the known hot-spot regions involved in autoactivation of the receptor in GISTs. Nevertheless, the possibility of identifying mutations by screening an expanded group of craniosynostosis patients and sequencing the complete PDGFRα gene remains. Moreover, activation of the PLC-γ pathway independent of PDGFRα mutations might be involved in human craniosynostosis.
We have shown that conditional expression of an autoactivated PDGFRα in NCCs and their derivatives induces craniosynostosis affecting the frontal and coronal sutures, which supports a role of PDGFRα in cranial NC development. In addition to the regulation of proliferation, PDGFRα signaling turned out to stimulate differentiation of NC-derived osteoblasts. We propose the activation of the PLC-γ pathway by PDGFRα as the molecular mechanism, as this pathway was highly upregulated in the mutant osteoblasts and inhibition of PLC-γ prevents mineralization of mutant and wild-type osteoblasts. However, further experiments that would establish the activation status of PLC-γ (phosphorylation and second messengers) are needed to fully support our conclusions. Our results not only reveal a role of PDGFRα in the development of cranial NCCs but show for the first time that PDGFRα is required for the differentiation of NC-derived osteoblasts into bone-forming cells by stimulating the PLC-γ pathway.
Supplementary Material
Acknowledgments
We thank Inge Heim for excellent technical assistance, Rainer Heuchel for the PDGFRα cDNA, and Andrea Vortkamp for intellectual and technical advice.
This work was supported by grants from the Mildred Scheel Stiftung Deutsche Krebshilfe (no. 10-2233 to H.S.) and Stem Cell Network NRW (to H.S.).
Footnotes
Published ahead of print on 1 December 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Akeno, N., J. Robins, M. Zhang, M. F. Czyzyk-Krzeska, and T. L. Clemens. 2002. Induction of vascular endothelial growth factor by IGF-I in osteoblast-like cells is mediated by the PI3K signaling pathway through the hypoxia-inducible factor-2alpha. Endocrinology 143420-425. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, H. C. 1995. Molecular biology of matrix vesicles. Clin. Orthop. Relat. Res. 314266-280. [PubMed] [Google Scholar]
- 3.Barnes, G. L., A. Javed, S. M. Waller, M. H. Kamal, K. E. Hebert, M. Q. Hassan, A. Bellahcene, A. J. Van Wijnen, M. F. Young, J. B. Lian, G. S. Stein, and L. C. Gerstenfeld. 2003. Osteoblast-related transcription factors Runx2 (Cbfa1/AML3) and MSX2 mediate the expression of bone sialoprotein in human metastatic breast cancer cells. Cancer Res. 632631-2637. [PubMed] [Google Scholar]
- 4.Bazenet, C. E., J. A. Gelderloos, and A. Kazlauskas. 1996. Phosphorylation of tyrosine 720 in the platelet-derived growth factor alpha receptor is required for binding of Grb2 and SHP-2 but not for activation of Ras or cell proliferation. Mol. Cell. Biol. 166926-6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellows, C. G., J. N. Heersche, and J. E. Aubin. 1992. Inorganic phosphate added exogenously or released from beta-glycerophosphate initiates mineralization of osteoid nodules in vitro. Bone Miner. 1715-29. [DOI] [PubMed] [Google Scholar]
- 6.Betsholtz, C., L. Karlsson, and P. Lindahl. 2001. Developmental roles of platelet-derived growth factors. Bioessays 23494-507. [DOI] [PubMed] [Google Scholar]
- 7.Bradley, J. P., J. P. Levine, C. Blewett, T. Krummel, J. G. McCarthy, and M. T. Longaker. 1996. Studies in cranial suture biology: in vitro cranial suture fusion. Cleft Palate Craniofac. J. 33150-156. [DOI] [PubMed] [Google Scholar]
- 8.Carinci, P., E. Becchetti, and M. Bodo. 2000. Role of the extracellular matrix and growth factors in skull morphogenesis and in the pathogenesis of craniosynostosis. Int. J. Dev. Biol. 44715-723. [PubMed] [Google Scholar]
- 9.Chai, Y., X. Jiang, Y. Ito, P. Bringas, Jr., J. Han, D. H. Rowitch, P. Soriano, A. P. McMahon, and H. M. Sucov. 2000. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 1271671-1679. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhary, L. R., A. M. Hofmeister, and K. A. Hruska. 2004. Differential growth factor control of bone formation through osteoprogenitor differentiation. Bone 34402-411. [DOI] [PubMed] [Google Scholar]
- 11.Cohen, M. M., Jr. 1988. Craniosynostosis update 1987. Am. J. Med. Genet. Suppl. 499-148. [DOI] [PubMed] [Google Scholar]
- 12.Cohen, M. M., Jr. 1995. Craniosynostosis: phenotypic/molecular correlations. Am. J. Med. Genet. 56334-339. [DOI] [PubMed] [Google Scholar]
- 13.Danielian, P. S., D. Muccino, D. H. Rowitch, S. K. Michael, and A. P. McMahon. 1998. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 81323-1326. [DOI] [PubMed] [Google Scholar]
- 14.Eriksson, A., E. Nanberg, L. Ronnstrand, U. Engstrom, U. Hellman, E. Rupp, G. Carpenter, C. H. Heldin, and L. Claesson-Welsh. 1995. Demonstration of functionally different interactions between phospholipase C-gamma and the two types of platelet-derived growth factor receptors. J. Biol. Chem. 2707773-7781. [DOI] [PubMed] [Google Scholar]
- 15.Garimella, R., X. Bi, N. Camacho, J. B. Sipe, and H. C. Anderson. 2004. Primary culture of rat growth plate chondrocytes: an in vitro model of growth plate histotype, matrix vesicle biogenesis and mineralization. Bone 34961-970. [DOI] [PubMed] [Google Scholar]
- 16.Ge, C., G. Xiao, D. Jiang, and R. T. Franceschi. 2007. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 176709-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutierrez, S., A. Javed, D. K. Tennant, M. van Rees, M. Montecino, G. S. Stein, J. L. Stein, and J. B. Lian. 2002. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J. Biol. Chem. 2771316-1323. [DOI] [PubMed] [Google Scholar]
- 18.Hall, B. K. 1999. Bone. Telford Press, Caldwell, NJ.
- 19.Hammacher, A., U. Hellman, A. Johnsson, A. Ostman, K. Gunnarsson, B. Westermark, A. Wasteson, and C. H. Heldin. 1988. A major part of platelet-derived growth factor purified from human platelets is a heterodimer of one A and one B chain. J. Biol. Chem. 26316493-16498. [PubMed] [Google Scholar]
- 20.Harada, H., S. Tagashira, M. Fujiwara, S. Ogawa, T. Katsumata, A. Yamaguchi, T. Komori, and M. Nakatsuka. 1999. Cbfa1 isoforms exert functional differences in osteoblast differentiation. J. Biol. Chem. 2746972-6978. [DOI] [PubMed] [Google Scholar]
- 21.Hart, C. E., M. Bailey, D. A. Curtis, S. Osborn, E. Raines, R. Ross, and J. W. Forstrom. 1990. Purification of PDGF-AB and PDGF-BB from human platelet extracts and identification of all three PDGF dimers in human platelets. Biochemistry 29166-172. [DOI] [PubMed] [Google Scholar]
- 22.Heinrich, M. C., C. L. Corless, A. Duensing, L. McGreevey, C. J. Chen, N. Joseph, S. Singer, D. J. Griffith, A. Haley, A. Town, G. D. Demetri, C. D. Fletcher, and J. A. Fletcher. 2003. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299708-710. [DOI] [PubMed] [Google Scholar]
- 23.Hirota, S., A. Ohashi, T. Nishida, K. Isozaki, K. Kinoshita, Y. Shinomura, and Y. Kitamura. 2003. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 125660-667. [DOI] [PubMed] [Google Scholar]
- 24.Horner, A., S. Bord, P. Kemp, D. Grainger, and J. E. Compston. 1996. Distribution of platelet-derived growth factor (PDGF) A chain mRNA, protein, and PDGF-alpha receptor in rapidly forming human bone. Bone 19353-362. [DOI] [PubMed] [Google Scholar]
- 25.Jager, R., J. Maurer, A. Jacob, and H. Schorle. 2004. Cell type-specific conditional regulation of the c-myc proto-oncogene by combining Cre/loxP recombination and tamoxifen-mediated activation. Genesis 38145-150. [DOI] [PubMed] [Google Scholar]
- 26.Jiang, X., S. Iseki, R. E. Maxson, H. M. Sucov, and G. M. Morriss-Kay. 2002. Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241106-116. [DOI] [PubMed] [Google Scholar]
- 27.Klinghoffer, R. A., B. Duckworth, M. Valius, L. Cantley, and A. Kazlauskas. 1996. Platelet-derived growth factor-dependent activation of phosphatidylinositol 3-kinase is regulated by receptor binding of SH2-domain-containing proteins which influence Ras activity. Mol. Cell. Biol. 165905-5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kono, S. J., Y. Oshima, K. Hoshi, L. F. Bonewald, H. Oda, K. Nakamura, H. Kawaguchi, and S. Tanaka. 2007. Erk pathways negatively regulate matrix mineralization. Bone 4068-74. [DOI] [PubMed] [Google Scholar]
- 29.Kratchmarova, I., B. Blagoev, M. Haack-Sorensen, M. Kassem, and M. Mann. 2005. Mechanism of divergent growth factor effects in mesenchymal stem cell differentiation. Science 3081472-1477. [DOI] [PubMed] [Google Scholar]
- 30.Lasota, J., A. Dansonka-Mieszkowska, L. H. Sobin, and M. Miettinen. 2004. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab. Investig. 84874-883. [DOI] [PubMed] [Google Scholar]
- 31.Liu, F., L. Malaval, and J. E. Aubin. 2003. Global amplification polymerase chain reaction reveals novel transitional stages during osteoprogenitor differentiation. J. Cell Sci. 1161787-1796. [DOI] [PubMed] [Google Scholar]
- 32.Lynch, S. E., S. B. Trippel, R. D. Finkelman, R. A. Hernandez, C. P. Kiritsy, and H. N. Antoniades. 1994. The combination of platelet-derived growth factor-BB and insulin-like growth factor-I stimulates bone repair in adult Yucatan miniature pigs. Wound Repair Regen. 2182-190. [DOI] [PubMed] [Google Scholar]
- 33.Matei, D., R. E. Emerson, Y. C. Lai, L. A. Baldridge, J. Rao, C. Yiannoutsos, and D. D. Donner. 2006. Autocrine activation of PDGFRalpha promotes the progression of ovarian cancer. Oncogene 252060-2069. [DOI] [PubMed] [Google Scholar]
- 34.Medeiros, F., C. L. Corless, A. Duensing, J. L. Hornick, A. M. Oliveira, M. C. Heinrich, J. A. Fletcher, and C. D. Fletcher. 2004. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am. J. Surg. Pathol. 28889-894. [DOI] [PubMed] [Google Scholar]
- 35.Mitlak, B. H., R. D. Finkelman, E. L. Hill, J. Li, B. Martin, T. Smith, M. D'Andrea, H. N. Antoniades, and S. E. Lynch. 1996. The effect of systemically administered PDGF-BB on the rodent skeleton. J. Bone Miner. Res. 11238-247. [DOI] [PubMed] [Google Scholar]
- 36.Morrison-Graham, K., G. C. Schatteman, T. Bork, D. F. Bowen-Pope, and J. A. Weston. 1992. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development 115133-142. [DOI] [PubMed] [Google Scholar]
- 37.Moss, M. L. 1958. Fusion of the frontal suture in the rat. Am. J. Anat. 102141-165. [DOI] [PubMed] [Google Scholar]
- 38.Newberry, E. P., T. Latifi, and D. A. Towler. 1998. Reciprocal regulation of osteocalcin transcription by the homeodomain proteins Msx2 and Dlx5. Biochemistry 3716360-16368. [DOI] [PubMed] [Google Scholar]
- 39.Orr-Urtreger, A., M. T. Bedford, M. S. Do, L. Eisenbach, and P. Lonai. 1992. Developmental expression of the alpha receptor for platelet-derived growth factor, which is deleted in the embryonic lethal Patch mutation. Development 115289-303. [DOI] [PubMed] [Google Scholar]
- 40.Paredes, R., G. Arriagada, F. Cruzat, A. Villagra, J. Olate, K. Zaidi, A. van Wijnen, J. B. Lian, G. S. Stein, J. L. Stein, and M. Montecino. 2004. Bone-specific transcription factor Runx2 interacts with the 1α,25-dihydroxyvitamin D3 receptor to up-regulate rat osteocalcin gene expression in osteoblastic cells. Mol. Cell. Biol. 248847-8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Payne, J., F. Shibasaki, and M. Mercola. 1997. Spina bifida occulta in homozygous Patch mouse embryos. Dev. Dyn. 209105-116. [DOI] [PubMed] [Google Scholar]
- 42.Peitz, M., R. Jager, C. Patsch, A. Jager, A. Egert, H. Schorle, and F. Edenhofer. 2007. Enhanced purification of cell-permeant Cre and germline transmission after transduction into mouse embryonic stem cells. Genesis 45508-517. [DOI] [PubMed] [Google Scholar]
- 43.Pfisterer, P., J. Ehlermann, M. Hegen, and H. Schorle. 2002. A subtractive gene expression screen suggests a role of transcription factor AP-2 alpha in control of proliferation and differentiation. J. Biol. Chem. 2776637-6644. [DOI] [PubMed] [Google Scholar]
- 44.Rice, D. P., T. Aberg, Y. Chan, Z. Tang, P. J. Kettunen, L. Pakarinen, R. E. Maxson, and I. Thesleff. 2000. Integration of FGF and TWIST in calvarial bone and suture development. Development 1271845-1855. [DOI] [PubMed] [Google Scholar]
- 45.Roca, H., M. Phimphilai, R. Gopalakrishnan, G. Xiao, and R. T. Franceschi. 2005. Cooperative interactions between RUNX2 and homeodomain protein-binding sites are critical for the osteoblast-specific expression of the bone sialoprotein gene. J. Biol. Chem. 28030845-30855. [DOI] [PubMed] [Google Scholar]
- 46.Rydziel, S., C. Ladd, T. L. McCarthy, M. Centrella, and E. Canalis. 1992. Determination and expression of platelet-derived growth factor-AA in bone cell cultures. Endocrinology 1301916-1922. [DOI] [PubMed] [Google Scholar]
- 47.Schatteman, G. C., K. Morrison-Graham, A. van Koppen, J. A. Weston, and D. F. Bowen-Pope. 1992. Regulation and role of PDGF receptor alpha-subunit expression during embryogenesis. Development 115123-131. [DOI] [PubMed] [Google Scholar]
- 48.Shirakabe, K., K. Terasawa, K. Miyama, H. Shibuya, and E. Nishida. 2001. Regulation of the activity of the transcription factor Runx2 by two homeobox proteins, Msx2 and Dlx5. Genes Cells 6851-856. [DOI] [PubMed] [Google Scholar]
- 49.Sierra, O. L., S. L. Cheng, A. P. Loewy, N. Charlton-Kachigian, and D. A. Towler. 2004. MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J. Biol. Chem. 27932913-32923. [DOI] [PubMed] [Google Scholar]
- 50.Smith, E. A., M. F. Seldin, L. Martinez, M. L. Watson, G. G. Choudhury, P. A. Lalley, J. Pierce, S. Aaronson, J. Barker, S. L. Naylor, et al. 1991. Mouse platelet-derived growth factor receptor alpha gene is deleted in W19H and patch mutations on chromosome 5. Proc. Natl. Acad. Sci. USA 884811-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 2170-71. [DOI] [PubMed] [Google Scholar]
- 52.Soriano, P. 1997. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 1242691-2700. [DOI] [PubMed] [Google Scholar]
- 53.Stephenson, D. A., M. Mercola, E. Anderson, C. Y. Wang, C. D. Stiles, D. F. Bowen-Pope, and V. M. Chapman. 1991. Platelet-derived growth factor receptor alpha-subunit gene (Pdgfra) is deleted in the mouse patch (Ph) mutation. Proc. Natl. Acad. Sci. USA 886-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun, T., D. Jayatilake, G. B. Afink, P. Ataliotis, M. Nister, W. D. Richardson, and H. K. Smith. 2000. A human YAC transgene rescues craniofacial and neural tube development in PDGFRalpha knockout mice and uncovers a role for PDGFRalpha in prenatal lung growth. Development 1274519-4529. [DOI] [PubMed] [Google Scholar]
- 55.Tallquist, M. D., and P. Soriano. 2003. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 130507-518. [DOI] [PubMed] [Google Scholar]
- 56.Valius, M., C. Bazenet, and A. Kazlauskas. 1993. Tyrosines 1021 and 1009 are phosphorylation sites in the carboxy terminus of the platelet-derived growth factor receptor β subunit and are required for binding of phospholipase Cγ and a 64-kilodalton protein, respectively. Mol. Cell. Biol. 13133-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wardelmann, E., A. Hrychyk, S. Merkelbach-Bruse, K. Pauls, J. Goldstein, P. Hohenberger, I. Losen, C. Manegold, R. Buttner, and T. Pietsch. 2004. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J. Mol. Diagn. 6197-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warren, S. M., L. J. Brunet, R. M. Harland, A. N. Economides, and M. T. Longaker. 2003. The BMP antagonist noggin regulates cranial suture fusion. Nature 422625-629. [DOI] [PubMed] [Google Scholar]
- 59.Warren, S. M., J. A. Greenwald, J. A. Spector, P. Bouletreau, B. J. Mehrara, and M. T. Longaker. 2001. New developments in cranial suture research. Plast. Reconstr. Surg. 107523-540. [DOI] [PubMed] [Google Scholar]
- 60.Werling, U., and H. Schorle. 2002. Transcription factor gene AP-2γ essential for early murine development. Mol. Cell. Biol. 223149-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilkie, A. O., and G. M. Morriss-Kay. 2001. Genetics of craniofacial development and malformation. Nat. Rev. Genet. 2458-468. [DOI] [PubMed] [Google Scholar]
- 62.Xie, J., M. Aszterbaum, X. Zhang, J. M. Bonifas, C. Zachary, E. Epstein, and F. McCormick. 2001. A role of PDGFRalpha in basal cell carcinoma proliferation. Proc. Natl. Acad. Sci. USA 989255-9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu, J., T. F. Deuel, and H. R. Kim. 2000. Platelet-derived growth factor (PDGF) receptor-alpha activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-beta-induced phenotypic transformation. J. Biol. Chem. 27519076-19082. [DOI] [PubMed] [Google Scholar]
- 64.Yu, X., S. C. Hsieh, W. Bao, and D. T. Graves. 1997. Temporal expression of PDGF receptors and PDGF regulatory effects on osteoblastic cells in mineralizing cultures. Am. J. Physiol. 272C1709-C1716. [DOI] [PubMed] [Google Scholar]
- 65.Zaidi, S. K., A. Javed, J. Y. Choi, A. J. van Wijnen, J. L. Stein, J. B. Lian, and G. S. Stein. 2001. A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J. Cell Sci. 1143093-3102. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, L., E. Leeman, D. C. Carnes, and D. T. Graves. 1991. Human osteoblasts synthesize and respond to platelet-derived growth factor. Am. J. Physiol. 261C348-C354. [DOI] [PubMed] [Google Scholar]
- 67.Zhen, X., J. P. Bonjour, and J. Caverzasio. 1997. Platelet-derived growth factor stimulates sodium-dependent Pi transport in osteoblastic cells via phospholipase Cgamma and phosphatidylinositol 3′-kinase. J. Bone Miner. Res. 1236-44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.