

Abstract
To elucidate the pathogenic mechanisms in Huntington's disease (HD) elicited by expression of full-length human mutant huntingtin (fl-mhtt), a bacterial artificial chromosome (BAC)-mediated transgenic mouse model (BACHD) was developed expressing fl-mhtt with 97 glutamine repeats under the control of endogenous htt regulatory machinery on the BAC. BACHD mice exhibit progressive motor deficits, neuronal synaptic dysfunction, and late-onset selective neuropathology, which includes significant cortical and striatal atrophy and striatal dark neuron degeneration. Power analyses reveal the robustness of the behavioral and neuropathological phenotypes, suggesting BACHD as a suitable fl-mhtt mouse model for preclinical studies. Additional analyses of BACHD mice provide novel insights into how mhtt may elicit neuropathogenesis. First, unlike previous fl-mhtt mouse models, BACHD mice reveal that the slowly progressive and selective pathogenic process in HD mouse brains can occur without early and diffuse nuclear accumulation of aggregated mhtt (i.e., as detected by immunostaining with the EM48 antibody). Instead, a relatively steady-state level of predominantly full-length mhtt and a small amount of mhtt N-terminal fragments are sufficient to elicit the disease process. Second, the polyglutamine repeat within fl-mhtt in BACHD mice is encoded by a mixed CAA-CAG repeat, which is stable in both the germline and somatic tissues including the cortex and striatum at the onset of neuropathology. Therefore, our results suggest that somatic repeat instability does not play a necessary role in selective neuropathogenesis in BACHD mice. In summary, the BACHD model constitutes a novel and robust in vivo paradigm for the investigation of HD pathogenesis and treatment.
Keywords: Huntington's disease, huntingtin, bacterial artificial chromosome, BAC, transgenic mouse, neurodegeneration, polyglutamine, repeat instability, preclinical model
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder characterized by late-onset motor, psychiatric, and cognitive deficits. Patients with adult-onset HD experience symptom onset around age 40 and death usually occurs 10–20 years after disease onset. HD is caused by a CAG repeat expansion (>35 repeats) in the huntingtin (htt) gene, encoding an expanded polyglutamine (polyQ) repeat in the widely expressed huntingtin protein (Huntington's Disease Collaborative Research Group, 1993; Sharp et al., 1995). Although the mutant protein is widely expressed, the most affected cell types in HD are the medium-sized spiny neurons (MSNs) in the striatum and pyramidal neurons in the cortex (Vonsattel and DiFiglia, 1998). The mechanisms underlying selective neuropathology in HD remain poorly understood.
Mouse models have been valuable in investigating the critical pathogenic mechanisms in HD (Zoghbi and Orr, 2000; Menalled and Chesselet, 2002). One group of mouse models expresses mouse (Hdh-KI models) (Wheeler et al., 2000; Menalled et al., 2002) or human fl-mhtt [yeast artificial chromosome (YAC)] (Hodgson et al., 1999; Slow et al., 2003). These models exhibit progressive motor and pathological deficits and, in YAC128 mice, culminate in selective cortical and striatal atrophy and neurodegeneration (Slow et al., 2003; Van Raamsdonk et al., 2005). A common feature of Hdh-KI and YAC128 mice is early and selective striatal localization of diffuse nuclear staining of mhtt with the EM48 antibody (Gutekunst et al., 1999). Thus, current fl-mhtt mouse models support the hypothesis that early nuclear localization of EM48+ mhtt may be a critical early pathogenic event in HD (Wheeler et al., 2000; Van Raamsdonk et al., 2005).
Another mechanism implicated in selective neuropathogenesis in HD is somatic CAG repeat instability. Somatic and germline repeat instability is present in multiple HD mouse models (Kennedy and Shelbourne, 2000; Ishiguro et al., 2001), and in postmortem HD brain tissues (Shelbourne et al., 2007). Somatic repeat expansion may be modified by the mismatch repair systems (Manley et al., 1999; Wheeler et al., 2003), and a recent study suggests it could be initiated by an age-dependent single-nucleotide excision DNA repair of oxidized nucleotides (Kovtun et al., 2007). Together, these studies promote the hypothesis that tissue-specific and age-dependent somatic repeat expansion of the CAG repeat in mhtt may play a crucial role in selective neuropathogenesis in HD.
To address how fl-mhtt elicits selective neuropathogenesis in HD, we applied the bacterial artificial chromosome (BAC)-mediated transgenic approach (Yang et al., 1997; Gong et al., 2002) to generate a BAC transgenic mouse model of HD (BACHD). BACHD mice express fl-mhtt, exhibit progressive motor deficits and selective late-onset neuropathology without somatic repeat instability in the aged brain. Moreover, BACHD mice reproduce a mhtt aggregation pattern reminiscent of that in adult-onset HD. Importantly, a relatively steady-state level of predominantly fl-mhtt and a small amount of mhtt N-terminal fragments, present in both the nucleus and cytoplasm, are responsible for the onset of neuropathology. Thus, BACHD mice represent a novel and robust in vivo paradigm to study HD pathogenesis and treatment.
Materials and Methods
Generation of BACHD transgenic mice.
A 240 kb RP11-866L6 BAC containing the 170 kb human htt locus was modified by adding an exon 1 with 97 mixed CAA-CAG repeats (Kazantsev et al., 1999) into the human htt gene. BAC DNA was prepared according to published protocols (Yang et al., 1997; Gong and Yang, 2005) and microinjected into FvB fertilized eggs. BACHD mice were maintained in the FvB/NJ background and bred and maintained under standard conditions consistent with National Institutes of Health guidelines and approved by the University of California, Los Angeles, Institutional Animal Care and Use Committees.
Transgenic copy number and RNA expression.
Genomic DNA was purified from the tails of three BACHD transgenic and three wild-type animals for copy number quantitation of exogenously derived fl-mhtt. Primers specific for fl-mhtt genomic DNA were designed (forward, 5′-GAG CCA TGA TTG TGC TAT CG-3′; reverse, 5′-AGC TAC GCT GCT CAC AGA AA-3′). Human genomic DNA was used as a two copy per diploid genome control to estimate copy number in BACHD mice. For RNA quantitation, total RNA was extracted using the RNaeasy mini-kit (Qiagen). A combination of random primer and oligo-dT20 (Invitrogen) primed cDNA synthesis was used to generate cDNA for RT-PCR using OmniScript RT-PCR kit (Qiagen). Primers for RT-PCR were designed so that the forward primer recognized both endogenous Hdh and exogenous mhtt, whereas the reverse primers were species-specific [forward, 5′-ATC TTG AGC CAC AGC TCC AGC CA-3′; reverse (human), 5′-GGC CTC CGA GGC TTC ATC AGG-3′; reverse (murine), 5′-TCT GAA AAC ATC TGA GAC TTC ACC AGA-3′]. Both primer sets generated products of approximately the same size (300 bp) and span the junction of exons 12–13. For RT-PCR, because we were directly comparing the levels of endogenous Hdh versus exogenous mhtt, standards were generated to determine primer set efficiencies. Standards were derived from PCR amplicons generated from each primer set, which were subcloned in pCR2.1 (Invitrogen). Genomic DNA samples were normalized using 18S. Samples of cDNA were normalized against β-actin. The quantitation of band intensities was done using Kodak 1D Image Analysis software.
Analyzing polyglutamine repeats.
Genomic DNA was prepared from tail using standard proteinase K digestion (10 mg/ml) followed by phenol/chloroform extraction. Genomic DNA was extracted from brain tissue using the Qiagen DNeasy Tissue kit. Primers flanking the polyQ repeat in exon 1 of mhtt were used to PCR amplify the polyQ regions, in which 5′ primer was labeled with FAM (carboxyfluorescein amidite). The PCR product was run on an automated Sequencer (Applied Biosystems 3730XL) with Bioventures' ROX1000 internal size standard. GeneMapper software version 4.0 (Applied Biosystems) was used to determine PCR fragment size, and the number of CAA-CAG repeats was calculated based on the size of the PCR products.
Preparation of protein lysates.
Mouse brains were dissected in ice-cold 100 mm PBS supplemented with Complete Protease Inhibitor Mixture tablets (Roche). Lysates were prepared by homogenizing brain tissue in a modified RIPA buffer using 10–12 strokes from a Potter–Elvehjem homogenizer followed by centrifugation at 4°C for 15 min at 16,100 × g. Resultant supernatants were used for all nonfractionation experiments. The pellet fraction was further processed to obtain the insoluble protein fractions as follows: Pellets were dissolved in 10% SDS and heated to 70°C for 10 min. The mixture was then spun at 2000 × g for 30 s, and the new soluble portion was taken to be the insoluble fraction.
Western blotting.
Protein samples were prepared for loading by heating in NuPAGE LDS buffer (Invitrogen) according to the manufacturer's protocol and heated for 10 min at 70°C. Proteins were resolved on either 10% Bis-Tris or 3–8% Tris-acetate NuPAGE gels (Invitrogen) using either MOPS [3-(N-morpholino)propanesulfonic acid] or Tris-acetate running buffer, respectively, and wet-transferred onto polyvinylidene difluoride membranes using NuPAGE transfer buffer (Invitrogen). Immunoblots were probed with anti-huntingtin 2166 and 1C2, S830 (G. Bates), and anti-histone H2B (Millipore Bioscience Research Reagents) at 1:3000 dilutions in 5% blocking solution, and anti-α-tubulin (Sigma-Aldrich) at a 1:10,000 dilution in 5% blocking solution. Chemiluminescent detection was accomplished using ECL Plus Western Blotting Detection reagents (GE Healthcare), Western Lightning Plus (Perkin-Elmer), or SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Subcellular fractionation.
Brain tissue was homogenized on ice in 9 vol (w/v) of 0.25 m sucrose/buffer A [50 mm triethanolamine, pH 7.5, 25 mm KCl, 5 mm MgCl2, and 0.5 mm dithiothreitol, supplemented with Complete protease inhibitor mixture tablets (Roche)] using eight to nine strokes from a Potter–Elvehjem homogenizer followed by centrifugation at 4°C for 15 min at 800 × g. The supernatants were further centrifuged at 4°C for 1 h at 100,000 × g, and the resultant supernatants were taken to be the cytosolic fractions. The crude nuclear pellets from the initial centrifugation step were washed in 0.25 m sucrose/buffer A and centrifuged at 4°C for 10 min at 800 × g. Pellets were resuspended in 2 ml of 2.3 m sucrose/buffer A and further homogenized on ice using five to six strokes from a Dounce homogenizer. Homogenates were each mixed with 2 ml of 0.25 m sucrose/buffer A and 2 ml of 2.3 m sucrose/buffer A, and each mixture was layered onto 4 ml of 2.3 m sucrose/buffer A. Nuclei were pelleted by centrifugation at 4°C for 1 h at 80,000 × g, and then resuspended in 0.25 m sucrose/buffer A and washed by centrifugation at 500 × g for 15 min. The final pellets were designated as the nuclear fraction and were resuspended in 0.25 m sucrose/buffer A.
Rotarod test.
BACHD and wild-type littermates were trained on an Ugo Basile 7650 Accelerating Rotarod for three trials per day for 2 d at 2 months of age adapted from existing protocol (Clark et al., 1997). Animals were tested for three trials per day for 3 d at 2, 6, and 12 months of age. Animals from the same cohort of mice were measured at 2, 6, and 12 months. All rotarod tests were performed during the light phase of the light cycle.
Immunohistochemistry with EM48.
Immunohistochemistry with rabbit polyclonal EM48 antibody (diluted 1:300) was performed using an established protocol to detect mhtt on 35-μm-thick cryostat brain sections (Menalled et al., 2002; Gu et al., 2005). EM48-stained sections from BACHD brains were stained with Hematoxylin QS solution to identify the nucleus (Vector Laboratories). Sections were imaged using a Zeiss Axioskop 2 microscope with an AxioCam HRc camera.
Electron microscopy.
Electron microscopy (EM) was performed on 12-month-old BACHD and wild-type littermates using an established protocol (Yu et al., 2003; Gu et al., 2005). Two BACHD and two wild-type littermates at 12 months were used for EM studies.
Stereology.
The optical fractionator method was used to determine the cortical and striatal volume and striatal neuronal number (Oorschot, 1996; Menalled et al., 2002). BACHD and wild-type animals were perfused with 4% paraformaldehyde. Thirty-five micrometer coronal brain sections were cut using a Leica CM 1850 cryostat. The first section was chosen at random from the first 10 sections containing striatum and every 10th section thereafter was stained with NeuN (1:500 dilution). Sections were analyzed using the Optical Fractionator Method using Stereo Investigator software (MicroBrightField). Striatal neuronal counts were obtained by counting NeuN-positive cells that fell within a 50 μm × 50 μm grid size at 100× and within 10 μm of the thickness of the section. The Cavalieri method was used to determine the total volume of the striatum and cortex at 6 and 12 months of age. The same sections used for cell counts were used to estimate the volume of the cortex and striatum.
Toluidine blue staining and quantitation of dark degenerating neurons.
The toluidine blue staining of semithin brain sections was modified from existing protocols (Hodgson et al., 1999; Turmaine et al., 2000). BACHD and wild-type littermates were perfused with 2% glutaraldehyde and 2% paraformaldehyde. The 1.5-μm-thick sections were cut at bregma level 0–1 mm. The striatum was cut into dorsal, medial, and lateral pieces. Each piece of tissue was fixed with 1% osmic acid solution for 1 h at room temperature. Tissue was embedded in Epon overnight at 60°C, and then 1.5-μm-thick sections were cut on a Leica RM 2165 microtome and every 10th section was mounted on slides. Sections were stained with 1% toluidine blue in 1% sodium borate for 1 min at 60°C. Three sections at a minimum of 150 μm apart for each striatal region were imaged using a MicroFire camera at 40× using a Zeiss Axioskop2 microscope. For each section, all neurons from five 1600 × 1200 pixel frames chosen at random were counted.
Electrophysiological recordings in slices.
Whole-cell patch-clamp recordings from MSNs were obtained using standard methods (Cepeda et al., 1998). Cells were identified by soma size, by basic membrane properties (input resistance, membrane capacitance, and time constant), and in some cases, by the addition of biocytin (0.2%) to the internal solution. The patch pipette (3–5 MΩ) contained the following solution (in mm): 140 K-gluconate, 10 HEPES, 2 MgCl2, 0.1 CaCl2, 1.1 EGTA, and 2 K2ATP, for voltage and current clamp, pH 7.25–7.3 and osmolality of 280–290 mOsm. Spontaneous postsynaptic currents (PSCs) were recorded in standard artificial CSF composed of the following (in mm): 130 NaCl, 26 NaHCO3, 3 KCl, 2 MgCl2, 1.25 NaHPO4, 2 CaCl2, and 10 glucose, pH 7.4 and osmolality of 300 mOsm. Cells were held at −70 mV to minimize the contribution of GABAA receptors and that of voltage-gated conductances. After characterizing the basic membrane properties of the neuron, spontaneous PSCs were recorded for variable periods of time (usually 3–6 min). The membrane current was filtered at 1 kHz and digitized at 200 μs using Clampex. Spontaneous EPSCs were analyzed off-line using the Mini Analysis Program (Jaejin Software). The threshold amplitude for the detection of an event was adjusted above root mean square noise level (generally 3–4 pA). This software was used to calculate PSC frequency, amplitude for each synaptic event, and to construct amplitude–frequency histograms. Frequencies were expressed as number of events per second (in hertz).
BDNF expression analyses. Quantitative reverse transcriptase-PCR.
RNA was extracted from mouse brain tissue according to the Trizol reagent (Invitrogen) protocol. Total RNA was treated with TURBO DNase (Ambion) followed by cDNA synthesis using SuperScript III Reverse Transcriptase kit (Invitrogen). PCR primers for quantitative reverse transcriptase-PCR (qRT-PCR) used were as follows: BDNF exon IV, forward, 5′-GCA GCT GCC TTG ATG TTT AC-3′; BDNF exon IV, reverse, 5′-CCG TGG ACG TTT ACT TCT TTC-3′; BDNF coding sequence, forward, 5′-ATT AGC GAG TGG GTC ACA GC-3′; BDNF coding sequence, reverse, 5′-TCA GTT GGC CTT TGG ATA CC-3′; GAPDH, forward, 5′-CTG AGT ATG TCG TGG AGT CTA CTG G-3′; GAPDH, reverse, 5′-GTC ATA TTT CTC GTG GTT CAC ACC-3′. Quantitative PCR was performed in an iCycler (Bio-Rad) with the SYBR Green Supermix (Bio-Rad). The threshold cycle for each sample was chosen from the linear range and converted to a starting quantity by interpolation from a standard curve run on the same plate for each set of primers. The BDNF exon IV mRNA levels and coding sequence mRNA levels were normalized for each well to the GAPDH mRNA levels.
Statistical analysis. Behavioral experiments.
All data are shown as the mean ± SEM. For rotarod analysis, SPSS 14.0 statistics software was used to perform all of the analysis. The significance level was set at 0.05. A general linear model with repeated measures was performed to assess within-subjects effects of age, the interaction of genotype and age, and between-subjects effect of genotype. Student's t test was used to compare the performance of BACHD and wild-type mice for each trial at all ages examined (comparing one variable between two genotypes). In some sets of data, one-way ANOVA was used to analyze the time effect on rotarod performance of BACHD mice and wild-type mice from 2, 6, and 12 months of age. Tukey's honestly significant difference (HSD) post hoc analysis was performed for pairwise comparisons among ages. For neuropathology and stereology studies, all comparisons between BACHD and wild-type mice are shown as mean ± SEM. Comparisons were made with an independent sample Student's t test with a significance level of 0.05. For electrophysiological studies, data in the text and figures are presented as mean ± SEM. Differences between groups were assessed with Student's t tests or appropriate designed ANOVAs. The significance level was set at 0.05.
Results
Generation and initial characterization of BACHD mice
We engineered a 240 kb human BAC (RP11-866L6) that contains the entire 170 kb human htt locus with ∼20 kb of the 5′ flanking genomic sequence and ∼50 kb of the 3′ flanking genomic sequence so that wild-type htt-exon 1 was replaced with mhtt-exon 1 containing 97 mixed CAA-CAG repeats encoding a contiguous polyQ repeat (Fig. 1A) (Yang et al., 1997; Kazantsev et al., 1999; Gong et al., 2002). Two LoxP sites are located in the 5′ untranslated region of mhtt and in intron 1 flanking mhtt-exon 1. Thus, these LoxP sites do not interfere with the expression of fl-mhtt but do allow for Cre-mediated excision of mhtt-exon 1. As a result, the BACHD model is designed to be a conditional inactivation model in which Cre can switch off fl-mhtt expression. We microinjected purified intact BACHD DNA into fertilized murine FvB eggs, and obtained 14 transgenic founders. Initial Western blot analyses with 1C2, an antibody against an expanded polyQ epitope, revealed that the majority of the founder lines expressed full-length mhtt (data not shown). We focused our phenotypic analyses on the BACHD-I line (referred to as BACHD in the remainder of the text), which expresses relatively high levels of intact mhtt. In addition, we also performed phenotypic analyses of another independent BACHD line (BACHD-L line) to confirm that the phenotypes observed in BACHD mice are not attributable to transgene integration effects. Quantitative PCR analyses of the genomic DNA revealed that BACHD mice have tandem integrates of approximately five copies of the BAC transgene, and qRT-PCR analyses using 2-month-old brain RNA revealed that BACHD mice express fl-mhtt at approximately threefold the mRNA level of the endogenous Hdh at the mRNA level. To assess mhtt protein expression, we first performed Western blot analyses using the 1C2 antibody, which showed that BACHD mice ubiquitously express fl-mhtt (∼350 kDa) at comparable levels in the cortex, striatum, and cerebellum (Fig. 1B). On longer exposure, we detected a small amount of mhtt N-terminal fragments containing the expanded polyQ repeat in the 2-month-old BACHD brains; however, these mhtt fragments were a minority species compared with fl-mhtt (Fig. 1C). To visualize expression of both fl-mhtt and the endogenous wild-type murine htt, we used the mAb2166 antibody, which is able to detect both proteins, with mhtt migrating as a slightly higher molecular weight band than the wild-type murine htt because of the glutamine repeat expansion. Because the mAb2166 antibody is known to bind more strongly to wild-type htt than to mhtt (Wheeler et al., 2000), we cannot precisely quantitate the mhtt protein level in BACHD mice. However, based on the relative affinity of this antibody to mutant and to wild-type htt in Hdh-Q111 knock-in mice (ibid), and from multiple mAb2166 Western blots (Fig. 1B,D), we estimate the expression level of mhtt protein in BACHD mice to be ∼1.5- to 2-fold the level of endogenous htt. In addition, the BACHD-L line mice also express the mhtt protein, although at a much lower level than the BACHD mice as revealed by both 1C2 and mAb2166 (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). Finally, to demonstrate that BACHD mice express functional mhtt in an endogenous-like expression pattern (at least during embryonic development), we bred BACHD mice onto the Hdh null background (Zeitlin et al., 1995). The BACHD transgene was able to rescue the embryonic lethality of the Hdh null mice in a Mendelian ratio: there are 6 rescued mice (BACHDTg/+/Hdh−/−) of a total of 53 mice born (an observed rate of 11.3% compared with the expected rate of 12.5%); and we did not observe any Hdh−/− mice born without the BACHD transgene (an observed rate of 0% compared with the expected rate of 12.5%). Furthermore, Western blot analyses with mAb2166 revealed that rescued mice expressed only mhtt and not murine wild-type htt (Fig. 1D). These results demonstrate that fl-mhtt is properly expressed in BACHD mice and it retains all of the essential functions of wild-type htt during development.
Figure 1.

Generation of the BACHD transgenic mouse model. A, Schematic representation of the modified 240 kb human BAC containing the entire 170 kb htt genomic locus, plus ∼20 kb 5′ flanking and 50 kb 3′ flanking genomic region. The BAC was engineered to replace the endogenous htt exon-1 with a mhtt exon-1 with mixed CAA-CAG repeats encoding 97 polyQ repeats. Two loxP sites were positioned to flank the mhtt exon-1, so that the expression of fl-mhtt can be switched off by Cre recombinase. B, Top panel, Western blot analysis of htt expression in BACHD and wild-type mice with 1C2. Middle panel, Western blot with htt specific mAb2166 reveals both wild-type endogenous murine htt (bottom band in all lanes) and mhtt (top band in BACHD lanes). Bottom panel, The same blot was probed with anti-α-tubulin antibody for the loading control. Cortex (ctx), striatum (str), and cerebellum (crb) all express comparable levels of mhtt. C, All panels, Overexposure of the blots in B. The 1C2 blot reveals the presence of 1C2-positive fragments containing the expanded polyQ repeat in BACHD transgenic mice but not wild-type littermates (arrows). D, Western blot analysis with mAb2166 using cortical and striatal protein extracts in the BACHD genetic rescue experiment. Plus and minus signs indicate the allele status at the Hdh locus. The rescue mice, with BACHD and two null Hdh alleles (−/−), contain only the mhtt band (top band) and lack the wild-type htt band (bottom band). The same blot was reprobed with anti-α-tubulin antibody for the loading control.
Polyglutamine repeats are stable in BACHD mice
Both germline and somatic repeat instability is a feature of triplet repeat disorders including HD (Pearson et al., 2005), and has been observed in mouse models of these disorders (Mangiarini et al., 1997; Kennedy et al., 2003; Watase et al., 2003; Wheeler et al., 2003). Somatic repeat expansion in HD is shown to be more pronounced in the vulnerable cortical and striatal neurons in HD mice (Kennedy and Shelbourne, 2000; Ishiguro et al., 2001) and in HD patients (Shelbourne et al., 2007), and may be dependent on an age-dependent oxidation–DNA damage repair cycle (Kovtun et al., 2007). Therefore, these results prompted the hypothesis that somatic repeat expansion in HD may be critical to disease onset and/or selective neuropathogenesis in the disorder (Kennedy and Shelbourne, 2000). One strategy to test whether somatic repeat instability is necessary for the pathogenesis of selective neuropathology is to develop a mouse model that expresses full-length mhtt with a stable polyQ repeat.
The polyQ repeat in mhtt in the BACHD mouse model was generated using a mixed CAA-CAG repeat from the reiteration of the CAA CAG CAG CAA CAG CAA sequence, which encodes a pure polyQ stretch (Kazantsev et al., 1999). Polyglutamine repeats with interruptions of the CAG repeat have been shown to be stable in vivo (Pearson et al., 1998; Choudhry et al., 2001; Dorsman et al., 2002). We used established methodology, GeneMapper scanning plus sequencing, to analyze the CAA-CAG repeat lengths in BACHD mice (Mangiarini et al., 1997; Manley et al., 1999; Kovtun et al., 2007). We demonstrated that BACHD mice consistently show repeat lengths of 97 CAA-CAG repeats in the germline from both maternal and paternal transmissions (Fig. 2A,E). These mice do not exhibit any repeat expansion or retraction as demonstrated by a single sharp peak obtained from GeneMapper analysis (Fig. 2A). Furthermore, when we examined repeat lengths in DNA from the cortex (Fig. 2B), striatum (Fig. 2C), and cerebellum (Fig. 2D) of BACHD mice at 12 months of age, we always detected a single peak with 97 CAA-CAG repeats, which is the same as the repeat length observed in the germline (Fig. 2E). Thus, we conclude that the polyQ repeat length in BACHD mice is stable both in the germline and in aged somatic brain tissues at 12 months of age. The stable polyQ repeat in the BACHD model, as predicted by the nature of its mixed CAA-CAG repeat, provide us an opportunity to address whether somatic repeat instability would be necessary to elicit HD-like disease pathogenesis by fl-mhtt in vivo.
Figure 2.

BACHD mice exhibit stable polyglutamine repeats. A–D, Images of scans generated from GeneMapper analysis. Only one peak with 97 polyglutamines was identified in the germline (tail) (A) from BACHD mice. DNA from the cortex (B), striatum (C), and cerebellum (D) of BACHD mice at 12 months examined by GeneMapper analysis shows that mhtt in BACHD mice contains 97 repeats without any contractions or retractions. E, Table demonstrating that BACHD mice do not have any repeat expansion in the germline from maternal or paternal transmissions with both parents containing 97 polyQ repeats. The cortex, striatum, and cerebellum do not have any contractions or expansions in the polyQ region at 12 months.
Progressive motor deficits in BACHD mice
An important clinical feature of HD is late-onset and progressive motor deficits. However, even years before the onset of overt motor deficits, such as chorea, HD patients exhibit subtle motor and cognitive deficits (Brandt et al., 1995; Smith et al., 2000). We performed repeated testing of BACHD and wild-type mice at 2, 6, and 12 months of age to assess their motor performance and learning using the accelerating rotarod paradigm (Clark et al., 1997). A general linear model ANOVA with repeated measures (SPSS 14.0) revealed an effect of genotype on performance (over the 3 d testing period) at 2 months (F(1,62) = 10.5; p < 0.003), 6 months (F(1,51) = 34.61; p < 0.0001), and 12 months of age (F(1,23) = 74.03; p < 0.0001). At 2 months of age, BACHD mice exhibited a subtle but significant decrease in rotarod performance in multiple trials when compared with wild-type mice (Fig. 3A) (p < 0.04, Student's t test). When the same cohort of mice were tested on the rotarod at 6 months of age, BACHD mice performed significantly worse than wild-type mice in all trials examined (p < 0.004, Student's t test) (Fig. 3B). The significant decrease in rotarod performance of BACHD mice compared with wild-type mice was maintained at 12 months of age for all trials examined (p < 0.003, Student's t test) (Fig. 3C). As shown in Figure 3D, when comparing rotarod performance over time (2, 6, and 12 months of age), BACHD mice showed a significant progressive decline in performance as they aged (one-way ANOVA, F(2,82) = 20.103; p < 0.0001). Tukey's HSD post hoc tests reveal a significant decrease in performance from 2 to 6 months (p < 0.0001) and 2 to 12 months (p < 0.0001). In contrast, there was no significant overall time effect on rotarod performance for wild-type controls (F(2,57) = 2.362; p = 0.104), suggesting the wild-type mice can maintain the same level of performance as they age. Repeated-measures ANOVA analysis (average of day 3 trials at 2, 6, and 12 months of age) revealed a significant effect of age (F(2,46) = 3.327; p < 0.05) and genotype (F(1,23) = 41.05; p < 0.0001) on rotarod performance; in addition, ANOVA analysis also revealed a significant genotype and age interaction (F(2,46) = 10.017; p < 0.0001). Together, these data demonstrate that BACHD mice have progressive and robust rotarod deficits.
Figure 3.

Progressive behavioral deficits in BACHD transgenic mice. A–C, Repeated rotarod tests of BACHD transgenic mice and wild-type littermates at 2, 6, and 12 months. Rotarod deficits are present at several testing points at 2 months (wild type, n = 26; BACHD, n = 37; *p < 0.04, Student's t test), but become more significant in BACHD mice at 6 months (wild type, n = 21; BACHD, n = 33; *p < 0.004) and 12 months (wild type, n = 13; BACHD, n = 12; *p < 0.003). D, When comparing rotarod performance at 2, 6, and 12 months of age, BACHD mice show a significant progressive decline in performance (one-way ANOVA, F(2,82) = 20.103; p < 0.0001). Post hoc tests reveal a significant decrease in performance from 2 to 6 months (p < 0.0001) and 2 to 12 months (p < 0.0001). In contrast, there is no significant overall time effect on rotarod performance for wild-type controls (F(2,57) = 2.362; p = 0.104), suggesting that the wild-type mice can maintain the same level of performance over time. Repeat-measure ANOVA analysis (average of day 3 trials at 2, 6, and 12 months) reveals a significant effect of age (F(2,46) = 3.327; p < 0.05) and genotype (F(1,23) = 41.05; p < 0.0001) on rotarod performance; in addition, ANOVA analysis also reveals a significant genotype and age interaction (F(2,46) = 10.017; p < 0.0001). Error bars indicate SEM.
Because previous studies revealed that overexpression of both mutant and wild-type htt in YAC transgenic mice leads to increased body weight (Van Raamsdonk et al., 2006), and body weight could be a factor influencing rotarod performance (McFadyen et al., 2003), we examined the relationship between weight and rotarod performance in BACHD mice. Similar to YAC mice, we found that BACHD mice have significant weight gain of an average of 20–30% compared with wild-type controls between 2 and 6 months of age (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) (p < 0.001, Student's t test), and they maintain this weight gain between 6 and 12 months. However, we found that the rotarod performance in this cohort of BACHD mice at 6 months is not significantly correlated with weight (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material) (R2 = −0.0275; p = 0.184). Furthermore, using an established method to identify the subset of mutant mice within the normal weight range (within 2 SDs of the wild-type mouse weight) (Kernie et al., 2000), we found that this subgroup of BACHD mice performed comparable with those BACHD mice with weight gain, and both subgroups performed significantly worse than the wild-type controls (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material) (p < 0.001, Student's t test). Therefore, we conclude that, although BACHD mice experience weight gain, the weight changes do not correlate with the rotarod deficits in this cohort of mice. In summary, our data demonstrate that BACHD mice exhibit progressive motor deficits, a key clinical feature of HD.
Progressive and selective neuropathology in BACHD mice
Although the mhtt protein is ubiquitously expressed in the brain, the neuropathology in HD is remarkably selective, in which dramatic and robust atrophy and neuronal cell loss is seen in the caudate/putamen and to a lesser extent in the cortex (Vonsattel et al., 1985; Vonsattel and DiFiglia, 1998). In addition, atrophy and cell loss have also been described in the hypothalamus and hippocampus (Kremer et al., 1991; Spargo et al., 1993; Kassubek et al., 2004; Petersen et al., 2005). We evaluated the neuropathology in BACHD brains at 6 and 12 months of age. At 6 months of age, using multiple assays, including brain weight and stereological volume measurement of the cortex and striatum, we did not detect any significant differences between BACHD mice and wild-type littermates (data not shown). At 12 months of age, the majority of BACHD brains were visibly atrophic compared with those of the wild-type littermate controls on gross examination (Fig. 4A,B). When we compared brain weight, we found that the forebrain (whole brain minus the cerebellum and olfactory bulb) of BACHD mice weighed 20% less than that of the controls (wild type, 0.446 g, n = 10; BACHD, 0.356 g, n = 13; p < 0.001, Student's t test) but the cerebellar weight was comparable between the two genotypes (wild type, 0.130; BACHD, 0.134; p = 0.25, Student's t test). Therefore, despite widespread expression of fl-mhtt, BACHD mice exhibit a pattern of selective brain atrophy similar to that of the HD patients.
Figure 4.

Selective and robust cortical and striatal atrophy in BACHD mice. A, Coronal brain section of wild-type mouse brain section stained with hematoxylin and eosin (H&E). B, A matched coronal brain section from a 12-month-old BACHD littermate stained with H&E reveals robust brain atrophy in the transgenic mouse at 12 months. ctx, Cortex; str, striatum.
To further quantify the magnitude of cortical and striatal atrophy in BACHD brains, we used unbiased stereology to determine the cortical and striatal volume in 6- and 12-month-old BACHD mice (Menalled et al., 2002). We did not find a significant difference in the cortical or striatal volumes between 6-month-old BACHD and wild-type controls (n = 5 per genotype). However, at 12 months of age, BACHD brains exhibited a robust decrease in both the cortical volume (BACHD, 3.6 × 1010 μm3; wild type, 5.2 × 1010 μm3; 32% of wild-type controls; p < 0.002, Student's t test; n = 7 per genotype) and striatal volume (BACHD, 1.25 × 1010 μm3; wild type, 1.74 × 1010 μm3; 28% of wild-type controls; p < 0.02, Student's t test; n = 7 per genotype). This result demonstrates that BACHD mice have late-onset atrophy of the cortex and striatum. HD neuropathology is also characterized by neurodegeneration primarily targeting the striatal MSNs (Vonsattel and DiFiglia, 1998). As a pathological marker of this neurodegenerative process, dying striatal neurons in the brains of HD patients and HD mice can be identified by EM as darkly stained (by osmium), atrophic neurons that lack clear morphology of the subcellular organelles (Davies et al., 1997; Yu et al., 2003). These dark degenerating neurons can also be identified by toluidine blue staining of semithin (1.5 μm) brain sections (Davies et al., 1997; Hodgson et al., 1999; Wheeler et al., 2002). We used these techniques to examine BACHD striata for evidence of neurodegeneration at 12 months of age. We first performed EM analyses using 12-month-old BACHD brains and wild-type controls (n = 2 per genotype), and consistently found the presence of degenerating dark neurons in the cortices and striata of the BACHD mice, but not in the wild-type control mice (Fig. 5A,B). To further quantify the extent of dark neuron degeneration in the striatum, we used toluidine blue staining of semithin striatal brain sections from 12-month-old BACHD and wild-type littermates (n = 5 per genotype). We examined neurons in the lateral, medial, and dorsal areas of the striata of all mice. Semithin striatal sections from the wild-type mice rarely had darkly stained neurons, with the vast majority of the cells exhibiting normal morphology (Fig. 5C). Semithin sections of BACHD mice consistently contained numerous neurons with dark toluidine blue staining and atrophic morphology (Fig. 5D). In BACHD brains, the lateral striatal region was the most affected, and the medial and dorsal regions exhibited only a few dark neurons. Remarkably, in the lateral striata of BACHD mice, ∼14% of the total neurons counted were dark neurons (387 of 2781; n = 5 BACHD mice), which was significantly higher than the number in wild-type control mice (0.34% or 10 of 2876; n = 5 wild-type mice; p < 0.003, Student's t test). Finally, to assess the striatal neuronal loss, we used unbiased stereology to count the number of NeuN+ striatal neurons in 12-month-old BACHD and control mice. We did not find any significant difference in the number of striatal NeuN-positive neurons between BACHD and control mice (BACHD, 2,129,025 ± 56,400; wild type, 2,037,701 ± 82,031; n = 5 per genotype; p = 0.39, Student's t test), suggesting that BACHD striatal medium spiny neurons are degenerating (i.e., atrophy and dark neurons) but have not yet been lost at this age. In summary, BACHD mice recapitulate HD neuropathology, as evidenced by selective cortical and striatal atrophy and significant striatal dark neuron degeneration.
Figure 5.
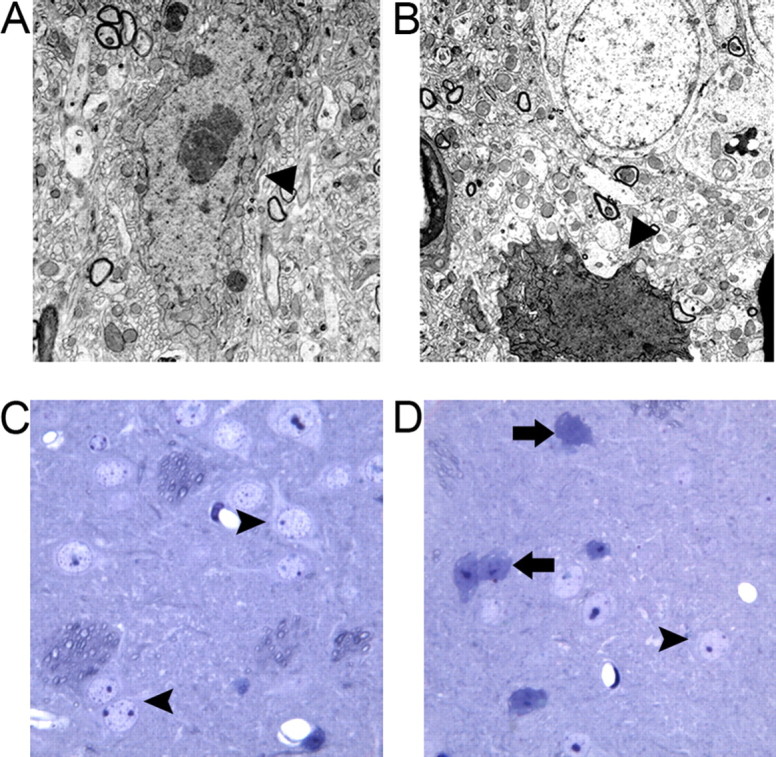
Neurodegenerative pathology in 12-month-old BACHD brains. A, B, EM reveals the presence of dark degenerating neurons in the cortex (A) and striatum (B) of BACHD mice but not in the control brains (data not shown). C, Toluidine blue staining of semithin striatal sections revealed healthy and lightly stained neurons (arrowheads) in the lateral striatum of 12-month-old wild-type mice. D, BACHD mice contain numerous darkly degenerating neurons (arrows) that are heavily stained with toluidine blue and exhibit atrophic morphology in the lateral striata.
BACHD mice recapitulate the adult-onset pattern of mhtt aggregation
Progressive aggregation of mhtt is another pathological hallmark of HD mice and HD patients (DiFiglia et al., 1997; Gutekunst et al., 1999; Kuemmerle et al., 1999). In adult-onset HD patients, mhtt aggregation patterns consist predominantly of large neuropil aggregates (NAs) (also known as dystrophic neurites; ibid), with only a small percentage of aggregates present in the nucleus [called nuclear inclusions (NIs); ibid]. Interestingly, this nuclear versus cytoplasmic distribution of mhtt aggregates in the cortex is opposite in juvenile-onset HD patients, with NIs being the predominant species (DiFiglia et al., 1997). Furthermore, a greater number of large aggregates are observed in the cortex of both juvenile- and adult-onset HD brains when compared with the striatum (DiFiglia et al., 1997; Gutekunst et al., 1999). Previous studies of mhtt aggregation in full-length mhtt expressing mouse models (i.e., YAC models and Hdh-KI mice) revealed early and selective diffuse nuclear accumulation of aggregated mhtt, as detected by the EM48 antibody, in the striatum and cortex by 6 months of age (Hodgson et al., 1999; Li et al., 2000; Wheeler et al., 2000; Menalled et al., 2002; Slow et al., 2003; Van Raamsdonk et al., 2005).
To determine whether mhtt aggregation patterns in BACHD brains reproduce the pattern seen in HD patient brains, we used the polyclonal EM48 antibody to stain 6-, 12-, and 18-month-old BACHD and wild-type brains. To our surprise, EM48 failed to detect obvious or diffuse nuclear mhtt accumulation in the striata or cortices of the BACHD mice at any age examined (Fig. 6A–F). However, under the same staining conditions, we could readily detect nuclear accumulation of mhtt by EM48 staining in 12-month-old YAC128 mice (Fig. 6I) and in an mhtt-exon1 mouse model developed in our laboratory (Gu et al., 2005, 2007). Instead, in BACHD brains at both 12 and 18 months but not at 6 months of age, we detected large mhtt inclusions (up to 2.5 μm in diameter) in the deep cortical layers, smaller inclusions (0.5–1.5 μm in diameter) in the upper cortical layers, and very few tiny aggregates (<1 μm in diameter) in the striatum (Fig. 6E,H). Overall, the frequency of aggregates is higher in the cortex than in the striatum: on average, we detected ∼1 aggregate per 1000 μm2 in the cortex and 0.3 aggregates per 1000 μm2 in the striatum. We quantified the relative subcellular distribution of these aggregates in the neuropil (NAs) versus the nucleus (NIs) by double staining the 12- and 18-month-old brain sections with hematoxylin (staining the nucleus) and EM48 (Fig. 6G,H). We found that, at both 12 and 18 months, ∼93% of the aggregates were NAs and only ∼7% were NIs in both the cortex and striatum (Fig. 6J,K). This subcellular distribution pattern of mhtt aggregation, particularly in the BACHD cortex, resembles what has been observed in adult-onset HD (DiFiglia et al., 1997; Gutekunst et al., 1999). In addition to the cortex and striatum, occasional EM48+ neuropil aggregates were also detected in the globus pallidus and substantia nigra of BACHD animals, but they were not detected in other brain regions such as the cerebellum and brainstem (data not shown). In summary, the BACHD model demonstrates progressive accumulation of mhtt aggregates, and it reproduces a mhtt aggregation pattern that is reminiscent of that in adult-onset HD. Because overall BACHD mice have a very low abundance of mhtt aggregation in the striatum, and lack striatal nuclear translocation of EM48+ mhtt, our data suggest that progressive and selective nuclear translocation and accumulation of EM48+ mhtt is not necessarily associated with the pathogenesis in BACHD mice.
Figure 6.
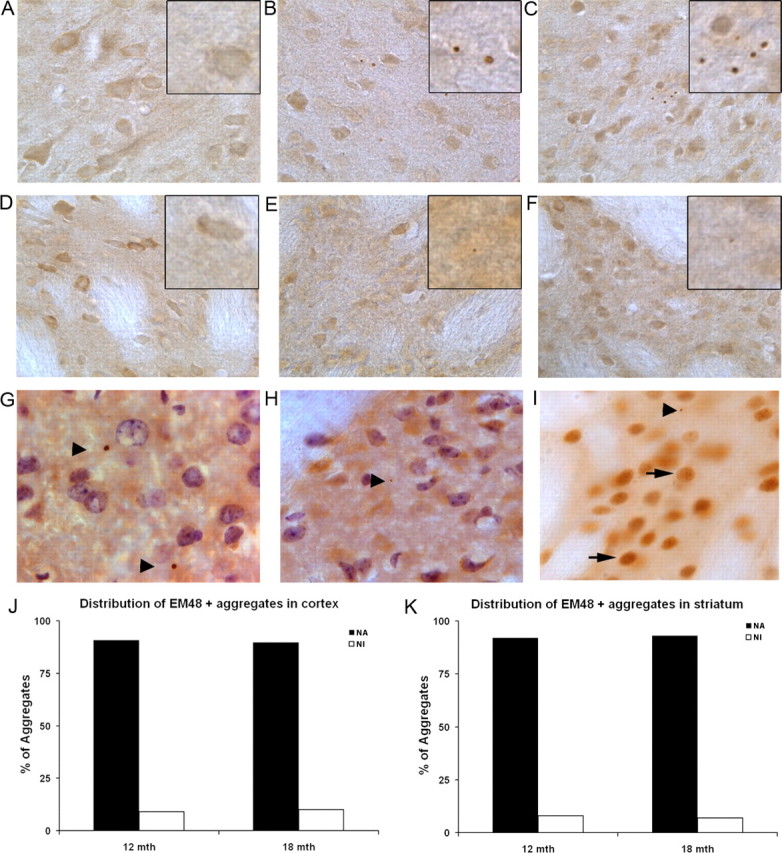
BACHD brains exhibit adult-onset HD pattern of mutant huntingtin aggregation. Polyclonal EM48 antibody staining of cortical sections (A–C) and striatal sections (D–F) from 6-, 12-, and 18-month-old BACHD brains. At 6 months, we do not detect any EM48+ aggregates or diffuse nuclear staining in BACHD cortices (A) or striata (D). At 12 months (B, E) and 18 months (C, F) of age, we detect large EM48+ inclusions in the cortex (see insets), and very tiny aggregates in the striata (G, H). We do not detect diffuse nuclear staining indicating the nuclear accumulation of EM48+ mhtt at any of these ages tested. EM48+ aggregates were never detected in wild-type littermate brains at these ages (data not shown). Combined nuclear staining with hematoxylin and EM48 staining reveal predominantly large neuropil aggregates (arrowheads) in BACHD cortices (G), and small striatal neuropil inclusions (H) at 12 months of age. I, YAC128 mice exhibit EM48+ staining in the nucleus at 12 months of age (arrows) and neuropil aggregates (arrowhead). J, K, Quantitation of EM48+ mhtt aggregate distribution in the neuropil (NA) versus nucleus (NI) in BACHD cortex (G) and striatum (H) at 12 and 18 months of age. Magnification: A–F, 40×; insets, 100×; G–I, 100×.
Analyses of BACHD-L line replicating phenotypic aspects of BACHD mice
When generating transgenic mice, it is important to characterize independent lines of mice to ensure that the phenotypes identified in one line are not merely attributable to the consequences of transgene integration. To that end, we performed behavioral and neuropathological analyses of another mouse line, the BACHD-L line. Western blot analyses with 1C2 and mAb2166 antibodies show that BACHD-L mice express predominantly full-length mhtt, but at a level that is about one-third of that of the BACHD line in three brain regions, the cortex, striatum, and cerebellum (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). To determine whether there were any motor deficits present in the BACHD-L mice, we performed rotarod analysis on these mice. At 2 months of age, rotarod performance of BACHD-L mice was comparable with their wild-type littermates. However, at 6 months of age, over the 3 d of repeated testing there are significant genotype differences between BACHD-L mice and wild-type littermates (F = 5.328; p = 0.035) (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material). We next performed EM48 analyses to determine whether the adult-onset pattern of mhtt aggregation, a key distinguishing phenotype in BACHD mice compared with the previous fl-mhtt mouse models, could be replicated in BACHD-L line. As shown in supplemental Figure S2, E–H (available at www.jneurosci.org as supplemental material), our examination shows 12-month-old BACHD-L brains have only a few small aggregates predominantly in the neuropil in the cortex and very tiny aggregates in the striatum, and we did not detect any diffuse EM48+ nuclear translocation of mhtt in either the cortex or the striatum. Thus, the EM48 staining pattern in BACHD-L line resembles the adult-onset HD pattern as seen in BACHD mice, albeit the size and load of mhtt aggregates is much less compared with BACHD as a result of lower level fl-mhtt expression in this line. Preliminary analyses of brain weight in BACHD-L line at 12 months did not yet reveal forebrain atrophy (data not shown), suggesting the neurodegenerative process in this model with lower level of mhtt expression is likely to be much slower than BACHD mice. In addition, the BACHD-L line exhibits an increase in weight when compared with wild-type siblings at 2 months of age (BACHD-L, 27.4 g; wild type, 25.3 g; p < 0.05) (supplemental Fig. S2I, available at www.jneurosci.org as supplemental material). However, the percentage weight gain in BACHD-L line mice (2 months, 8%; 6 months, 14% increase over wild-type siblings) is less than what is seen in BACHD mice (2 months, 21%; 6 months, 33% increase over wild type). Together, these data suggest that, although the BACHD-L line of mice expresses a much lower level of the full-length mhtt protein, these mice exhibit progressive rotarod deficits and replicate the adult-onset pattern of mhtt aggregation displayed by the BACHD mice, thus providing independent validation of several key phenotypes in the BACHD mice.
Biochemical analyses demonstrate the lack of progressive accumulation of polyQ-containing mhtt N-terminal fragments in the BACHD brain
Next, we performed biochemical analyses to determine whether BACHD mice have comparable levels of soluble mhtt fragments compared with one of the previous fl-mhtt models, YAC128 mice; and furthermore, whether progressive accumulation of such nonaggregated mhtt N-terminal fragments is a pathogenic feature of BACHD mice. Previous studies revealed that transfected primary neurons containing diffusely expressed mhtt fragments had an increased risk of death compared with transfected neurons containing mhtt inclusions (Arrasate et al., 2004). PolyQ-containing mhtt N-terminal fragments, as detected by 1C2 antibody, have been shown to progressively accumulate in an Hdh-KI mouse model in vivo (Zhou et al., 2003). We first compared mhtt fragments in 12-month-old BACHD and YAC128 mice. As shown in supplemental Figure S3 (available at www.jneurosci.org as supplemental material), with the 1C2 antibody, BACHD and YAC128 mice both express predominantly fl-mhtt, but they both have a small amount of mhtt fragments. This result is consistent with the interpretation that the mhtt N-terminal polyQ fragments may contribute to disease pathogenesis in both BACHD and YAC128 mice.
We then performed a series of Western blot analyses to examine whether BACHD mice exhibit progressive nuclear or cytoplasmic accumulation of mhtt N-terminal fragments over the course of disease, as previously observed in Hdh-KI mice (ibid). We performed Western blot analyses on cortical and striatal extracts using the S830 antibody, an anti-mhtt-exon 1 antibody that detects both soluble and certain aggregated mhtt N-terminal fragments, but not wild-type murine htt on Western blots (Benn et al., 2005). Analyses were performed on both the soluble fraction and the 10% SDS-soluble fraction. In the latter fraction, 10% SDS was shown to solubilize a substantial amount of mhtt aggregates from patient brain tissue (Huang et al., 1998). At both 2 and 12 months of age, we detected predominantly fl-mhtt and a small amount of mhtt N-terminal fragments in the BACHD cortex and striatum (Fig. 7A,B). We did not detect a significant amount of aggregated mhtt in the wells, which is consistent with the low mhtt aggregate load in these mice (Fig. 6B,E). We next performed nuclear and cytoplasmic fractionation of the BACHD brains at 2, 6, and 12 months of age followed by Western blotting with the 1C2 antibody to determine whether any polyQ-containing mhtt N-terminal fragments accumulated over time. As shown in Figure 7, at all ages tested and in both the cortex and striatum, we detected predominantly fl-mhtt in the cytoplasm and a small amount of fl-mhtt in the nucleus. We did not detect progressive nuclear or cytoplasmic accumulation of any polyQ-containing mhtt N-terminal fragments up to 12 months of age with the 1C2 antibody. When we reprobed the same blot with mAb2166 (Fig. 7E,F; supplemental Fig. S4, available at www.jneurosci.org as supplemental material), we were unable to detect any mhtt fragments unique to BACHD mice. We could detect some bands in the range of 55–64 kDa in both the wild-type and BACHD cortices and striata, which may change in abundance between 2 and 12 months in the mutant cortices (Fig. 7E). However, our interpretation is that these fragments are not polyQ-containing mhtt N-terminal fragments for the following reasons. First, the abundance of these fragments is comparable between wild-type and BACHD cortices at 12 months (ibid). Second, we do not detect any accumulation of such mhtt polyQ fragments in the same molecular range with the 1C2 antibody over time (Fig. 7C). However, our current analyses do not permit us to definitely address whether these mAb2166-positive fragments may have any pathogenic significance in BACHD mice.
Figure 7.
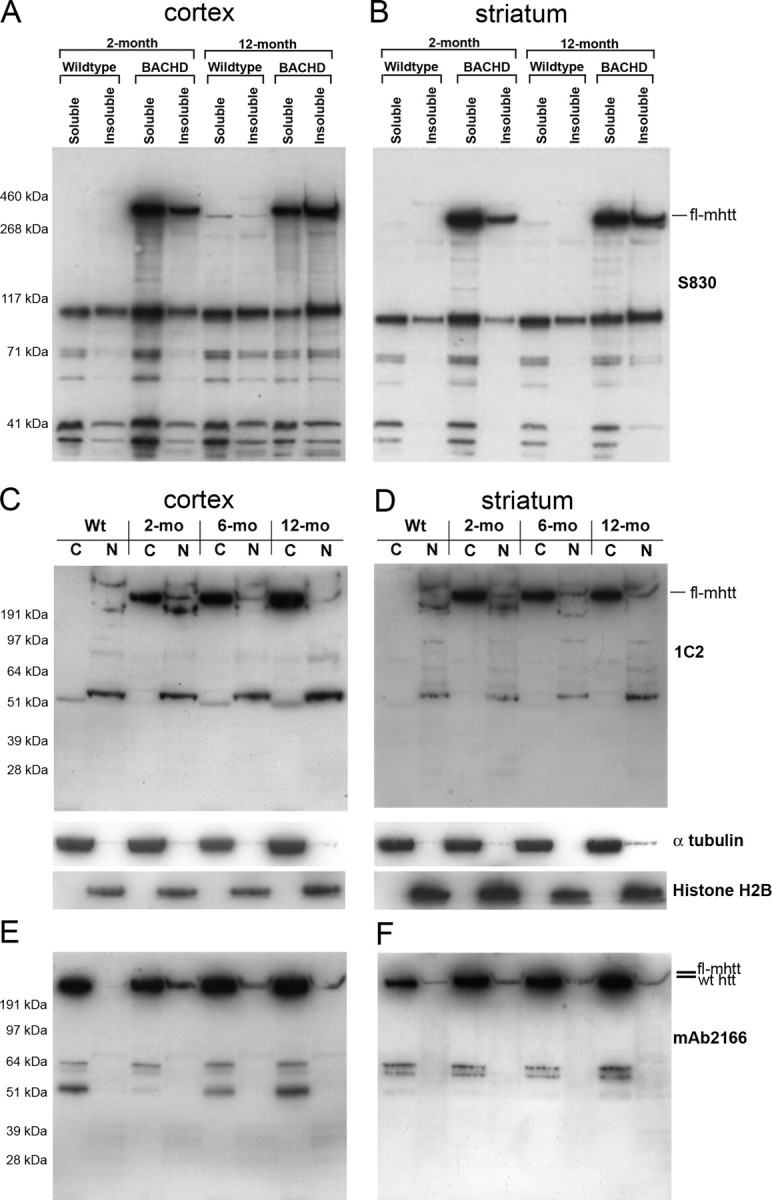
Western blot analysis of htt expression BACHD mice. A, B, Western blot analyses with anti-mhtt-exon1 antibody, S830, reveal lack of significant increases in mhtt N-terminal fragments in both soluble and insoluble fraction in the cortex (A) and striatum (B) of BACHD mice between 2 and 12 months of age. C, D, Subcellular fractionations were performed to isolate protein extracts from cytosolic (C) and nuclear (N) fractions from the cortex (C) and striatum (D) of BACHD mice at 2, 6, and 12 months of age and a wild-type mouse. The blots were first probed with the 1C2 antibody (top panels), which reveals a lack of significant accumulation of any 1C2+ mhtt N-terminal fragments in the nuclear or cytosolic fraction between 2, 6, and 12 months. Control for the fractionation process using anti-α-tubulin (cytoplasmic fraction) and anti-histone H2B (nuclear fraction). The same blots were probed with mAb2166 (E, F), to label both wild-type htt and mhtt. Both 1C2 and mAb2166 blots reveal that fl-mhtt is predominantly in the cytoplasm at all ages tested, but a small amount of fl-mhtt is also present in the nucleus at all ages.
In summary, our biochemical analyses provided evidence that BACHD mice have a small amount of polyQ-containing mhtt N-terminal fragments that may contribute to disease pathogenesis, but these polyQ-containing toxic mhtt fragments do not exhibit progressive nuclear or cytoplasmic accumulation before the onset of selective neuropathology. Because our data demonstrated a relatively small amount of fl-mhtt and wild-type htt in the nucleus, it suggests that fl-htt may have a normal nuclear function, and the relatively steady-state low level of nuclear mhtt could also play a pathogenic role in HD.
Neuronal dysfunction precedes selective neurodegeneration in BACHD brains
Existing mouse models of neurodegeneration suggest that neuronal dysfunction often precedes neurodegeneration and may provide important clues regarding the nature of early pathogenic mechanisms that trigger late-onset degeneration. In BACHD mice, we already demonstrated robust motor deficits in rotarod performance at 6 months of age that precede the onset of selective neuropathology at 12 months. We examined basic electrophysiological and synaptic properties in 6-month-old BACHD mice for direct evidence of neuronal dysfunction. We performed voltage- and current-clamp recordings of striatal MSNs using patch pipettes in acute brain slices from wild-type and BACHD mice. There were no significant differences in passive membrane properties (capacitance, input resistance, or time constant) or resting membrane potentials in MSNs from wild-type (n = 22) and BACHD (n = 19) mice (data not shown). Neurons from both groups of mice displayed typical inward rectification (Fig. 8A). In contrast, there were significant differences in the occurrence of spontaneous inward currents. The overall frequency of spontaneous EPSCs (holding potential, −70 mV) was slightly reduced in MSNs from BACHD compared with wild-type mice (1.8 ± 0.21 and 2.3 ± 0.26 Hz, respectively) (Fig. 8B), but the difference was not statistically significant (p = 0.16). However, analysis of amplitude–frequency histograms of spontaneous currents revealed that a number of medium- to large-amplitude bins displayed significantly reduced frequencies in cells from BACHD mice (Fig. 8C). When grouped together, the difference in frequency of events >20 pA was significantly reduced in BACHD compared with wild-type mice (Fig. 8C, inset) (p < 0.01). Cumulative interevent interval distributions, a measure of release probability, were not different between cells from wild-type and BACHD mice (Fig. 8D). In contrast, the relative probability of occurrence of small-amplitude events was significantly greater in cells from BACHD compared with wild-type mice (Fig. 8E) which is expected because there was a significant decrease in larger amplitude events. These results suggest that the excitatory input to the striatal MSNs is altered very early in the BACHD brains, and it is possible that this early neuronal dysfunction may underlie some of the motor deficits observed in this model.
Figure 8.
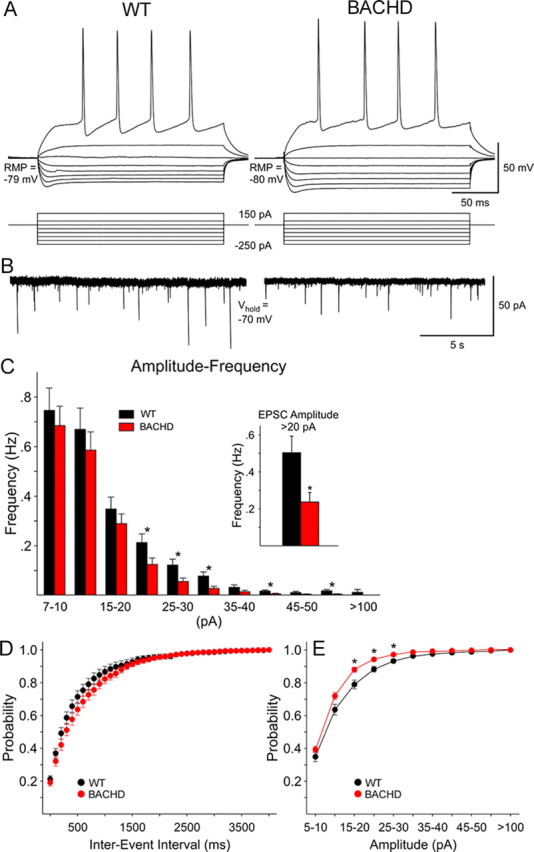
BACHD mice demonstrate decreases in synaptic activity of medium-sized spiny neurons at 6 months. A, Current-clamp recordings show the voltage deflections induced by hyperpolarizing and depolarizing current pulses in wild-type (WT) and BACHD striatal neurons. At 150 pA, both cells displayed repetitive action potentials. B, Voltage-clamp recordings from the same cells indicated that the overall frequency of spontaneous synaptic currents was not different in cells from WT or BACHD animals. However, medium- and large-amplitude (>20 pA) synaptic currents occurred more frequently in WT cells. C, Graph shows the amplitude–frequency distributions of spontaneous synaptic events. When grouped together, the average number of medium- to large-amplitude synaptic currents was significantly reduced in cells from BACHD mice (p < 0.01, inset). D, Cumulative interevent interval distributions, a measure of release probability, overlapped in cells from WT and BACHD mice. E, In contrast, relative cumulative amplitude distributions revealed that small-amplitude events had higher probability in cells from BACHD compared with cells from WT mice. The asterisks indicate the differences were statistically significant (p < 0.01 to p < 0.001). Error bars indicate SEM.
Because BDNF is a well known molecular player in modulation of synaptic transmission and plasticity (Lu, 2003) and axonal transport (Baquet et al., 2004; Gauthier et al., 2004) and cortical BDNF expression (Zuccato et al., 2001) is affected in existing HD mice and HD patients, we examined whether an alteration of cortical BDNF expression might be an early phenotype in the BACHD brains. We performed quantitative RT-PCR analyses using RNA derived from BACHD cortices at 6 months of age, and we found a robust 50% reduction of cortical BDNF transcripts, which is significant compared with that in the wild-type controls (Fig. 9) (p < 0.05, Student's t test). Our results suggest that a reduction in cortical BDNF transcription is an early and robust feature of the BACHD model. Together, both the striatal electrophysiological studies and cortical BDNF expression analyses provide direct evidence that early cortical and striatal neuronal dysfunction coincides with the occurrence of motor deficits, and may contribute to the late-onset selective neuropathogenesis in BACHD mice.
Figure 9.

Reduced BDNF expression in BACHD mice. Quantitative RT-PCR analyses reveal a reduction of cortical BDNF expression in 6-month-old BACHD mice compared with wild-type control mice for the entire coding region of BDNF (*p < 0.05, Student's t test) and for exon IV (***p < 0.0001). Error bars indicate SEM.
BACHD as a robust preclinical mouse model expressing full-length mutant huntingtin
We next asked whether BACHD could be used as a robust preclinical mouse model of HD expressing fl-mhtt. To facilitate preclinical studies in HD, a mouse model should have phenotypes that not only recapitulate key features of disease (i.e., selective neuropathology) but are also robust with low phenotypic variability. Using the data obtained from the characterization of the BACHD mice, we performed power analyses to estimate the number of mice that would be required to observe a rescue of some of the disease-relevant parameters identified in the BACHD mice. BACHD mice demonstrated significant rotarod deficits at 6 months of age without overt neuropathological changes. Power analysis showed that a sample size of only 22 BACHD mice per treatment group (i.e., drug treatment or vehicle controls) is necessary to have 80% power of detecting a modest therapeutic benefit (i.e., a 30% rescue of the rotarod deficit at 6 months of age) (Fig. 10A,E). In addition to the behavioral assessment after therapeutic intervention, neuropathological assessment is also necessary. Power analysis demonstrated that to have an 80% chance to detect a 30% rescue of the striatal dark neuron degeneration phenotype we would need to use only 14 BACHD mice per treatment group (Fig. 10B,E). Furthermore, to have an 80% chance to observe a 30% rescue of the cortical and striatal volumes, we would need to use only 5 and 11 mice per treatment group, respectively (Fig. 10C–E). In the neuropathological measures, a SD of 15–20% can be expected. Thus, the power analyses reveal that BACHD mice exhibit robust behavioral and neuropathological outcomes, demonstrating BACHD is a suitable fl-mhtt mouse model for HD preclinical studies.
Figure 10.

BACHD mice for therapeutic testing. Power analyses of BACHD mice. A, Chart of power analysis for rotarod data at 6 months of age. B–D, Dark neuron degeneration (B), cortical volume (C), and striatal volume (D) power analyses at 12 months of age. The red line indicates number of experimental mice used in each study (BACHD plus controls). The other lines (blue, pink, gray, and green) indicate number of total experimental mice that could be used for therapeutic studies. E, Table of power analyses, indicating number of experimental mice needed to have an 80% chance of observing a 30, 50, or 70% rescue of phenotypes at the significance level of p < 0.05.
Discussion
We developed a novel human fl-mhtt expressing transgenic mouse model of HD to study disease pathogenesis and to test preclinical candidates. BACHD mice express functional human fl-mhtt under the control of its endogenous regulatory machinery and recapitulate several key phenotypic features of HD, including progressive motor deficits, late-onset and relatively selective neuropathology in the cortex and striatum with no change in cerebellum, and a pattern of mhtt aggregation reminiscent of that in adult-onset HD. Several salient phenotypic features of BACHD mice, including progressive motor deficits and an adult-onset pattern of mhtt aggregation, can be replicated in the independent BACHD-L line. The BACHD-L mice have a milder phenotype than BACHD mice because of their lower level of fl-mhtt expression. Our power analyses on the key behavioral and neuropathological outcomes provide strong evidence that BACHD mice could be used as a novel and robust preclinical fl-mhtt mouse model for HD. One phenotype of BACHD mice, the body weight gain, is not a clinical feature of HD because HD patients often exhibit weight loss after disease onset. Because overexpression of both human mutant and wild-type htt in the YAC models (i.e., YAC128 and YAC18) also result in weight gain as in BACHD mice (Van Raamsdonk et al., 2006), the weight gain phenotype in these mice is likely attributable to the dose-dependent increase of htt function. This is further supported by the comparison of the BACHD and BACHD-L lines in which BACHD mice weigh more than the lower expressing BACHD-L line mice. Previous analyses of the YAC mice showed that HD-related neuropathology only occurs in the mice expressing mhtt but not those expressing the wild-type htt (ibid), demonstrating the weight gain is not causally related to the HD-like phenotypes in these mice. Consistent with this interpretation, our current analyses reveal that the rotarod deficits in BACHD mice are not correlated with weight gain, and significant and selective HD-like neuropathology (including selective brain atrophy and adult-onset HD pattern of mhtt aggregation) can occur in aged BACHD brains despite the body weight phenotype. Therefore, we conclude that the HD-like phenotypes in BACHD mice are attributable to the pathogenic effects of mhtt, and are not attributable to the dosage-dependent effects of increasing htt function (i.e., weight gain). In summary, our phenotypic analyses demonstrate that BACHD mice are a novel and robust paradigm to study the pathogenic effects of fl-mhtt in vivo and to test preclinical candidates.
The analyses of BACHD mice provided novel insights into the role of somatic repeat instability in selective neuropathogenesis in HD. Previous studies of Hdh knock-in mice (Kennedy and Shelbourne, 2000; Ishiguro et al., 2001) and a recent study in HD patients (Shelbourne et al., 2007) have implicated somatic CAG repeat expansion in selective neuronal vulnerability in HD, because the expansion is more dramatic in the striatum and less so in the cerebellum. A recent study also reveals that the CAG repeat expansion in the mouse brains appears to be attributable to an age-dependent repair of oxidized DNA base pairs by the excision repair enzyme, OGG1 (Kovtun et al., 2007). This latter study provides a mechanism to support the role of somatic repeat instability in age-dependent disease pathogenesis in HD. Despite these appealing results, more direct genetic studies are needed to address the role of somatic repeat instability in mhtt in the pathogenesis of selective neuropathology in vivo. We believe the BACHD model provides some novel insights into this question. Our results clearly demonstrate that somatic repeat instability is not required for the onset of progressive motor deficits and selective neuropathology in the BACHD model. Our study is consistent with previous studies from other polyQ disease mouse models (i.e., SCA1 and SCA7 mice) suggesting somatic repeat instability in a given region is not correlated with neuropathology (Orr et al., 1993; Libby et al., 2003; Watase and Zoghbi, 2003; Jackson et al., 2005). Although our model argues against the role of somatic instability in selective neuropathogenesis in HD, our data could not exclude the possibility that somatic repeat expansion could be a modifier of disease pathogenesis in vivo.
Our analyses of the BACHD mice also provided novel molecular insights into how mhtt may elicit selective neuropathogenesis in vivo. Studies of previous fl-mhtt mouse models consistently demonstrate an early and selective striatal and cortical nuclear accumulation of aggregated form of mhtt that is stained positive by EM48 (Li et al., 2000; Wheeler et al., 2000; Menalled et al., 2002; Slow et al., 2003). These studies prompted the hypothesis that nuclear accumulation of EM48+ mhtt may be a critical early event required for selective neuropathogenesis in HD (Leavitt et al., 1999; Ross, 2002). Analyses of BACHD and BACHD-L mice do not reveal any diffuse nuclear accumulation of EM48+ mhtt in the cortex or striatum up to 18 months of age. This is not attributable to any technical issues related to EM48 immunostaining because we can readily detect EM48 nuclear staining in the striata of our mhtt exon1 model (Gu et al., 2005, 2007) and YAC128 mice (Fig. 6I). EM48 staining revealed that BACHD mice exhibit a distribution pattern of mhtt aggregates (i.e., more cortical than striatal and predominantly neuropil and less nuclear) reminiscent of that in adult-onset HD (DiFiglia et al., 1997; Gutekunst et al., 1999). Thus, BACHD is the first fl-mhtt mouse model demonstrating that early striatal and cortical nuclear accumulation of EM48+ mhtt is not necessary for selective neuropathogenesis. This result clearly emphasizes the important contribution of cytoplasmic full-length and fragmented mhtt in HD pathogenesis, but our biochemical analyses consistently reveal a small fraction of mhtt in the nucleus, hence suggesting the nuclear mhtt may also be pathogenic in our model.
Proteolysis of fl-mhtt to generate toxic mhtt N-terminal fragments has been postulated as a critical mechanism for HD pathogenesis (Ross, 2004; Graham et al., 2006). Our analyses of BACHD mice, particularly the comparison of mhtt fragments in this model with YAC128 mice (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), is consistent with the model that the mhtt N-terminal fragments may contribute to the disease pathogenesis in BACHD mice. Because recent studies suggest that the caspase 6 cleavage of fl-mhtt can generate a particular toxic mhtt fragment in vivo (Graham et al., 2006), we initiated collaboration [with R. K. Graham and M. R. Hayden (University of British Columbia, Vancouver, British Columbia, Canada) and with L. M. Ellerby (Buck Institute for Age Research, Novato, CA)] to characterize mhtt proteolysis in BACHD mice. Our initial analyses reveal that BACHD brains also have a small amount of caspase-6 cleaved mhtt toxic fragments (data not shown). Although our study is consistent with the potential role of mhtt fragments in HD pathogenesis, our biochemical analyses consistently reveal relatively steady-state levels of mhtt N-terminal fragments present in BACHD brains between 2 and 12 months of age. Hence, our study supports a model in which a relatively steady-state level of predominantly fl-mhtt and small amount of mhtt toxic fragments are sufficient to initiate selective neuropathogenesis in HD.
Analyses of BACHD mice provide strong evidence that aberrant corticostriatal neuronal interactions may underpin selective neuropathogenesis in HD. Our electrophysiological studies revealed that spontaneous excitatory neurotransmission, in the form of AMPA receptor-mediated spontaneous EPSCs, is significantly reduced in BACHD striatal neurons at 6 months of age. Although our current study could not precisely determine the cellular origin for such deficits (i.e., cortical neurons vs striatal neurons), it is apparent that abnormal reduction of the glutamatergic communication between the cortical and striatal neurons is an early dysfunction in BACHD mice. This result is consistent with previous studies revealing early dysfunction in cortical inputs and NMDA receptors in striatal neurons long before the onset of neurodegenerative changes in a variety of HD mouse models (Levine et al., 1999; Cepeda et al., 2001, 2003; Zeron et al., 2002; Starling et al., 2005). As additional support of aberrant corticostriatal interactions in BACHD mice, our study reveals a robust reduction of BDNF expression in BACHD cortex at 6 months of age. BDNF is an important neurotrophic factor in regulating neuronal transmission (Lu, 2003), striatal neuronal survival (Baquet et al., 2004), and has previously been implicated in HD pathogenesis (Zuccato et al., 2001; Gauthier et al., 2004). Our study provides valuable confirmation on the potential role of cortical BDNF reduction in selective striatal pathogenesis in HD. Based on our previous studies in a mhtt fragment model that pathological cell–cell interactions are likely to be important in HD pathogenesis (Gu et al., 2005, 2007), future studies are needed to more precisely define the role of pathological cell–cell interactions in a fl-mhtt mouse model. To this end, the BACHD model is specifically designed to be the first Cre/LoxP conditional mouse model of HD, in which the expression of fl-mhtt can be switched off in any cell type expressing Cre recombinase (data not shown). Future analyses of BACHD mice with fl-mhtt overexpression switched off in specific cell types (i.e., cortical pyramidal neurons or striatal medium spiny neurons) will help to define the roles of cell-autonomous mhtt toxicities and pathological cell–cell interactions in selective neuropathogenesis in HD.
Footnotes
This work was supported by the Hereditary Disease Foundation and National Institute of Neurological Disorders and Stroke (NINDS)/National Institutes of Health Grants 5R01NS049501 and 1R21NS047391 (X.W.Y.). M.G. is supported by the University of California, Los Angeles (UCLA) Mental Retardation Research Center Training Grants 5T32HD007032 (from the National Institute of Child Health and Human Development) and by UCLA Center for Neurobehavioral Genetics Training Grant 5T32NS048004 (from NINDS). We thank N. S. Wexler and C. Johnson for support and discussions, Scott Zeitlin (University of Virginia, Charlottesville, VA) for the Hdh knock-out mice, G. Bates (University College London, London, UK) for the S830 antibody, M. A. Hickey and M. F. Chesselet for advice on mouse behavioral studies, T. Fielder (University of California, Irvine, Irvine, CA) for performing the pronuclear microinjections, and members of the Yang Laboratory for discussions and comments on this manuscript. We also thank R. K. Graham, M. R. Hayden, and L. M. Ellerby for sharing unpublished data.
References
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn CL, Landles C, Li H, Strand AD, Woodman B, Sathasivam K, Li SH, Ghazi-Noori S, Hockly E, Faruque SM, Cha JH, Sharpe PT, Olson JM, Li XJ, Bates GP. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet. 2005;14:3065–3078. doi: 10.1093/hmg/ddi340. [DOI] [PubMed] [Google Scholar]
- Brandt J, Bylsma FW, Aylward EH, Rothlind J, Gow CA. Impaired source memory in Huntington's disease and its relation to basal ganglia atrophy. J Clin Exp Neuropsychol. 1995;17:868–877. doi: 10.1080/01688639508402436. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry S, Mukerji M, Srivastava AK, Jain S, Brahmachari SK. CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum Mol Genet. 2001;10:2437–2446. doi: 10.1093/hmg/10.21.2437. [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dorsman JC, Bremmer-Bout M, Pepers B, van Ommen GJ, Den Dunnen JT. Interruption of perfect CAG repeats by CAA triplets improves the stability of glutamine-encoding repeat sequences. Biotechniques. 2002;33:976–978. doi: 10.2144/02335bm01. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gong S, Yang XW. Modification of bacterial artificial chrmosomes (BACs) and preparation of intact BAC DNA for generation of transgenic mice. In: Crawley JN, Gerfen CR, Rogawski MA, Sibley DR, Skolnick P, Wray S, editors. Current protocols in neuroscience. New York: Wiley; 2005. p. 5.21.21. [DOI] [PubMed] [Google Scholar]
- Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 2002;12:1992–1998. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Gu X, Andre VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME, Gusella JF. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat Cell Mol Genet. 1998;24:217–233. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Yamada K, Sawada H, Nishii K, Ichino N, Sawada M, Kurosawa Y, Matsushita N, Kobayashi K, Goto J, Hashida H, Masuda N, Kanazawa I, Nagatsu T. Age-dependent and tissue-specific CAG repeat instability occurs in mouse knock-in for a mutant Huntington's disease gene. J Neurosci Res. 2001;65:289–297. doi: 10.1002/jnr.1153. [DOI] [PubMed] [Google Scholar]
- Jackson SM, Whitworth AJ, Greene JC, Libby RT, Baccam SL, Pallanck LJ, La Spada AR. A SCA7 CAG/CTG repeat expansion is stable in Drosophila melanogaster despite modulation of genomic context and gene dosage. Gene. 2005;347:35–41. doi: 10.1016/j.gene.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Kassubek J, Gaus W, Landwehrmeyer GB. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2004;62:523–524. doi: 10.1212/wnl.62.3.523-a. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy L, Shelbourne PF. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum Mol Genet. 2000;9:2539–2544. doi: 10.1093/hmg/9.17.2539. [DOI] [PubMed] [Google Scholar]
- Kennedy L, Evans E, Chen CM, Craven L, Detloff PJ, Ennis M, Shelbourne PF. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. 2003;12:3359–3367. doi: 10.1093/hmg/ddg352. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer HP, Roos RA, Dingjan GM, Bots GT, Bruyn GW, Hofman MA. The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington's disease. Neurosci Lett. 1991;132:101–104. doi: 10.1016/0304-3940(91)90443-w. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- Leavitt BR, Wellington CL, Hayden MR. Recent insights into the molecular pathogenesis of Huntington disease. Semin Neurol. 1999;19:385–395. doi: 10.1055/s-2008-1040853. [DOI] [PubMed] [Google Scholar]
- Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- Libby RT, Monckton DG, Fu YH, Martinez RA, McAbney JP, Lau R, Einum DD, Nichol K, Ware CB, Ptacek LJ, Pearson CE, La Spada AR. Genomic context drives SCA7 CAG repeat instability, while expressed SCA7 cDNAs are intergenerationally and somatically stable in transgenic mice. Hum Mol Genet. 2003;12:41–50. doi: 10.1093/hmg/ddg006. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- Manley K, Pugh J, Messer A. Instability of the CAG repeat in immortalized fibroblast cell cultures from Huntington's disease transgenic mice. Brain Res. 1999;835:74–79. doi: 10.1016/s0006-8993(99)01451-1. [DOI] [PubMed] [Google Scholar]
- McFadyen MP, Kusek G, Bolivar VJ, Flaherty L. Differences among eight inbred strains of mice in motor ability and motor learning on a rotorod. Genes Brain Behav. 2003;2:214–219. doi: 10.1034/j.1601-183x.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Chesselet MF. Mouse models of Huntington's disease. Trends Pharmacol Sci. 2002;23:32–39. doi: 10.1016/s0165-6147(00)01884-8. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Wu Y, Olivieri M, Li XJ, Li H, Zeitlin S, Chesselet MF. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996;366:580–599. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Eichler EE, Lorenzetti D, Kramer SF, Zoghbi HY, Nelson DL, Sinden RR. Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation. Biochemistry. 1998;37:2701–2708. doi: 10.1021/bi972546c. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- Petersen A, Gil J, Maat-Schieman ML, Bjorkqvist M, Tanila H, Araujo IM, Smith R, Popovic N, Wierup N, Norlen P, Li JY, Roos RA, Sundler F, Mulder H, Brundin P. Orexin loss in Huntington's disease. Hum Mol Genet. 2005;14:39–47. doi: 10.1093/hmg/ddi004. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ross CA. Huntington's disease: new paths to pathogenesis. Cell. 2004;118:4–7. doi: 10.1016/j.cell.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A, Hedreen J, Sisodia S, Snyder SH, Dawson TM, Ryugo DK, Ross CA. Widespread expression of Huntington's disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Shelbourne PF, Keller-McGandy C, Bi WL, Yoon SR, Dubeau L, Veitch NJ, Vonsattel JP, Wexler NS, Arnheim N, Augood SJ. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. 2007;16:1133–1142. doi: 10.1093/hmg/ddm054. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Smith MA, Brandt J, Shadmehr R. Motor disorder in Huntington's disease begins as a dysfunction in error feedback control. Nature. 2000;403:544–549. doi: 10.1038/35000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spargo E, Everall IP, Lantos PL. Neuronal loss in the hippocampus in Huntington's disease: a comparison with HIV infection. J Neurol Neurosurg Psychiatry. 1993;56:487–491. doi: 10.1136/jnnp.56.5.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling AJ, Andre VM, Cepeda C, de Lima M, Chandler SH, Levine MS. Alterations in N-methyl-d-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington's disease. J Neurosci Res. 2005;82:377–386. doi: 10.1002/jnr.20651. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Gibson WT, Pearson J, Murphy Z, Lu G, Leavitt BR, Hayden MR. Body weight is modulated by levels of full-length huntingtin. Hum Mol Genet. 2006;15:1513–1523. doi: 10.1093/hmg/ddl072. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Watase K, Venken KJ, Sun Y, Orr HT, Zoghbi HY. Regional differences of somatic CAG repeat instability do not account for selective neuronal vulnerability in a knock-in mouse model of SCA1. Hum Mol Genet. 2003;12:2789–2795. doi: 10.1093/hmg/ddg300. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Gutekunst CA, Vrbanac V, Lebel LA, Schilling G, Hersch S, Friedlander RM, Gusella JF, Vonsattel JP, Borchelt DR, MacDonald ME. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet. 2002;11:633–640. doi: 10.1093/hmg/11.6.633. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12:273–281. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]