

Abstract
Mlh1 is an essential factor of mismatch repair (MMR) and meiotic recombination. It interacts through its C-terminal region with MutL homologs and proteins involved in DNA repair and replication. In this study, we identified the site of yeast Mlh1 critical for the interaction with Exo1, Ntg2, and Sgs1 proteins, designated as site S2 by reference to the Mlh1/Pms1 heterodimerization site S1. We show that site S2 is also involved in the interaction between human MLH1 and EXO1 or BLM. Binding at this site involves a common motif on Mlh1 partners that we called the MIP-box for the Mlh1 interacting protein box. Direct and specific interactions between yeast Mlh1 and peptides derived from Exo1, Ntg2, and Sgs1 and between human MLH1 and peptide derived from EXO1 and BLM were measured with Kd values ranging from 8.1 to 17.4 μM. In Saccharomyces cerevisiae, a mutant of Mlh1 targeted at site S2 (Mlh1-E682A) behaves as a hypomorphic form of Exo1. The site S2 in Mlh1 mediates Exo1 recruitment in order to optimize MMR-dependent mutation avoidance. Given the conservation of Mlh1 and Exo1 interaction, it may readily impact Mlh1-dependent functions such as cancer prevention in higher eukaryotes.
Mismatch repair (MMR) is a powerful, evolutionary conserved, mutation avoidance mechanism. Inactivation of MMR in human causes genetic instability that has been associated with the development of hereditary nonpolyposis colorectal cancer (HNPCC) and a fraction of sporadic tumors that occur in a number of tissues (23). MMR corrects base-base mismatches and insertion/deletion loops arising during DNA replication. Thus, MMR increases the fidelity of DNA synthesis 100- to 1,000-fold. In eukaryotic cells, the MMR process begins when a MutS-homolog recognizes and binds to a mismatch (17, 32). In the subsequent step, the mismatch bound MutSα or MutSβ recruits a MutL homolog (22). In addition to MutS and MutL homologs, eukaryotic MMR requires several other factors, most of which are involved in DNA replication such as EXO1, PCNA, RFC, RPA, DNA polymerases, and DNA ligase (17, 22, 32).
The MutLα complex, composed of Mlh1 and Pms1 proteins, has an essential role in the Saccharomyces cerevisiae MMR pathway. MutLα has been proposed to act as a key molecular matchmaker that coordinates mismatch recognition with downstream functions in the course of the MMR process. Recent studies revealed that MutLα also contains a latent endonuclease activity that is activated in the presence of DNA with a mismatch, MutSα, PCNA, RFC, and ATP (18). Mutation at the endonuclease active-site motif of Pms1 (pms1-E707K) abolishes the activity of MutLα in vitro and confers a strong mutator phenotype in vivo, such as pms1Δ or mlh1Δ (9, 19). In higher eukaryotes, inactivation of MLH1 results in genetic instability and cancer predisposition; ca. 50% of mutations predisposing to HNPCC in human affects MLH1 (23). In addition to its essential role in MMR, Mlh1 also plays important roles in meiotic recombination (4). MLH1-deficient mice display a severe crossover defect, and male and female are sterile, whereas MSH2-deficient mice are fertile (7).
In S. cerevisiae, Mlh1 has been shown to physically interact with DNA and proteins such as Msh2, Pms1, Mlh2, Mlh3, Exo1 (5′-3′ exonuclease and flap endonuclease), Sgs1 (DNA helicase of the RecQ family), Ntg2 (DNA N-glycosylase and apurinic or apyrimidinic lyase), or PCNA (12, 15, 35, 51, 52). Mlh1 interacts with DNA, MutSα and the N-terminal region of Pms1 by its N-terminal region (15, 37, 50). In contrast, binding to Pms1, Mlh2, and Mlh3 and to Exo1, Sgs1 and Ntg2 requires the C-terminal region of Mlh1 (3, 12, 25, 35, 51, 53). It was recently proposed that this region of Mlh1 also contributes to the interaction with PCNA (27). However, many aspects of the interaction network involving the C-terminal region of Mlh1 remain unresolved, including the number of binding sites present in this region, their structural characteristics, and their functional role. Recent studies have proposed several Mlh1 positions critical for the binding to MutL homologs Pms1, Mlh2, and Mlh3 (3, 21) or for interaction with PCNA (27). With regard to Exo1, Ntg2, and Sgs1, we previously showed that these proteins have a common five amino acids motif R-S-K-[Y/F]-F, called the MIP-box (for Mlh1 interacting protein box), that is critical for Exo1 and Ntg2 binding to Mlh1 (12). The binding sites of these three proteins on Mlh1 and the biological function of the corresponding interactions remained largely unknown.
To characterize Mlh1 binding site(s) for Exo1, Ntg2, and Sgs1, we developed an approach coupling two-hybrid assays, site-directed mutagenesis, molecular modeling, and biochemical and biophysical methods. First, the conservation among eukaryotes of the interaction between Mlh1 and proteins containing a MIP-box was assessed by characterizing the impact of substitutions on the MIP-box of human EXO1 on its interaction with human MLH1. The subsequent conservation analysis was used as a guide to mutate 28 positions in Mlh1 and evaluate their impact on Pms1, Ntg2, and Exo1 interaction. Ten positions on Mlh1 were found to be essential for the interactions with Exo1 and Ntg2, although neutral for that with Pms1. Molecular modeling provided a first structural representation of the new binding site that we named S2, by reference to the Mlh1/Pms1 heterodimerization site S1. The affinities of the different interactions between Mlh1 and its partners at site S2 was questioned by measuring the binding affinities of each MIP box motif using isothermal titration calorimetry (ITC). Finally, to investigate the biological impact of interactions at the S1 and S2 sites, we constructed the chromosomal mutants mlh1-R547A and mlh1-E682A, respectively.
MATERIALS AND METHODS
Yeast strains, plasmids, and microbiological methods.
S. cerevisiae strains and plasmids used in the present study are listed in Table 1 (see also Table S1 in the supplemental material). Yeast strains were grown at 30°C in YPD medium (1% yeast extract, 1% Bacto peptone, and 2% glucose, with 2% agar for the plates) or YNBD medium (0.17% yeast nitrogen base without amino acids and 2% glucose, with 2% agar for the plates) supplemented with appropriate amino acids and bases. Strains were obtained after genetic crossing and tetrad analysis. Presporulation and sporulation procedures were performed as previously described (39). Micromanipulation and dissection of asci were performed using a Singer MSM system (45). Gene deletions were performed by a PCR-mediated one-step replacement technique (5, 28). Point mutations were performed by PCR-mediated mutagenesis (QuikChange site-directed mutagenesis kit; Stratagene, La Jolla, CA) using relevant plasmids and specific primers. For glutathione S-transferase (GST) pull-down assays, plasmid pcDNA3.1A(−)-hEXO1b expressing a COOH-terminal His-Myc-tagged hEXO1 protein was used as a template to construct hEXO1b-S504A, hEXO1b-F506A, hEXO1b-F507A, and hEXO1b-FF506AA. The wild-type hEXO1 allele used in the present study has been described previously (16, 29, 34, 38, 40). Mutations in hEXO1 were introduced by using a QuikChange site-directed mutagenesis kit. The entire coding region of the constructs containing hEXO1 was verified by DNA sequencing (Macrogen, Korea). Target plasmids and mutants are listed in Table S1 in the supplemental material.
TABLE 1.
Yeast strains used in this study
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| RKY3109 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2Δ1 ade8 met− | R. Kolodner |
| BG301 | RKY3109 with mlh1Δ::kanMX6 | 12 |
| BG320 | RKY3109 with exo1Δ::kanMX6 | 12 |
| FDB1 | RKY3109 with mlh1-R547A | This study |
| FDB2 | RKY3109 with mlh1-E682A | This study |
| FDB3 | RKY3109 with mlh1-R547A exo1Δ::kanMX6 | This study |
| FDB4 | RKY3109 with mlh1-E682A exo1Δ::kanMX6 | This study |
| GCY35 | MATahis3Δ200 hom3-10 ura3-52 ade2-101 trp1 met13 met4 | M. Liskay |
| PTY301 | GCY35 with pms1-G97A | M. Liskay |
| FDB5 | GCY35 with pms1-G97A exo1Δ::kanMX6 | This study |
| FDB6 | MATaura3-52 hom3-10 pms1-G97A mlh1-E682A | This study |
Mutations in the chromosomal MLH1 were obtained by the pop-in/pop-out method using the integrative pRS306 (URA3) plasmid harboring a mutant version of the MLH1 gene. Yeast transformation assays were performed using the polyethylene glycol-lithium chloride method (13). Bacterial strains XL-10, DH5α, and JM105 were used for plasmid construction, preparation, and mutagenesis (30). Gene disruptions and mutations were confirmed by PCR on genomic or plasmid DNA and sequencing analysis. Details of strains, plasmids, and primers are available upon request.
GST pull-down assays.
The GST-hMLH1 and GST-hMSH2 fusion proteins were purified as described previously (16). A total of 1 μg of GST-hMLH1 (110 μg/ml) or 1 μg of GST-hMSH2 (90 μg/ml) was bound to 20 μl of GST beads (glutathione-Sepharose 4B; GE Healthcare) prepared in a 50% slurry with binding buffer (20 mM Tris-HCl [pH 7.5], 10% glycerol, 300 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol, 0.1% Tween 20, 0.75 mg of bovine serum albumin/ml, and protease inhibitors [Complete; Roche Diagnostics]) by incubation for 2 h at 4°C on a rocking platform. The protein-bound beads were washed three times with 500 μl of binding buffer. Samples were diluted with binding buffer to 83 μl of GST beads/ml, followed by incubation for 30 min at 4°C on a rocking platform. In vitro transcription/translation (IVTT) protein products were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and quantified using software VisionWorksLS (UVP). Equal amounts of each IVTT protein were added (10 μl of hEXO1-WT) to the reaction mixture, followed by incubation for 2 h at 4°C on a rocking platform. Samples were washed four times with binding buffer, resolved on 7.5% sodium dodecyl sulfate-polyacrylamide gels (Lonza), and visualized using phosphorimaging (Storm 840; GE Healthcare). Band intensities from pull-down experiments were quantified by using software VisionWorksLS.
Yeast two-hybrid assay.
Yeast two-hybrid assays were performed in S. cerevisiae strain Y190 [MATa gal4 gal80 his3 trp1 ade2 ura3 leu2 URA3::GAL1::lacZ LYS2::GAL4(UAS)::HIS3 cyhR]. The yeast two-hybrid plasmid vectors were pGBT9 (2μ pADH1Δ GAL4-BD TRP1 AR; Stratagene) and pAS2ΔΔ (2μ pADH1 GAL4-BD TRP1 AR) (11) for bait constructs and pACT2 (2-μ pADH1Δ GAL4-AD LEU2 AR; Stratagene) for prey constructs (12). Y190 was transformed by a pair of plasmids expressing Gal4-BD-fusion TRP1 (bait)/Gal4-AD-fusion LEU2 (prey), plated onto selective media for 3 to 4 days at 30°C, and tested for β-galactosidase production in an overlay plate assay (12).
3D modeling of the C-terminal region of yeast Mlh1.
The amino acid sequence of the C-terminal region of Escherichia coli MutL (residues 428 to 615) was used as a query to search the nonredundant database (nr70) using PSI-BLAST (1). After four iterations, Pms1 from S. cerevisiae was found with an E-value of 10−24 within 376 homologous sequences. Realignment of these sequences was carried out using MUSCLE (8). In parallel, PSI-BLAST allowed to retrieve about 50 eukaryotic Mlh1 and Pms1 sequences using S. cerevisiae Mlh1 and Pms1 sequences as input. The Mlh1 and Pms1 homologs were independently realigned using MUSCLE. The resulting alignments were used to predict the secondary structures of the C-terminal regions of S. cerevisiae Pms1 and Mlh1 using PSIPRED (31) and to identify evolutionary conserved positions in Mlh1 and Pms1 using CONSURF (24). The Mlh1 sequences were aligned to the Pms1 sequences by matching (i) the secondary structure elements predicted by PSIPRED for Mlh1 and Pms1 and observed in the three-dimensional (3D) structure for MutL (14) and (ii) the conserved hydrophobic residues within these secondary structure elements, as deduced from the Mlh1 and Pms1 multiple alignments. The resulting Mlh1/Pms1 alignment is similar to that proposed by Kosinski et al. (21) (see Fig. S2 in the supplemental material). This alignment was used to build a model of the C-terminal region of Mlh1 (from E528 to R768) on the basis of the X-ray structure of E. coli MutL C-terminal domain with Modeler program (10). The region of yeast Mlh1 from amino acids 483 to 527 present in two-hybrid assays was not modeled since the corresponding region in E. coli was not crystallized (14). This model together with the Mlh1 alignments were submitted to Consurf server version 3.0 (24) in order to obtain a normalized evolutionary rate for each position of the alignment: low rates of divergence correspond to high sequence conservation.
Protein and peptide preparation.
The sequences coding for the C-terminal region of wild-type Mlh1, mutant R547A, and mutant E682A of Mlh1 were cloned into vectors pETM30 (G. Stier, EMBL, Heidelberg, Germany) with a His6-GST tag in the N terminus and a tobacco etch virus (TEV) protease site. Proteins were expressed in E. coli, Rosetta (DE3)/pLysS. Cells were grown at 37°C until reaching an optical density at 600 nm of 1.0 and induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) overnight at 20°C. The cells were lysed by sonication in buffer (50 mM Tris-HCl [pH 8], 300 mM NaCl, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM β-mercaptoethanol), 60 U of Benzonase (Novagen) was added per ml of lysate, and MgCl2 was added to a final concentration of 10 mM. After centrifugation, the supernatant fraction was applied to glutathione-Sepharose FF (GE Healthcare) equilibrated with lysis buffer. The Sepharose was washed with lysis buffer, the quantity of fusion protein bound was estimated by the Bradford assay, and 1% (wt/wt) TEV protease was added directly to the Sepharose resuspended with 15 ml of buffer overnight at 4°C. Mlh1 was collected in the flowthrough, concentrated, and applied to a HiLoad 16/60 Superdex (GE Healthcare) size exclusion chromatography column. The same protocol was used to purify the C-terminal region of human MLH1 (amino acids 486 to 756). The synthetic peptides used in the present study were purchased from Genecust at 95% purity.
Circular dichroism spectroscopy.
The purified C-terminal regions of Mlh1 were dialyzed against buffer (25 mM Tris-SO4 [pH 8], 75 mM NaF, 0.5 mM PMSF) to minimize chloride anion signal. Circular dichroism experiments were performed on a JASCO J-815 spectrometer equipped with a Peltier temperature control unit. Far-UV spectra were recorded between 180 and 250 nm, at 20°C, using 1-mm quartz cells. The spectra were acquired as average of 10 scans, with a scan speed of 50 nm/min and a response time of 1 s. Temperature denaturation curves were recorded between 5 and 95°C, with a temperature increase rate of 1°C/min.
ITC.
The binding between C-terminal regions of Mlh1 and different peptides was measured by using VP-ITC and isothermal titration calorimeters (Microcal, Northampton, MA). Prior to the measurements, all of the solutions were degassed under vacuum for elimination of bubbles. The reaction cell was loaded with 10 μM Mlh1 protein, and the syringe contained 150 to 200 μM peptide solutions. Thirty 10-μl injections of were made at 210-s intervals. To correct for the heat effects of dilution, control experiments were performed at the same concentration of peptides with buffer in the cell. The thermodynamic parameters ΔH (enthalpy change), n (stoichiometry), and Ka (association constant) were obtained by nonlinear least-squares fitting of the experimental data using the single set of independent binding sites model of the Origin software provided with the instrument. The free energy of binding (ΔG) and entropy (ΔS) were obtained by using the classical thermodynamic formulae ΔG = −RT × ln Ka and ΔG = ΔH − TΔS, where R is the gas constant, and T is the absolute temperature in kelvins. The experiments were performed at 303 K. The titrations were carried out in 50 mM phosphate (pH 7.7)-300 mM NaCl-0.5 mM PMSF-10 mM β-mercaptoethanol. Titrations were made with 150 mM NaCl for hMLH1 and peptides derived from EXO1 and BLM.
Spontaneous mutation rates.
Yeast strains were grown at 30°C in YPD or YNBD medium supplemented with the appropriate bases and amino acids. For each strain, 11 independent cultures of 2 ml each were inoculated with about 5 × 102 cells and grown at 30°C for 2 to 3 days. Cell density was measured by plating dilutions on YPD or YNBD plates and counting the colonies after 2 to 3 days at 30°C. The quantification of canavanin-resistant mutants (Canr) was determined after plating aliquots of each culture onto YNBD plates containing 60 mg of l-canavanin (Sigma)/liter. The quantification of Hom+ revertants was determined after plating onto supplemented YNBD without threonine. Colonies were counted after 4 to 5 days at 30°C. All experiments were carried out independently two to five times. The Canr mutation assay detects all kinds of mutation that inactivates the CAN1 gene. The hom3-10 reversion assay detects mutations that revert a +1 frameshift mutation in the HOM3 gene. Mutation rates were determined from the number of Canr colonies by the method of the median (26).
RESULTS
The interaction between the C-terminal region of Mlh1 and proteins containing a MIP-box is conserved in human proteins.
We previously identified a five-amino-acid motif, R-S-K-[Y/F]-F, common to Exo1 and Ntg2, whose serine and the two aromatic residues are essential for interaction with Mlh1 in S. cerevisiae (12). A motif, K-S-K-F-F, was also found in position 1382 of Sgs1, another protein known to interact with Mlh1 in yeast (35). Figure 1A shows that Ser1383, F1385, and F1386 of Sgs1, substituted in alanine in the present study, were also required for interaction with yeast Mlh1. Thus, the ([R/K]-S-K-[Y/F]-F)-motif shared by three proteins—Exo1, Ntg2, and Sgs1—appears as a pivotal element, allowing physical interaction with Mlh1 in S. cerevisiae. We refer to this motif as the MIP-box for the Mlh1 interacting protein box. Sequences analysis revealed that the MIP-box in Exo1 is conserved among most eukaryotes (Fig. 2 and see Fig. S1A in the supplemental material). The MIP-box is not conserved in all Sgs1 homologs, although it is present in different yeasts and metazoans, including human BLM protein (see Fig. S1B in the supplemental material). Interestingly, in the Ntg2 family, the MIP-box is only observed in a subset of yeasts closely related to S. cerevisiae (see Fig. S1C in the supplemental material). The evolutionary conservation of the MIP-box in Exo1 and Sgs1 prompted us to analyze interactions between yeast proteins containing a MIP-box and human MLH1 protein. Figure 1B shows that yeast Sgs1, Exo1, and Ntg2 interact with human MLH1. The MIP-box is critical for these interactions since Ala mutation at conserved Ser, Tyr/Phe, and Phe sites abolishes binding to human MLH1.
FIG. 1.

Sgs1, Exo1, and Ntg2 interact with yeast and human Mlh1 through their MIP-box. Yeast two-hybrid assays were performed using either pGBT9-Ntg2(1-380), pAS2ΔΔ-Exo1(400-702), or pAS2ΔΔ-Sgs1(785-1447) as baits. The MIP-box of Sgs1, Exo1, and Ntg2 proteins were mutated at critical amino acids in the motif (Ser, Tyr/Phe, and Phe) toward Ala on the bait constructs. Preys were pACT2-yMlh1(1-769) or pACT2-hMLH1(1-756). Transformants were tested for the production of β-galactosidase in an overlay plate assay. (A) Two-hybrid assays with yeast Mlh1; (B) two-hybrid assays with human MLH1. A representative patch is shown. Yeast two-hybrid assays with bait alone or prey alone were realized and gave white patches.
FIG. 2.

Histogram indicating sequence conservation of Exo1 MIP-box among eukaryotic sequences. The height of the amino acid symbols within a stack indicates the relative frequency of each residue. The figure was generated with the logo HMM program (42) with 27 Exo1 sequences taken from various eukaryotic phyla. This histogram illustrates the high conservation of the MIP-box motif in a region of Exo1 proteins with little overall conservation (see also Fig. S1A in the supplemental material for sequence alignments).
Human EXO1 interacts with human MLH1 C-terminal region and with MSH2 (38, 41). EXO1 presents a MIP-box (R-S-R-F-F) in a region important for its interaction with hMLH1. GST pull-down assays showed that substitutions on the MIP-box (S504A, F506A, F507A, and FF506AA) significantly weakened the interaction with MLH1 and have no impact on the interaction with MSH2 proteins (Fig. 3). This result suggests that the MIP-box present in EXO1 contribute significantly to the interaction with MLH1, although we could not exclude some additional contributions of other EXO1 regions to the interaction with MLH1.
FIG. 3.
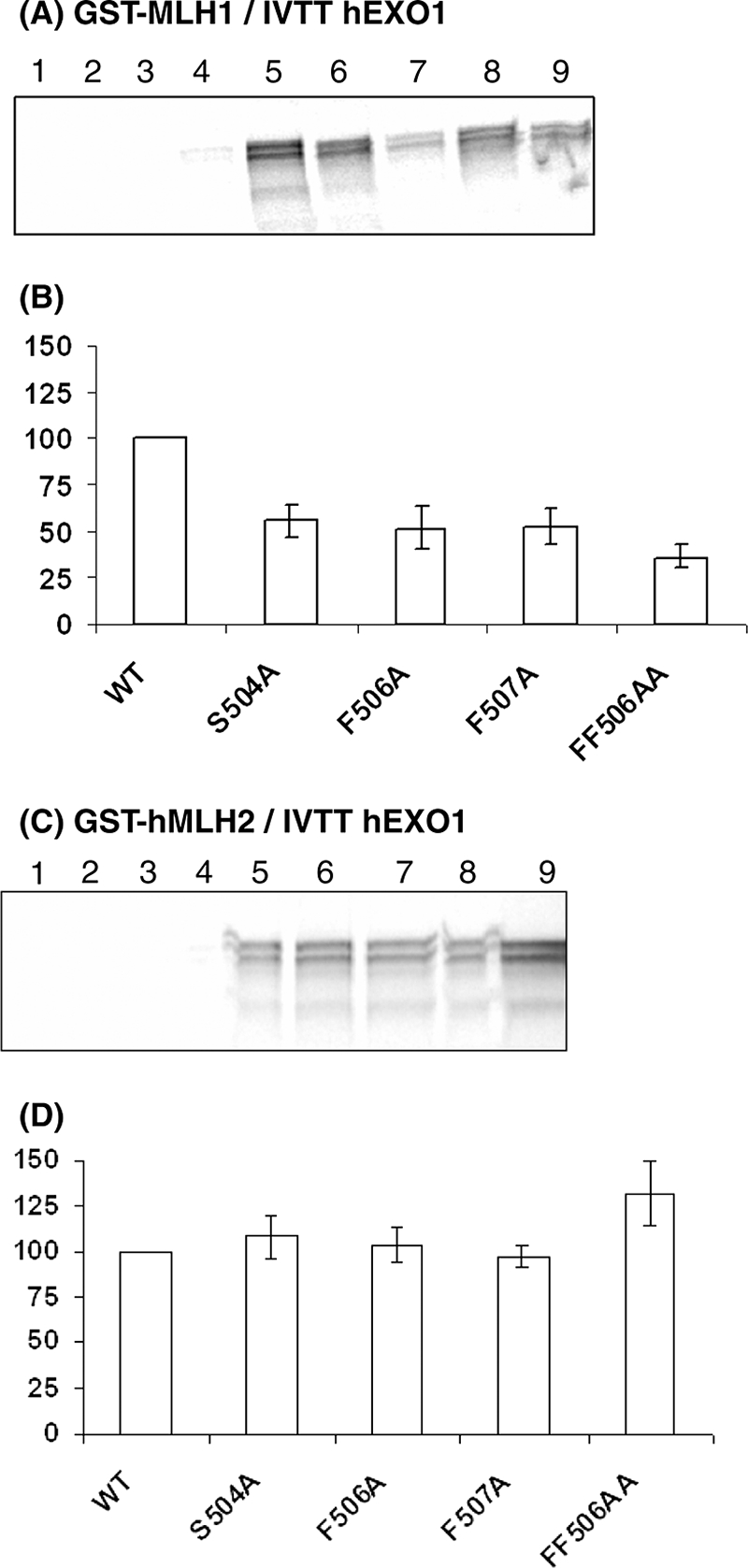
Interaction between hEXO1 MIP-box mutants and hMLH1 or hMSH2. (A) GST pull-down results between hMLH1 and hEXO1. Lane 1, beads and IVTT hEXO1-WT; lane 2, GST and IVTT hEXO1-WT; lane 3, GST-hMLH1 and IVTT from pcDNA3.1A(−); lane 4, GST-hPMS2 and IVTT hEXO1-WT; lane 5, GST-hMLH1 and IVTT hEXO1-WT; lane 6, GST-hMLH1 and IVTT hEXO1-S504A; lane 7, GST-hMLH1 and IVTT hEXO1-F506A; lane 8, GST-hMLH1 and IVTT hEXO1-F507A; lane 9, GST-hMLH1 and IVTT hEXO1-FF506AA. (B) Band intensities were analyzed with software VisionWorksLS, where the band intensity for each mutant protein was determined relative to the intensity of hEXO1-WT. The relative interactions between GST-hMLH1 and hEXO1 MIP-box mutants shown in panel B were determined from four independent experiments. (C) GST pull-down results between hMLH1 and hMSH2. Lane 1, beads and IVTT hEXO1-WT; lane 2, GST and IVTT hEXO1-WT; lane 3, GST-hMSH2 and IVTT from pcDNA3.1A(−); lane 4, GST-hPMS2 and IVTT hEXO1-WT; lane 5, GST-hMSH2 and IVTT hEXO1-WT; lane 6, GST-hMSH2 and IVTT hEXO1-S504A; lane 7, GST-hMSH2 and IVTT hEXO1-F506A; lane 8, GST-hMSH2 and IVTT hEXO1-F507A; lane 9, GST-hMSH2 and IVTT hEXO1-FF506AA. (D) The band intensities in panel C were analyzed with software VisionWorksLS, where the band intensity for each mutant protein was determined relative to the intensity of hEXO1-WT. The relative interaction between GST-hMSH2 and hEXO1 MIP-box mutants shown in panel 3C was determined from two independent experiments, except for data regarding hEXO1-FF506AA, which is based on three independent experiments.
The C-terminal part of the human BLM also interacts with yeast and human Mlh1 (25, 35). BLM possesses a degenerated MIP-box (S-S-H-Y-F) at its C terminus, with the canonical serine being separated from the two aromatic residues by a basic amino acid. The C-terminal part of BLM(1034-1378) interacted with both yeast and human Mlh1 proteins (see Fig. S3 in the supplemental material). The two aromatic residues (Y1325 and F1326) in the MIP-box of BLM were required for interactions with yeast and human Mlh1 proteins. The identification of functional MIP-boxes in yeast Exo1, Sgs1, and Ntg2 and in human EXO1 and BLM proteins and their capacity to bind both human and yeast Mlh1 proteins point to the presence of a highly conserved binding site on the C-terminal region of Mlh1.
Identification of Mlh1 residues involved in the interaction with the MIP-box motif.
To define the limits of the Mlh1 site(s) involved in the interaction with Exo1, Ntg2, and Sgs1, we first performed a deletion mapping study on the Mlh1 fragment from amino acids 483 to 769. Figure 4 shows that the deletion of amino acids from 483 to 527 did not affect the binding to Pms1 but abolished the binding to Exo1 and Ntg2. In contrast, deletion of amino acids from 483 to 559 abolished binding to both classes of partner proteins (Fig. 4). Deletion of amino acids 749 to 769 on the C-terminal side show that all binding is disrupted simultaneously. The possibility to dissociate Pms1 and Exo1/Ntg2 binding using Mlh1(528-769) suggests two independent binding sites on the C-terminal region of Mlh1. It should be noted that none of the deletions used in the present study was able to dissociate the binding sites for Exo1 and Ntg2. Thus, we propose that the C-terminal region of Mlh1 exhibits a binding site S1 for MutL homologs and a binding site S2 for MIP-box proteins.
FIG. 4.

Deletion mapping of yeast Mlh1 protein domains required for binding of Exo1, Ntg2, or Pms1. Yeast two-hybrid assays were performed with either pGBT9-Ntg2(1-380), pAS2ΔΔ-Exo1(400-702), or pAS2ΔΔ-Pms1(661-873) as baits. Preys are pACT2-Mlh1(483-769) or deletion mutants as indicated. The results of two-hybrid assays are reported with “+” for blue colonies and “−” for white colonies after 24 h at 30°C. Expression of the various truncated Mlh1 proteins was monitored using an antibody against HA epitope of the GAL4-AD-fusion proteins. All proteins were similarly expressed (data not shown).
To identify critical amino acids involved in the formation of the S2 site, we undertook an extensive site-directed mutagenesis analysis of the conserved positions in Mlh1 C-terminal region. We monitored with two-hybrid assays the remaining interaction with Pms1, Exo1, and Ntg2. Since the interaction Mhl1/MIP box is conserved in yeast and humans, we hypothesized that residues of Mlh1 required for a functional site S2 would be conserved among eukaryotic sequences. Twenty-eight point mutated variants of Mlh1(483-769) were tested by two-hybrid assay for their interaction with Ntg2, Exo1, and Pms1 (Table 2). For all of the mutants, at least one type of interaction was observed, suggesting that these mutations did not significantly impair the production nor disrupt the 3D structure of the C-terminal region of Mlh1 (Table 2). Seven variants on positions S513, F578, M626, E629, Y630, E682, and I689 exhibited no detectable two-hybrid signal with Exo1 and/or Ntg2 but still normally responded in assays with Pms1 (Table 2). Three additional substitutions—D577A, N580A, and Y631A—exhibited weak two-hybrid signal with Exo1 and/or Ntg2 and wild-type signal with Pms1 (Table 2). For comparison, we also constructed a point mutation that specifically impaired interactions at site S1. We generated the R547A mutation, since R547 is part of a cluster of five charged amino acids whose simultaneous substitution was reported to disrupt the interaction with Pms1 (3). Our results showed that Mlh1-R547A efficiently interacted with Exo1 and Ntg2 but not with Pms1 (Table 2). The positions critical for the interactions with Exo1 and Ntg2 are distributed within four separate Mlh1 sequence regions (Fig. 5A). These positions are remote from residues proposed as involved in the heterodimerization site S1 (Fig. 5A, magenta star) (21).
TABLE 2.
Site-directed mutagenesis on the C-terminal region of yeast Mlh1 and two-hybrid assays for interaction with Ntg2, Exo1, and Pms1
| Mlh1a | Interactionb
|
||
|---|---|---|---|
| Ntg2 | Exo1 | Pms1 | |
| WT | + | + | + |
| S513A | − | − | + |
| Y538A | + | + | + |
| R547A | + | + | − |
| Q552A | + | + | + |
| F558A | + | + | + |
| C566S | + | + | + |
| Y571A | + | ND | + |
| Q572A | + | ND | + |
| I573E | + | + | + |
| D577A | +/− | ND | + |
| F578A | − | − | + |
| N580A | +/− | ND | + |
| F581A | + | ND | + |
| M626A | − | ND | + |
| E629A | − | ND | + |
| Y630A | − | − | + |
| Y631A | +/− | ND | + |
| S632A | + | ND | + |
| L655E | + | + | + |
| E682A | − | − | + |
| C685A | + | + | + |
| I689A | − | ND | + |
| Y697A | + | ND | + |
| H733A | + | + | + |
| K740A | + | + | + |
| P746A | + | + | + |
| E755A | + | + | + |
| C769S | + | + | + |
pACT2-Mlh1(483-769) (either wild type [WT] or mutant as indicated) was used as the prey construct.
For Ntg2, Exo1, and Pms1, pGBT9-Ntg2(1-380), pAS2ΔΔ-Exo1(400-702), and pAS2ΔΔ-Pms1(661-873), respectively, were used as the bait constructs. Results are expressed as follows: +, blue colonies; +/−, light-blue colonies; −, white colonies; ND, not determined. The expression of the Mlh1 variants (R547A, F578A, Y630A, and E682A) was monitored by using an antibody against yeast Mlh1(483-769) and proved to be similarly expressed (data not shown).
FIG. 5.

Localization of heterodimerization site S1 and Exo1/Ntg2 interaction site S2 in the C-terminal region of Mlh1 from S. cerevisiae. (A) The 28 positions mutated on Mlh1 and tested for interaction with Exo1/Ntg2 are schematically represented over the Mlh1 sequence. Mutations that disrupt or weaken Mlh1 interactions with Exo1/Ntg2 and not with Pms1 are, respectively, in blue or light blue. Mutation R547A that disrupts Mlh1 interaction with Pms1 and not with Exo1/Ntg2 is in magenta. Mutations with no effect on both interactions are in gray. Magenta stars indicate residues proposed to be involved in heterodimerization site S1 (21). Dashed lines represent MutL regions that are not crystallized (14). (B) Surface representation of the Mlh1 model colored according to site-directed mutagenesis results with the same color code as in panel A. (C) Surface representation of the Mlh1 model colored according to the sequence conservation from low (red) to high (blue) conservation. This representation shows two conserved patches that coincide with regions proposed for heterodimerization site S1 (21) and Exo1/Ntg2 interaction site S2 (the present study). (See Fig. S2 in the supplemental material for ribbon representations of the model.)
A 3D model of the C-terminal region of Mlh1 was built with the program Modeler (10) from the alignment of S. cerevisiae Mlh1 C-terminal sequence with different eukaryotes and E. coli sequences (see Fig. S1D and S2 in the supplemental material) and from the X-ray structure of E. coli MutL (14). Interestingly, 9 of 10 positions that disrupt or weaken the two-hybrid signal with Exo1 and Ntg2 are clustering in a specific region at the surface of the model. These positions are contiguous and define the so-called binding site S2 (Fig. 5B). The tenth position, S513 corresponds to an E. coli position located upstream the crystallized MutL C-terminal region (N terminus of our model is E528, Fig. 5A) (14). It plays an important role in agreement with our results obtained with deletion constructs of Mlh1 that underline the critical role of the region spanning residues 483 to 518 for the interaction with Exo1 and Ntg2 (Fig. 4).
Figure 5C shows the distribution of conserved residues on the surface of Mlh1 model. Interestingly, site S2 of Mlh1 coincides with one of the two most conserved patches proposed by Consurf program (24). Remarkably, sequence alignment of 30 Mlh1 eukaryotic sequences show that all residues critical for site S2 are highly conserved or changed for conservative amino acid substitutions (see Fig. S1D in the supplemental material). The second conserved patch corresponds to the N terminus region (positions 537 to 564) and the C terminus region (positions 752 to 766) proposed to be involved in Mlh1 heterodimerization (Fig. 5A) (21). Taken together, these data point to a structure-function conservation of site S2 among eukaryotic Mlh1 proteins. To challenge this hypothesis, we generated two mutants of the human MLH1 (Y625A and E669A), the human counterparts of essential yeast positions (Y630 and E682), respectively. Table 3 shows that hMLH1-Y625A and hMLH1-E669A did not interact with Ntg2, Sgs1 and BLM and interacted weakly with Exo1, although they efficiently bound human PMS2. In conclusion, our results delineate a new, highly conserved site S2 in the C-terminal region of Mlh1 essential for interactions with Exo1, Ntg2, Sgs1, or BLM that is distinct from the site S1 that mediates interactions with Pms1 or PMS2.
TABLE 3.
Interspecies conservation of interactions at site S2 of Mlh1
| Construct | Interactiona
|
|||
|---|---|---|---|---|
| yMlh1 (WT) | hMLH1 (WT) | hMLH1 (Y625A) | hMLH1 (E669A) | |
| pGBT9-Ntg2(1-380) | + | + | − | − |
| pAS2-Exo1(400-702) | + | + | +/− | +/− |
| pAS2-Sgs1(785-1447) | + | + | − | − |
| pAS2-Pms1(661-873) | + | − | − | − |
| pAS2-hBLM(1034-1378) | + | + | − | − |
| pGBT9-hPMS2(637-862) | ND | + | + | + |
For yMlh1(WT) and hMLH1(WT), full-length pACT2-yMlh1(1-769) or pACT2-hMLH1(1-756) were use as prey partner constructs in two-hybrid assays. For hMLH1(Y625A) and hMLH1(E669A), full-length pACT2-hMLH1(1-756) with an alanine substitution at Y625 or E669, respectively, the counterparts of the yeast Y630 or E682 mutations that are essential for interactions at S2, were used. Bait partner constructs are indicated. Results are expressed as follows: +, blue; +/−, light blue; −, white; ND, not done.
The C-terminal region of yeast and human Mlh1 interacts directly and specifically with the Exo1/Ntg2/Sgs1 and EXO1/BLM peptides, respectively, containing the MIP-box.
To evaluate the strength of the interactions between the different proteins that target Mlh1 at site S2, we characterized the relative affinity of their MIP-box motif at Mlh1 site S2 with direct biophysical methods. We produced the C-terminal regions of yeast Mlh1 (amino acids 485 to 769) and of human MLH1 (amino acids 486 to 756). We also produced two mutants of the C-terminal region of yeast Mlh1: a mutant from site 2, Mlh1-E682A, that presents no interaction with Exo1 or Ntg2 (Table 2) and a mutant from site 1, Mlh1-R547A, that interacts with Exo1 or Ntg2 but not with Pms1. These fragments were purified to homogeneity, and their secondary structure content and thermal stability were analyzed by circular dichroism (see Fig. S4 in the supplemental material). The wild-type and mutant Mlh1 fragments present spectra with two minima at 208 and 222 nm that are characteristic of α+β proteins in agreement with the X-ray structure of E. coli MutL C-terminal region used to build our model (14). The stability of the variant Mlh1-E682A was estimated by thermal denaturation; it presented a cooperative denaturation profile and a melting temperature close that of the wild-type fragment. These results indicate that the wild-type and variants Mlh1 fragments used in the present study have an overall correct 3D structure, although we cannot exclude for the variants some local rearrangements introduced by the mutation.
To have access to the thermodynamic parameters that characterize interactions at site S2, we performed ITC studies. We measured the interactions of purified wild-type and mutant versions of yeast Mlh1 with peptides (10- to 14-mer) derived from Exo1, Ntg2, Sgs1, and BLM proteins. All of the peptides used contain a MIP-box either wild type or modified (Table 4). The binding reactions with peptides Exo1, Ntg2, Sgs1, and BLM gave exothermic heat exchange (negative enthalpy). Integrated thermograms were fitted to a one-site binding model and gave similar micromolar dissociation constants (Kd = 1/Ka) for pExo1, pNtg2, and pSgs1 peptides between 8.1 and 9.3 μM (Table 4 and Fig. 6A to C). We analyzed the interaction between the C-terminal region of hMLH1 with peptides derived from human EXO1 and BLM proteins and also observed micromolar dissociation constants for between 9.0 and 17.4 μM (Table 4, Fig. 6D, and see Fig. S5F in the supplemental material).
TABLE 4.
ITC analysis of yeast Mlh1 C-terminal region binding to Ntg2, Exo1, Sgs1, EXO1, and BLM peptides
| Proteina | Peptide | Sequenceb | ΔH° (kcal/mol) | Ka (105 M−1) | Kd (mM) | ΔG° (kcal/mol) | −TΔS° (kcal/mol) |
|---|---|---|---|---|---|---|---|
| Mlh1 WT | pNtg2 | 20VEVRSKYFKKNER | −20.5 ± 1.5 | 1.11 ± 0.08 | 9.0 ± 0.6 | −7.0 | +13.5 |
| Mlh1 WT | pExo1 | 441KDTRSKFFNKPS | −2.6 ± 0.7 | 1.07 ± 0.34 | 9.3 ± 2.9 | −7.0 | −4.4 |
| Mlh1 WT | pSgs1 | 1377QSTGTKSKFFGAN | −6.5 ± 0.9 | 1.24 ± 0.21 | 8.1 ± 1.4 | −7.05 | −0.55 |
| Mlh1 WT | pBLM | 1317VSSHYFASKT | −2.2 ± 0.2 | 1.20 ± 0.14 | 8.3 ± 1.0 | −7.05 | −4.85 |
| hMLH1 | pEXO1 | 498VVPGTRSRFFCSSDST | −8.5 ± 0.2 | 0.57 ± 0.03 | 17.4 ± 0.9 | −6.6 | +1.9 |
| hMLH1* | pBLM | 1317VSSHYFASKT | −21.0 ± 1.3 | 1.11 ± 0.07 | 9.0 ± 1.2 | −7.0 | +14.0 |
| Mlh1 WT (LS)* | pNtg2 | −30.5 ± 0.4 | 2.9 ± 0.11 | 3.4 ± 0.1 | −7.55 | +22.95 | |
| Mlh1 R547A* | pNtg2 | −18.8 ± 1.5 | 1.9 ± 0.20 | 5.3 ± 0.5 | −7.3 | +11.5 | |
| Mlh1 E682A | pNtg2 | NIc | NI | NI | NI | NI | |
| Mlh1 WT | pExo1(FF447AA) | KDTRSKAANKPS | NI | NI | NI | NI | NI |
| Mlh1 WT* | pNtg2(S24A) | VEVRAKYFKKNER | NI | NI | NI | NI | NI |
| Mlh1 WT* | pNtg2(S24[P]) | VEVRS(P)KYFKKNER | NI | NI | NI | NI | NI |
| Mlh1 WT* | pNtg2(Y26A) | VEVRSKAFKKNER | NI | NI | NI | NI | NI |
*, thermograms for these experiments are reported in Fig. S4 in the supplemental material. WT, wild type.
Numbers before peptide sequences correspond to the position in the protein sequence of the first amino acid of the peptide. The MIP-box is underscored. Modified amino acids are in boldface.
No interaction (NI) indicates weak and/or constant signal under the condition used (15 μM protein and 150 μM peptide).
FIG. 6.

Physical interactions between the C-terminal region of yeast and human Mlh1 and different peptides containing a MIP-box. The thermograms and binding isotherms of the calorimetric titrations of wild-type or mutant Mlh1 by peptides at 303 K are presented. The corresponding thermodynamic parameters are reported in Table 4. (A) Mlh1(WT) and pNtg2; (B) Mlh1(WT) and pSgs1; (C) Mlh1(WT) and pExo1; (D) hMLH1(WT) and pEXO1; (E) Mlh1(WT) with peptide Exo1 mutant on the MIP-box, pExo1 (FF447AA); (F) Mlh1 mutant on the critical position E682 of site S2 and pNtg2.
The interaction for Ntg2 peptide was also measured in the presence of lower salt (100 mM NaCl instead of 300 mM for all other interactions). In this case, the affinity was significantly enhanced, suggesting the contribution of electrostatic interaction to the binding (Table 4). We also analyzed by ITC the interactions with a peptide substituted at both aromatics in the MIP-box of Exo1 (FF447AA). This peptide showed no interaction, in agreement with a recent work that revealed that this double mutation on whole Exo1 protein was deleterious for the interaction with Mlh1 (49) (Table 4 and Fig. 6E). These results highlight the specificity of the interactions measured with the MIP-box peptides and confirm the major role suggested by two-hybrid assays for residues F447 and F448 in the Exo1-Mlh1 interaction (12). A similar result was obtained with two Ntg2 peptides containing Ala substitutions in the MIP-box: peptides S24A and Y26A. The peptide variants show no interaction (Table 4). A peptide containing a phosphoryl group on S24 was tested by ITC, and neither bind the C-terminal region of Mlh1 (Table 4). These results confirm by a direct method the critical role of the serine and of both aromatic residues in the MIP-box in Exo1-Mlh1 or Ntg2-Mlh1 interactions and highlight the conservation of this interaction in humans.
Finally, two Mlh1 variants (R547A and E682A) were purified and titrated with the Ntg2 peptide. Mlh1-R547A interacts with Exo1 or Ntg2 but not with Pms1 in two-hybrid assay (Table 2). ITC analysis shows that Mlh1-R547A interacts with Ntg2, as did the wild type (Table 4). On the other hand, Mlh1-E682A, which does not interact with Exo1 or Ntg2 in the two-hybrid assay, also does not interact with the Ntg2 peptide in the ITC assay (Table 4 and Fig. 6F). In conclusion, ITC results show a direct interaction between the C-terminal region of Mlh1 and peptides containing the MIP-box with a similar affinity in the micromolar range. Calorimetry data also confirm the role of amino acids from the MIP-box and the role of Mlh1 position E682 as an essential component of the site S2, as proposed by two-hybrid and modeling analysis.
Site S2 impacts on MMR-dependent mutation avoidance.
The evolutionary conservation of interactions between Mlh1 and proteins containing a MIP-box suggests important biological function(s). Since Mlh1 is an essential component of the MMR-dependent mutation avoidance process, the alteration of interaction between Mlh1 and MIP-box partner proteins could impact on mutation rates. Spontaneous mutation rates were determined by using two assays: (i) the Canr forward mutation assay and (ii) the hom3-10 reversion (Hom+) assay. Hom+ essentially detects a (−1) frameshift in a run of seven A-T base pairs; therefore, it can be used as an indicator of MMR-dependent mutagenesis (47, 49). To investigate the impact of mutations at site S2, we could measure mutation rates in mlh1Δ cells hosting plasmids that express wild-type and mutant versions of Mlh1. However, the mutator phenotype caused by nonphysiological levels of wild-type Mlh1 level dictates the use of chromosomal mutations at the MLH1 locus to measure the impact of interactions at S2 on the mutation rates in S. cerevisiae (43, 44). Thus, we generated two mutants of Mlh1, mlh1-R547A and mlh1-E682A, which are affected in their binding properties at sites S1 and S2, respectively. The results show that mlh1-R547A exhibited enhanced mutation rates for both Canr (2.8-fold) and Hom+ (30-fold) compared to the wild type. This result was expected since the R547A mutation affects the interaction between Mlh1 and Pms1 to form MutLα, which is required for functional MMR-dependent mutation avoidance.
On the other hand, mlh1-E682A exhibited only a weakly enhanced mutation rate for Hom+ (5.3-fold) compared to the wild type, like an exo1Δ mutant (Table 5). However, exo1Δ but not mlh1-E682A presented significant increase in Canr events (6.7-fold), which is primarily due to the inactivation of a subpathway in the course of postreplication repair (47, 49). We also measured mutation rates in mlh1-R547A exo1Δ and mlh1-E682A exo1Δ double mutants. The results show that mlh1-R547A synergistically interacted with exo1Δ, yielding high Canr and Hom+ mutation rates, whereas mlh1-E682A did not (Table 5). These results clearly show that mlh1-R547A and mlh1-E682A delineate two different classes of mutants of MLH1 in S. cerevisiae. These two classes differ in their capacity to synergistically interact with exo1Δ and in their capacity to physically interact with Exo1 or Pms1 proteins.
TABLE 5.
Spontaneous mutation rates in mlh1 mutants affected in interactions at the S1 and S2 sites in S. cerevisiaea
| Relevant genotype | Canr mutants
|
Hom+ revertants
|
||
|---|---|---|---|---|
| No. of mutations/107 (95% CI) | Ratio | No. of mutations/107 (95% CI) | Ratio | |
| Wild type | 1.6 (1.2-2.1) | 1.0 | 0.08 (0.07-0.12) | 1.0 |
| mlh1-R547A | 4.4 (3.2-7.6) | 2.8 | 2.40 (1.93-4.22) | 30.0 |
| mlh1-R547A exo1Δ | 34.7 (21.6-73.8) | 21.7 | 29.60* (17.21-44.86) | 370 |
| mlh1-E682A | 2.2 (1.4-2.8) | 1.4 | 0.42 (0.25-0.51) | 5.3 |
| mlh1-E682A exo1Δ | 8.3 (5.6-11.1) | 5.2 | 0.51 (0.45-0.89) | 6.4 |
| pms1-G97A | 2.6 (2.1-3.3) | 1.6 | 1.2 (0.8-1.6) | 15 |
| pms1-G97A exo1Δ | 15.8 (10.6-22.8) | 9.9 | 28.5* (19.0-38.1) | 360 |
| pms1-G97A mlh1-E682A | 6.4 (4.5-10.1) | 4.0 | 13.9* (8.8-18.1) | 169 |
| exo1Δ | 10.7 (8.1-11.7) | 6.7 | 0.40 (0.32-0.53) | 5.0 |
| mlh1Δ | 34.3 (26.6-44.3) | 21.4 | 49.28 (40.36-62.29) | 616 |
Mutation rates per cell division were determined from the number of Canr mutants and Hom+ revertants by the method of the median (26). Each value represents ≥22 independent cultures. The numbers in parentheses indicate the low and high values for the 95% confidence interval (CI) for each rate. The ratios represent the fold induction relative to the wild-type strain. Asterisks indicate the occurrence of synergistic interactions for Hom+ revertants (MMR-dependent mutagenesis).
Our results suggest that mlh1-E682A mimics a hypomorphic allele of Exo1 at least partially deficient in its MMR-dependent mutation avoidance function. In S. cerevisiae, the impact of Exo1 on MMR-dependent mutagenesis is more readily appreciated in combination with hypomorphic alleles of pms1 or mlh1 (2, 49). Therefore, our hypothesis predicts a synergistic interaction between mlh1-E682A and a hypomorphic allele of pms1 for MMR-dependent mutagenesis. We chose to use pms1-G97A, which is mutated in the ATP binding motif of Pms1 and that exhibits a moderate (15-fold) increase in Hom+ mutagenesis (50, 51) (Table 5). (Amino acids of Pms1 are numbered according to the SGD database [http://db.yeastgenome.org]; in previous studies, the Pms1 G97A mutant was G128A due to a wrong estimation of the start codon position.) Table 5 confirms that pms1-G97A synergized for Hom+ mutagenesis (360-fold) when combined with exo1Δ (51). Table 5 also shows that pms1-G97A also synergized for Hom+ (169-fold) when combined with mlh1-E682A. Importantly, the Hom+ mutation rate in the pms1-G97A mlh1-E682A double mutant was significantly lower than that in the pms1-G97A exo1Δ mutant. This last result indicates that the suppression of the physical interaction between Mlh1 and Exo1 in the mlh1-E682A mutant significantly reduces, but does not abolish, the impact of Exo1 on MMR-dependent mutation avoidance. Therefore, the properties of mlh1-E682A strongly suggest that one of the biological functions of S2 is to recruit Exo1 to optimize MMR-dependent mutation avoidance.
The capacity of Mlh1 to bind multiple partners at sites S1 and S2 may result in the formation of a variety of binary and ternary complexes, whose biological impact must be diverse. In order to modify the equilibrium between Mlh1-containing complexes, we overproduced Ntg2 in yeast cells that express either wild-type or mutant Mlh1. Table 6 shows that the overexpression of Ntg2 moderately enhanced Canr and Hom+ mutation rates in a wild-type background (Table 6). In an mlh1-R547A background, Ntg2 overexpression greatly enhanced Canr (10-fold) and Hom+ (523-fold) mutation rates. In contrast, the overexpression of Ntg2 did not modify spontaneous mutation rates in the mlh1-E682A context (Table 6). These results suggest that through an overexpression of Ntg2, the site S2 is saturated by Ntg2 preventing the formation of Mlh1-Exo1 complex on this site. These results are in agreement with the previous hypothesis that links enhanced MMR-dependent mutagenesis and disruption of the Exo1 recruitment process at S2 site in combination with hypomorphic alleles of pms1 or mlh1 (Table 5).
TABLE 6.
Impact of Ntg2 protein overexpression on spontaneous mutation rates in mlh1 mutants affected in interaction at sites S1 or S2 in S. cerevisiaea
| Relevant genotype | Canr mutants
|
Hom+ revertants
|
||
|---|---|---|---|---|
| No. of mutations/107 (95% CI) | Ratio | No. of mutations/107 (95% CI) | Ratio | |
| WT/pAS2 | 2.7 (1.7-3.5) | 1.0 | 0.09 (0.06-0.13) | 1.0 |
| WT/pAS2-NTG2 | 4.6 (2.9-6.3) | 1.7 | 4.3* (3.2-5.6) | 47.8 |
| mlh1-R547A/pAS2 | 4.3 (3.5-5.0) | 1.6 | 15.1 (13.7-19.5) | 167.8 |
| mlh1-R547A/pAS2-NTG2 | 26.9 (18.8-33.6) | 10.0 | 47.1* (38.8-54.1) | 523.3 |
| mlh1-E682A/pAS2 | 1.8 (1.4-4.1) | 0.7 | 0.5 (0.3-0.6) | 5.6 |
| mlh1-E682A/pAS2-NTG2 | 2.0 (1.5-3.8) | 0.7 | 0.4 (0.2-0.7) | 4.4 |
| mlh1D/pAS2 | 53.6 (42.8-65.4) | 19.9 | 82.6 (65.4-102.3) | 917.8 |
Mutation rates per cell division were determined from the number of Canr mutants and Hom+ revertants by the method of the median (26). Each value represents ≥22 independent cultures. The numbers in parentheses indicate the low and high values for the 95% confidence interval (CI) for each rate. The ratios represent the fold induction relative to the wild-type (WT) strain. Asterisks indicate the occurrence of synergistic interactions for Hom+ revertants (MMR-dependent mutagenesis).
DISCUSSION
The Mlh1 protein interacts with numerous partners to form complexes presumably involved in a variety of biological transactions (12, 17, 32, 35, 51, 52). Interactions involve both the N-terminal and the C-terminal parts of Mlh1. In the present study, we focused on interactions that occur at the C-terminal part of Mlh1 that spans amino acids 483 to 769 in S. cerevisiae. At least two independent binding sites are present in the C-terminal region of the Mlh1 protein: (i) site S1, which binds Pms1, Mlh2, or Mlh3 and (ii) site S2, characterized in the present study, that binds Exo1, Ntg2, and Sgs1. Interactions of Exo1, Ntg2, and Sgs1 with yeast and human Mlh1 at site S2 involve a common motif ([R/K]-S-K-[Y/F]-F). This motif, which we named the MIP-box, is highly conserved in the C-terminal part of most eukaryotic homologs of Exo1 in a region with little overall conservation (Fig. 2). Multiple sequence alignments of Sgs1 and Ntg2 underline that the MIP-box is less conserved among eukaryotic homologs of these two proteins (see Fig. S1B and C in the supplemental material). We found this motif in the C-terminal region of numerous yeast and metazoan Sgs1 homologs. Interactions assays realized in the present study between human helicase BLM, a homolog of Sgs1, and yeast and human Mlh1 show that the MIP-box present in BLM is required for its interaction with Mlh1. In Ntg2 protein, the MIP-box is located in the N-terminal region of the protein and is not conserved among eukaryotic members of Endo III family, whereas the catalytic site and the DNA-binding motif are highly conserved. Only four yeasts, closely related to S. cerevisiae, contain a MIP-box surrounded by nonconserved positions. Some of these yeasts have diverged after a common genome duplication event and may have acquired the MIP-box for a function that remains to be elucidated. Recently, Tap-Tag and immunoaffinity approaches have identified new partners for human MLH1 (6, 36). Some of these proteins present a potential MIP-box in their sequence that could contribute to their interaction with Mlh1. For example, FancJ has an SRHF motif in position 105 inside the region from amino acids 1 to 145 required for its interaction with MLH1 (36), and angiomentin has an SQYF motif in its N-terminal region in position 116 (6). It will be interesting to evaluate whether these motifs are functional and thus are critical for their interaction with MLH1.
Characteristics of the MIP-box are reminiscent of the properties reported for another well-characterized motif, the PIP-box (named for the PCNA interacting protein) (33). It is also constituted of a small number of amino acids comprising two consecutive aromatics; the motif is “Qxxhxaa,” with “h” representing a hydrophobic residue and “a” representing an aromatic residue. The PIP-box mediates PCNA interaction with various partners, and binding of these factors to PCNA is often competitive (33). The common features observed for the MIP-box and PIP-box may correspond to properties that facilitate interactions with multiple partners (20).
In the present study, we characterized the S2 binding site of Mlh1 through an approach coupling two-hybrid assay, site-directed mutagenesis, molecular modeling, and biochemical and biophysical methods. We identified 10 amino acids important for the interaction with Exo1 and Ntg2 but not for the interaction with Pms1. These positions are distributed in four regions of Mlh1 C-terminal sequence (positions 483 to 769) (Fig. 5A and see Fig. S1D and 2B in the supplemental material). (i) A first position, S513, conserved in 29 over 30 eukaryotic sequences aligned, is located in a region (positions 483 to 518) shown to be important by two-hybrid assay (Fig. 4). The first position observed in the X-ray structure of E. coli MutL C-terminal region (14) corresponds to position E528 in the yeast Mlh1 model. The distance between E528 and site S2 (23 Å) suggests that the S513 position could be located in close proximity to site S2 since a 14-amino-acid linker separates these two positions. (ii) Next, positions D577, F578, and N580 would be located according to the model at the hinge between the external and internal domains in the E. coli MutL structure. The model suggests that these positions are exposed on the surface Mlh1 (Fig. 5B). (iii) Four positions—M626, E629, Y630, and Y631—are located in a region predicted as a helix in the middle of the internal domain of MutL C-terminal region. In our model, the E629 residue is solvent exposed and M626, Y630, and Y631 are more buried. (iv) Finally, E682 and I689 are also located in the internal domain and in a region predicted as a helix with E682 exposed at the surface of Mlh1. From our model, we conclude that the positions identified define the structurally consistent site S2, which is composed of a contiguous patch on the surface of Mlh1 with two aromatic (F578 and Y630) and two hydrophobic residues (M626 and I689) that are good candidates to interact with the two aromatic amino acids of the MIP-box. E629 and E682 residues could contribute to electrostatic interactions at the interface, in agreement with the ITC measurements that showed salt-dependent interaction probably accounted for an electrostatic contribution (Table 4). Interestingly, the model reveals two main conserved patches on the surface of Mlh1 (Fig. 5C). In this model, site S2 is, apart from the heterodimerization region (S1), proposed from the X-ray structure of E. coli MutL homodimer and biochemical studies (21). Site S1 was proposed to be located in the external domain of E. coli structure and to involve positions inside two segments in the N terminus (positions 537 to 564) and C terminus (positions 752 to 766) of the Mlh1 C-terminal region (Fig. 5A). These segments are apart from the 10 positions identified in the present study for site S2. The separate positions of sites S1 and S2 are also consistent with the possibility of making a ternary complex with these proteins as reported for Exo1, Mlh1, and Pms1 (51). Interestingly, position F578 from S2 site was also proposed to be involved, with the two other Mlh1 positions Q574 and L576, in the interaction between S. cerevisiae Mlh1 and PCNA (27). Substitution of the QLF motif significantly reduces the interaction with PCNA and causes a strong mutator phenotype. This result suggests that PCNA may interact in a competitive manner with protein containing an MIP-box.
To investigate the biological impact of interactions at sites S1 and S2, we isolated mutants of Mlh1 specifically affected in either of these two sites. We measured the impact of these mutations upon one of the functions that requires Mlh1 in S. cerevisiae, the mitotic MMR-dependent mutation avoidance pathway. First, we isolated mlh1-R547A that specifically impairs interactions between Mlh1 and Pms1 at site S1. This mutant exhibits significantly enhanced mutation rates, Canr and Hom+, and synergizes with exo1Δ. Thus, mlh1-R547A behaves in the same way as an already-described class of mutants such as mlh1-E31A or mlh1-R547K (2, 49). However, mlh1-R547A provides novel information, since it is the first mutant of that class where a measured alteration in the physical interaction between Mlh1 and Pms1 is associated with an MMR defect. Second, we isolated mlh1-E682A that is the prototype of a novel class of Mlh1 mutants deficient in their capacity to bind Mlh1 partners containing an MIP-box. Our results showed that mlh1-E682A mutants, like exo1Δ mutants, exhibited a small deficiency in MMR-dependent mutation avoidance. Furthermore, mlh1-E682A did not present synergy with exo1Δ for MMR-dependent mutagenesis. Finally, the combination of mlh1-E682A with pms1-G97A, a hypomorphic allele of pms1, resulted in a synergistic increase in Hom+ mutagenesis, as in a pms1-G97A exo1Δ double mutant (Table 5). Taken together, these results lead us to propose that mlh1-E682A behaves as a hypomorphic allele of Exo1 for MMR-dependent mutagenesis. This hypothesis is in agreement with the fact that the contribution of Exo1 to MMR in yeast is modest in physiological conditions; however, it becomes extremely important in pathological conditions such as the alteration of the 3′ to 5′ proofreading activity of DNA polymerase δ or ɛ (48) or the functional alteration of MMR compounds such as Mlh1, Pms1, Msh2, Msh6, or PCNA (2, 51). According to this hypothesis, mutations affecting site S2 in Mlh1 should be isolated by using weak pms1 mutators and screening for hypermutators. Indeed, using pms1-A99V, two mutations that affect the C terminus of Mlh1 (mlh1-L511F and mlh1-M623I) were isolated, and these positions are close to residues critical for site S2 (S513 and M626, respectively) (2). Moreover, the exo1-FF447AA allele, with alanine substitutions on the two aromatic residues of the MIP-box, is partially defective in the MMR-dependent mutation avoidance in the presence of the hypomorphic alleles of pms1 or mlh1 (49). In conclusion, we propose that one of the biological roles of Mlh1 site S2 is to allow the physical association with Exo1 and to optimize MMR-dependent mutation avoidance. One must note that the impact of mutations such as mlh1-E682A becomes dramatic in situations that weaken or overload MMR in S. cerevisiae.
In a recent functional analysis, 101 human MLH1 variants that covered most of HNPCC missense mutations were examined for dominant mutator effects and in an in vitro MMR assay (46). We identified six MLH1 variants that are close to yeast site S2 and to human site S2 positions Y625 and E669 shown to be critical for interaction with BLM. These variants are V506A (close to yeast S513 position from site S2), I565F and L574P (close to yF578), K618T and L622H (close to yM626), and L676R (corresponds to yI689) (see Fig. S1D in the supplemental material). It will be interesting to determine whether these variants are affected in their interactions with EXO1 and/or BLM proteins.
In higher eukaryotes, the biological impact of Exo1 is much more pronounced than in yeast, since Exo1−/− mice show major defects in MMR with an increase in the mutation rate at the hprt locus, microsatellite instability, increased cancer susceptibility, and male and female sterility (54). Since the interactions between Mlh1 and Exo1 described here are evolutionarily conserved, the impact of mutations that affect site S2 may have major consequences in mice or humans.
Supplementary Material
Acknowledgments
We thank Carine Tellier and Floriana Londino for technical assistance for this project and Gunter Stier (EMBL, Heidelberg, Germany) for the pETM30 and TEV protease vectors. We thank Lionel Gellon, Pascale Bertrand, Josef Jiricny, and Mike Liskay for biological materials and discussions. We thank Xavier Veaute and Raphael Guerois for careful reading of the manuscript.
We acknowledge the support of the Centre National de la Recherche Scientifique and the Commissariat à l'Energie Atomique and the Association pour la Recherche sur le Cancer (contract 3480 to S.B.).
Footnotes
Published ahead of print on 17 November 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amin, N. S., M. N. Nguyen, S. Oh, and R. D. Kolodner. 2001. exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol. Cell. Biol. 215142-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Argueso, J. L., A. W. Kijas, S. Sarin, J. Heck, M. Waase, and E. Alani. 2003. Systematic mutagenesis of the Saccharomyces cerevisiae MLH1 gene reveals distinct roles for Mlh1p in meiotic crossing over and in vegetative and meiotic mismatch repair. Mol. Cell. Biol. 23873-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker, S. M., A. W. Plug, T. A. Prolla, C. E. Bronner, A. C. Harris, X. Yao, D. M. Christie, C. Monell, N. Arnheim, A. Bradley, T. Ashley, and R. M. Liskay. 1996. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 13336-342. [DOI] [PubMed] [Google Scholar]
- 5.Baudin, A., O. Ozier-Kalogeropoulos, A. Denouel, F. Lacroute, and C. Cullin. 1993. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 213329-3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cannavo, E., B. Gerrits, G. Marra, R. Schlapbach, and J. Jiricny. 2007. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2822976-2986. [DOI] [PubMed] [Google Scholar]
- 7.Edelmann, W., P. E. Cohen, M. Kane, K. Lau, B. Morrow, S. Bennett, A. Umar, T. Kunkel, G. Cattoretti, R. Chaganti, J. W. Pollard, R. D. Kolodner, and R. Kucherlapati. 1996. Meiotic pachytene arrest in MLH1-deficient mice. Cell 851125-1134. [DOI] [PubMed] [Google Scholar]
- 8.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 321792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erdeniz, N., M. Nguyen, S. M. Deschenes, and R. M. Liskay. 2007. Mutations affecting a putative MutLα endonuclease motif impact multiple mismatch repair functions. DNA Repair 61463-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiser, A., and A. Sali. 2003. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 374461-491. [DOI] [PubMed] [Google Scholar]
- 11.Fromont-Racine, M., J. C. Rain, and P. Legrain. 1997. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet. 16277-282. [DOI] [PubMed] [Google Scholar]
- 12.Gellon, L., M. Werner, and S. Boiteux. 2002. Ntg2p, a Saccharomyces cerevisiae DNA N-glycosylase/apurinic or apyrimidinic lyase involved in base excision repair of oxidative DNA damage, interacts with the DNA mismatch repair protein Mlh1p. Identification of a Mlh1p binding motif. J. Biol. Chem. 27729963-29972. [DOI] [PubMed] [Google Scholar]
- 13.Gietz, D., A. St. Jean, R. A. Woods, and R. H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 201425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guarne, A., S. Ramon-Maiques, E. M. Wolff, R. Ghirlando, X. Hu, J. H. Miller, and W. Yang. 2004. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J. 234134-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall, M. C., P. V. Shcherbakova, J. M. Fortune, C. H. Borchers, J. M. Dial, K. B. Tomer, and T. A. Kunkel. 2003. DNA binding by yeast Mlh1 and Pms1: implications for DNA mismatch repair. Nucleic Acids Res. 312025-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jager, A. C., M. Rasmussen, H. C. Bisgaard, K. K. Singh, F. C. Nielsen, and L. J. Rasmussen. 2001. HNPCC mutations in the human DNA mismatch repair gene hMLH1 influence assembly of hMutLα and hMLH1-hEXO1 complexes. Oncogene 203590-3595. [DOI] [PubMed] [Google Scholar]
- 17.Jiricny, J. 2006. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 7335-346. [DOI] [PubMed] [Google Scholar]
- 18.Kadyrov, F. A., L. Dzantiev, N. Constantin, and P. Modrich. 2006. Endonucleolytic function of MutLα in human mismatch repair. Cell 126297-308. [DOI] [PubMed] [Google Scholar]
- 19.Kadyrov, F. A., S. F. Holmes, M. E. Arana, O. A. Lukianova, M. O'Donnell, T. A. Kunkel, and P. Modrich. 2007. Saccharomyces cerevisiae MutLα is a mismatch repair endonuclease. J. Biol. Chem. 28237181-37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keskin, O., A. Gursoy, B. Ma, and R. Nussinov. 2008. Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem. Rev. 1081225-1244. [DOI] [PubMed] [Google Scholar]
- 21.Kosinski, J., I. Steindorf, J. M. Bujnicki, L. Giron-Monzon, and P. Friedhoff. 2005. Analysis of the quaternary structure of the MutL C-terminal domain. J. Mol. Biol. 351895-909. [DOI] [PubMed] [Google Scholar]
- 22.Kunkel, T. A., and D. A. Erie. 2005. DNA mismatch repair. Annu. Rev. Biochem. 74681-710. [DOI] [PubMed] [Google Scholar]
- 23.Lagerstedt Robinson, K., T. Liu, J. Vandrovcova, B. Halvarsson, M. Clendenning, T. Frebourg, N. Papadopoulos, K. W. Kinzler, B. Vogelstein, P. Peltomaki, R. D. Kolodner, M. Nilbert, and A. Lindblom. 2007. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J. Natl. Cancer Inst. 99291-299. [DOI] [PubMed] [Google Scholar]
- 24.Landau, M., I. Mayrose, Y. Rosenberg, F. Glaser, E. Martz, T. Pupko, and N. Ben-Tal. 2005. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33W299-W302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langland, G., J. Kordich, J. Creaney, K. H. Goss, K. Lillard-Wetherell, K. Bebenek, T. A. Kunkel, and J. Groden. 2001. The Bloom's syndrome protein (BLM) interacts with MLH1 but is not required for DNA mismatch repair. J. Biol. Chem. 27630031-30035. [DOI] [PubMed] [Google Scholar]
- 26.Lea, D. E., and D. A. Coulson. 1949. The distribution of the numbers of mutants in bacterial populations. J. Genet. 49264-285. [DOI] [PubMed] [Google Scholar]
- 27.Lee, S. D., and E. Alani. 2006. Analysis of interactions between mismatch repair initiation factors and the replication processivity factor PCNA. J. Mol. Biol. 355175-184. [DOI] [PubMed] [Google Scholar]
- 28.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14953-961. [DOI] [PubMed] [Google Scholar]
- 29.Lutzen, A., S. E. Liberti, and L. J. Rasmussen. 2004. Cadmium inhibits human DNA mismatch repair in vivo. Biochem. Biophys. Res. Commun. 32121-25. [DOI] [PubMed] [Google Scholar]
- 30.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 31.McGuffin, L. J., K. Bryson, and D. T. Jones. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16404-405. [DOI] [PubMed] [Google Scholar]
- 32.Modrich, P. 2006. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 28130305-30309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moldovan, G. L., B. Pfander, and S. Jentsch. 2007. PCNA, the maestro of the replication fork. Cell 129665-679. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen, F. C., A. C. Jager, A. Lutzen, J. R. Bundgaard, and L. J. Rasmussen. 2004. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene 231457-1468. [DOI] [PubMed] [Google Scholar]
- 35.Pedrazzi, G., C. Perrera, H. Blaser, P. Kuster, G. Marra, S. L. Davies, G. H. Ryu, R. Freire, I. D. Hickson, J. Jiricny, and I. Stagljar. 2001. Direct association of Bloom's syndrome gene product with the human mismatch repair protein MLH1. Nucleic Acids Res. 294378-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng, M., R. Litman, J. Xie, S. Sharma, R. M. Brosh, Jr., and S. B. Cantor. 2007. The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J. cells. EMBO J. 263238-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plotz, G., C. Welsch, L. Giron-Monzon, P. Friedhoff, M. Albrecht, A. Piiper, R. M. Biondi, T. Lengauer, S. Zeuzem, and J. Raedle. 2006. Mutations in the MutSα interaction interface of MLH1 can abolish DNA mismatch repair. Nucleic Acids Res. 346574-6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen, L. J., M. Rasmussen, B. Lee, A. K. Rasmussen, D. M. Wilson III, F. C. Nielsen, and H. C. Bisgaard. 2000. Identification of factors interacting with hMSH2 in the fetal liver utilizing the yeast two-hybrid system. In vivo interaction through the C-terminal domains of hEXO1 and hMSH2 and comparative expression analysis. Mutat. Res. 46041-52. [DOI] [PubMed] [Google Scholar]
- 39.Resnick, M. A., J. C. Game, and S. Stasiewicz. 1983. Genetic effects of UV irradiation on excision-proficient and -deficient yeast during meiosis. Genetics 104603-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmutte, C., R. C. Marinescu, M. M. Sadoff, S. Guerrette, J. Overhauser, and R. Fishel. 1998. Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. 584537-4542. [PubMed] [Google Scholar]
- 41.Schmutte, C., M. M. Sadoff, K. S. Shim, S. Acharya, and R. Fishel. 2001. The interaction of DNA mismatch repair proteins with human exonuclease I. J. Biol. Chem. 27633011-33018. [DOI] [PubMed] [Google Scholar]
- 42.Schuster-Bockler, B., J. Schultz, and S. Rahmann. 2004. HMM Logos for visualization of protein families. BMC Bioinform. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shcherbakova, P. V., M. C. Hall, M. S. Lewis, S. E. Bennett, K. J. Martin, P. R. Bushel, C. A. Afshari, and T. A. Kunkel. 2001. Inactivation of DNA mismatch repair by increased expression of yeast MLH1. Mol. Cell. Biol. 21940-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shcherbakova, P. V., and T. A. Kunkel. 1999. Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol. Cell. Biol. 193177-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherman, F., and J. Hicks. 1991. Micromanipulation and dissection of asci. Methods Enzymol. 19421-37. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi, M., H. Shimodaira, C. Andreutti-Zaugg, R. Iggo, R. D. Kolodner, and C. Ishioka. 2007. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 674595-4604. [DOI] [PubMed] [Google Scholar]
- 47.Tishkoff, D. X., A. L. Boerger, P. Bertrand, N. Filosi, G. M. Gaida, M. F. Kane, and R. D. Kolodner. 1997. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. USA 947487-7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran, H. T., D. A. Gordenin, and M. A. Resnick. 1999. The 3′→5′ exonucleases of DNA polymerases delta and epsilon and the 5′→3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol. Cell. Biol. 192000-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran, P. T., J. P. Fey, N. Erdeniz, L. Gellon, S. Boiteux, and R. M. Liskay. 2007. A mutation in EXO1 defines separable roles in DNA mismatch repair and post-replication repair. DNA Repair 61572-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran, P. T., and R. M. Liskay. 2000. Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLα. Mol. Cell. Biol. 206390-6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tran, P. T., J. A. Simon, and R. M. Liskay. 2001. Interactions of Exo1p with components of MutLα in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 989760-9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Umar, A., A. B. Buermeyer, J. A. Simon, D. C. Thomas, A. B. Clark, R. M. Liskay, and T. A. Kunkel. 1996. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 8765-73. [DOI] [PubMed] [Google Scholar]
- 53.Wang, T. F., N. Kleckner, and N. Hunter. 1999. Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc. Natl. Acad. Sci. USA 9613914-13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei, K., A. B. Clark, E. Wong, M. F. Kane, D. J. Mazur, T. Parris, N. K. Kolas, R. Russell, H. Hou, Jr., B. Kneitz, G. Yang, T. A. Kunkel, R. D. Kolodner, P. E. Cohen, and W. Edelmann. 2003. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 17603-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.