

Abstract
We have investigated the assembly of FtsZ from Mycobacterium tuberculosis (MtbFtsZ). Electron microscopy confirmed the previous observation that MtbFtsZ assembled into long, two-stranded filaments at pH 6.5. However, we found that assembly at pH 7.2 or 7.7 produced predominantly short, one-stranded protofilaments, similar to those of Escherichia coli FtsZ (EcFtsZ). Near pH 7, which is close to the pH of M. tuberculosis cytoplasm, MtbFtsZ formed a mixture of single- and two-stranded filaments. We developed a fluorescence resonance energy transfer assay to measure the kinetics of initial assembly and the dynamic properties at steady state. Assembly of MtbFtsZ reached a plateau after 60−100 s, about 10 times slower than EcFtsZ. The initial assembly kinetics were similar at pH 6.5 and 7.7, despite the striking difference in the polymer structures. Both were fit with a cooperative assembly mechanism involving a weak dimer nucleus, similar to EcFtsZ but with slower kinetics. Subunit turnover and GTPase at steady state were also about 10 times slower for MtbFtsZ than for EcFtsZ. Specifically, the half-time for subunit turnover in vitro at pH 7.7 was 42 s for MtbFtsZ compared with 5.5 s for EcFtsZ. Photobleaching studies in vivo showed a range of turnover half-times with an average of 25 s for MtbFtsZ as compared with 9 s for EcFtsZ.
FtsZ, a bacterial homologue of tubulin, is the major cytoskeletal protein in bacterial cell division. FtsZ from most bacterial species assembles into single straight protofilaments in vitro, and these assemble further in vivo to form the cytokinetic Z-ring. FtsZ of Escherichia coli (EcFtsZ)2 shows rapid dynamics both in vitro (1) and in vivo (2, 3), with a turnover half-time of about 9 s. The turnover rate of the Z-ring in the Gram-positive Bacillus subtilis is identical, so the rapid assembly dynamics may be a universal part of the bacterial division mechanism.
Recently we developed two assays to study the kinetics of E. coli FtsZ in vitro. One is based on the tryptophan mutant, FtsZ-L68W, which showed a 2.5-fold increase in fluorescence upon assembly (4). The other is a FRET (fluorescence resonance energy transfer)-based assay (1). Both assays showed that assembly of EcFtsZ is a cooperative process with a weak dimer nucleus. Initial assembly was very fast and reached a plateau within 5 to 10 s. The FRET assay was used to monitor the rate of subunit exchange at steady state by mixing preassembled donor and acceptor EcFtsZ. The half-time for exchange was about 7 s, very close to the 8−9 s in vivo turnover measured by FRAP (fluorescence recovery after photobleaching) (3). Thus every protofilament in the Z ring, or in a steady state mixture in vitro, is disassembling and reassembling with a 7−9 s half-time.
Tuberculosis is the single greatest cause of death in humans by a bacterium. The World Health Organization estimates that nearly 3 million people each year die from tuberculosis. Several reports have already studied FtsZ as a promising target for new antibacterial drug discovery, some of them with a particular interest in the FtsZ of Mycobacterium tuberculosis (MtbFtsZ) (5-7). MtbFtsZ has been studied previously and was reported to differ from EcFtsZ in several respects (8, 9). In contrast to the short, one-stranded protofilaments of EcFtsZ, MtbFtsZ assembled into long, two-stranded filaments. The kinetics of initial assembly, assayed by light scattering, were reported to be much slower than those of EcFtsZ. Dynamics at steady state were not examined. Because our FRET assay has several important advantages over light scattering, we used it to study the kinetics and assembly dynamics of MtbFtsZ.
EXPERIMENTAL PROCEDURES
MtbFtsZ Purification
pSAR1 is a pET15 vector expressing a His-tagged MtbFtsZ (10). We expressed the protein in BL21-pLysS and prepared a bacterial lysate as described previously (4). The His-tagged MtbFtsZ protein in Talon buffer (20 mm Tris-HCl, 300 mm KCl, pH 7.4) was passed over a Talon metal affinity column (Clontech Laboratories) and eluted with the same buffer containing 150 mm imidazole. After dialysis against the Talon buffer, 2 units/ml thrombin was added to remove the His tag. The reaction was incubated for 2 h at 4 °C. The sample was mixed with Talon beads again to remove any remaining His-tagged MtbFtsZ. The sample was dialyzed into column buffer (50 mm Tris, pH 7.7, 50 mm KCl, 10% glycerol) and further purified on a Source 15Q 10/10 column (GE Healthcare), eluted with a linear gradient of 50−500 mm KCl in column buffer. Peak fractions were identified by SDS-PAGE and stored at −80 °C.
Protein Labeling
MtbFtsZ has a single cysteine, Cys-155, at the position of E. coli Val-157. This location is not near any known protofilament interface, and so we used this site to attach a fluorescent label. Donor FtsZ was prepared by reacting with fluorescein-5-maleimide (Molecular Probes) and acceptor with tetramethylrhodamine-5-maleimide (TMR; Molecular Probes). Reactions contained a 5-fold excess of probe and were incubated for 2 h at room temperature in a pH 7.9 Tris buffer. Samples were dialyzed three times against 50 mm HEPES, pH 7.7, 100 mm KAc, 2.5 mm MgAc, and 1 mm EGTA to remove free probe. A cycle of calcium assembly and disassembly was done to remove any inactive protein and remaining free probe, as described by Chen and Erickson (1). Protein was resuspended in the desired buffer and stored at −80 °C. The labeling efficiency was 30−40% for each fluorophore.
Most of the buffers used for assembly contained 100 mm KAc, 5 mm MgAc, and 1 mm EGTA. The pH 6.5 and 6.7 buffers contained 50 mm MES; pH 6.9 and 7.2 buffers had 50 mm MOPS; and pH 7.4 and 7.7 buffers had 50 mm HEPES.
Electron Microscopy
Negative stain electron microscopy was used to visualize MtbFtsZ filaments. About 10 μl of MtbFtsZ in the appropriate buffer was incubated with GTP for 5−10 min and applied to a carbon-coated copper grid. Grids were negatively stained with 2% uranyl acetate, and specimens were imaged with a Philips 301 electron microscope at ×50,000 magnification.
GTPase Activity Measurement
GTPase activity was determined by an improved sensitivity malachite green assay (11) as described in detail by Redick et al. (12).
Fluorescence and Light-scattering Measurements
Kinetics data were measured using a Shimadzu RF-5301 PC spectrofluorometer. For fluorescence measurements, the samples were excited at 450 nm and detected at 515 nm. For light scattering, the samples were both excited and measured at 350 nm. Because the assembly was slow we did not use a stopped flow device. For kinetic measurements assembly was initiated by adding GTP to 200 μm and pipetting to mix rapidly. The measurement started immediately after GTP was added. To assay for FtsZ filament exchange, donor- and acceptor-labeled FtsZ were assembled separately for 2 min in 0.5 mm GTP. Equal volumes of the two polymer samples were mixed, and measurement was begun. To measure GDP-induced disassembly, a mixture of acceptor and donor protein was first assembled in 0.1 mm GTP, and the fluorescence measurement was begun after adding 2 mm GDP.
Kinetic Modeling
The MtbFtsZ assembly kinetics data were fit using the minimal kinetic model described previously for EcFtsZ (1, 4). This model has three steps: monomer activation, formation of a dimer nucleus, and elongation. The data were fit using the programs KINSIM40 and FITSIM40 (13-16). The kinetic mechanism we used is as follows.
| SCHEME 1 |
k1 and k−1 describe a step of monomer activation when assembly is initiated by the addition of GTP (perhaps the time to release GDP and bind GTP; this step is independent of the FtsZ concentration). k2 and k−2 are for the formation of the dimer nucleus. k3 and k−3, and all subsequent rate constant pairs, are equal to ke and k−e and describe the steps of elongation when we use a dimer nucleus model to fit the data.
Growth of Mycobacterium smegmatis Cultures
Merodiploid M. smegmatis strain mC2-79 carries the plasmid pJFR79 (Pami:ftsZsmeg-GFP), which expresses MsmFtsZ-GFP (17). Strain mC2-11 carries plasmid pJFR11 (Pami:ftsZtb-GFP), which expresses MtbFtsZ-GFP (18). Strains were maintained as freezer stocks at −80 °C and streaked onto plates of 7H10 Middlebrook agar supplemented with albumin-dextrose complex and 50 μg/ml kanamycin 4 days prior to use in a FRAP experiment. After 3 days, one colony of M. smegmatis cells was transferred into liquid medium (7H9 Middlebrook broth supplemented with albumin-dextrose complex and 0.05% Tween 80 plus 50 μg/ml kanamycin) and grown overnight at 37 °C with shaking. The following day, cultures were diluted to OD ∼ 0.1 and grown for 4−6 h before undergoing FRAP microscopy.
Microscopy and in Vivo FRAP Measurements
Culture samples of 4−5 μl were applied to the surface of 1% agarose pads and then covered with a coverslip, as in our most recent FRAP studies (3). Cells were imaged, and FRAP experiments were performed on a Nikon TE300 inverted fluorescence microscope equipped with a laser source and driven by Metamorph software as described previously (2, 3, 19). All images and FRAP measurements were taken at room temperature. Exposures were typically 500 ms, and laser bleach pulses were 100 ms. Measurements of integrated fluorescence intensity in the bleached and other regions at each time point were made in Metamorph and exported to Microsoft Excel. For each measured region, background intensity was subtracted, and a correction factor was applied for overall photobleaching of the sample during observation (determined from intensity over time of a control cell in the same field). Recovery half-times were determined by performing a least-squares fit to a single-exponential equation as described previously (3).
RESULTS
MtbFtsZ Assembles into Different Filamentous Structures Depending on pH
The previous study of MtbFtsZ reported assembly into long, two-stranded filaments (8), in contrast to the much shorter, one-stranded protofilaments of EcFtsZ (1). The study of MtbFtsZ used an assembly buffer at pH 6.5. We confirmed that MtbFtsZ assembly gave a predominantly long, two-stranded filaments at pH 6.5 (Fig. 1a). Even in the early stages, the assembly consisted predominantly of short two-stranded filaments. Single protofilaments were also found, but they were rare. However, we found that at pH 7.7 MtbFtsZ assembled predominantly into short, one-stranded protofilaments, very similar to those of EcFtsZ (Fig. 1c). At intermediate pH, between 6.7 and 7.4, MtbFtsZ formed a mixture of two-stranded and one-stranded filaments (Fig. 1b). If we added 10 mm calcium to the pH 7.7 assembly buffer, we obtained long, two-stranded filaments or multistranded bundles. If we increased the KAc from 100 to 350 mm at pH 7.7, about half of the filaments were two-stranded (not shown).
FIGURE 1. Electron microscopy of MtbFtsZ polymers.

a, at pH 6.5 MtbFtsZ assembled into long, two-stranded filaments. b, at pH 6.9, MtbFtsZ assembled into a mixture of two- and one-stranded filaments. c, at pH 7.7 MtbFtsZ assembled into short, one-stranded filaments. d, when 10 mm calcium was added to the pH 7.7 buffer, MtbFtsZ formed predominantly two-stranded filaments and small bundles but with some one-stranded filaments visible. All experiments contained 10 μm MtbFtsZ and 500 μm GTP. Bar = 100 nm.
Kinetics of Initial Assembly of MtbFtsZ
We labeled MtbFtsZ with fluorescein as donor and with TMR as acceptor. We first tested whether the fluorophore labeling had any effect on assembly using the light-scattering assay at pH 6.5. Fig. 2 shows that there was only a small decrease in the assembly plateau measured by light scattering after labeling with fluorescein or TMR, and there was no significant change in kinetics. We then mixed equal parts donor and acceptor and assayed assembly by FRET using the decrease in donor fluorescence as the measure of FRET. Donor fluorescence showed a rapid 3−4% decrease after mixing and before adding GTP. This may reflect a weak dimer formation. This decrease occurred within 1 s (data not shown). Following the addition of GTP, donor fluorescence slowly dropped by 20%, which we attributed to the assembly of filaments (Fig. 2, curve e). The kinetics measured by FRET are very similar to those measured by light scattering. We also checked the labeled protein by electron microscopy and found similar structures assembled by labeled and unlabeled protein (data not shown).
FIGURE 2.
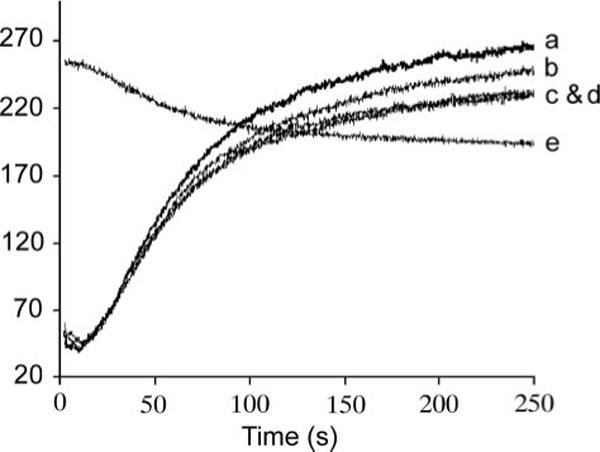
a−d, light scattering was used to compare the assembly of 10 μm MtbFtsZ before and after labeling with fluorophores. a, unlabeled; b, fluorescein-labeled; c, TMR-labeled; d, a mixture of 5 μm MtbFtsZ-fluorescein and 5 μm MtbFtsZ-TMR. Curve e shows this mixture assayed for assembly by FRET. Light-scattering assays were excited and detected at 350 nm; the FRET assay was excited at 450 nm and detected at 515 nm (assembly is measured by the decrease in donor emission at 515 nm). The FRET signal was normalized to the light-scattering signal. Both light-scattering and fluorescence measurement started immediately after adding GTP.
Fig. 3a shows the FRET assay of assembly kinetics of 7 μm total MtbFtsZ in three different pH buffers: pH 6.5, pH 6.9, and pH 7.7. The initial donor fluorescence before assembly was higher at pH 7.7 than at pH 6.5, consistent with the well established sensitivity of fluorescein emission to pH (20). We therefore normalized the fluorescence of each curve to the value before adding GTP. The kinetics of MtbFtsZ are slow at all three values of pH, requiring 60−100 s to reach a plateau. This is 10 times slower than assembly of EcFtsZ. The total change in donor fluorescence was significantly larger at pH 6.5 than at pH 7.7. This may be related to the structures of the polymers. In the two-stranded filaments assembled at pH 6.5, FRET can occur from one strand to the other, as well as along each strand.
FIGURE 3.
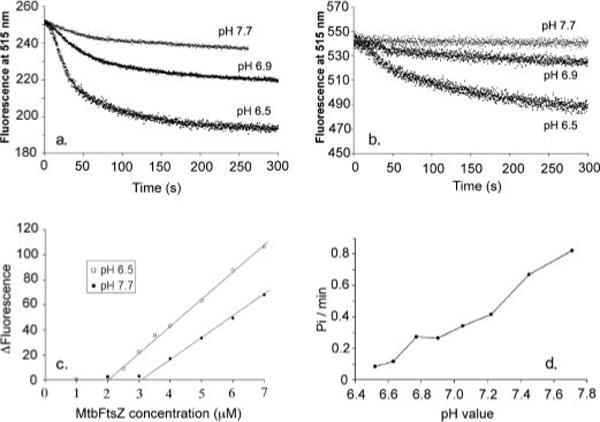
a, MtbFtsZ assembly kinetics measured by FRET with 3.5 μm fluorescein-labeled protein (donor) plus 3.5 μm tetramethylrhodamine-labeled protein (acceptor). Fluorescence at time zero was normalized to the same value, 250. The extent of fluorescence change was greater at lower pH. However, the kinetics of assembly were similar at each pH. b, the homo-FRET signal of fluorescein-labeled MtbFtsZ without acceptor. The protein concentration was 7 μm. c, the equilibrium FRET signal (the absolute change in donor fluorescence from 0 to 120 s) is plotted as a function of FtsZ concentration. The critical concentration is 2 μm in MMK buffer (50 mm MES, pH 6.5, 100 mm KAc, 5 mm MgAc, 1 mm EGTA) and 3.2 μm in HMK buffer (50 mm HEPES, pH 7.7, 100 mm KAc, 5 mm MgAc, 1 mm EGTA). d, MtbFtsZ has different GTPase activity at different pH. The GTPase activity is about 0.08 GTP/min/FtsZ at pH 6.5 and increases to about 0.8 at pH 7.7. The results show an approximately linear increase in GTPase with increasing pH. All measurements were done at room temperature.
Because the excitation and emission spectra of fluorescein overlap, fluorescein-labeled MtbFtsZ in the absence of acceptor can produce a homo-FRET or quenching signal. This is shown in Fig. 3b. The homo-FRET signal is barely detectable at pH 7.7 but is substantial (9% decrease) at pH 6.5. The Förster distance is 4.0 nm for fluorescein homo-FRET versus 5.5 nm for fluorescein-rhodamine FRET. Because the subunit spacing is 4.3 nm along the filament, homo-FRET may be small for one-stranded filaments and stronger across the two-stranded filament. This signal would be especially strong if the labeled Cys-155 residues were facing each other across the center of the two-stranded filament.
We concluded that some fraction of the total FRET signal in Fig. 3a was due to homo-FRET of the fluorescein donor, but this did not affect our interpretation. We used the drop in donor fluorescence as a measure of relative assembly, and it is not important whether the decrease was generated by hetero- or homo-FRET.
Fig. 3c shows the total change in donor fluorescence from time zero to the plateau, measured at 120 s. (The curves in Fig. 3a show a continuous slow decrease after 120 s. This may be because of an enhanced FRET produced by bundling protofilaments or by bleaching. For the present purpose we have ignored this and defined 120 s as the end point of the primary assembly reaction.) The plots show the characteristic feature of a critical concentration, which is 2 μm at pH 6.5 and 3.2 μm at pH 7.7. The different slopes of the lines reflect the difference in absolute FRET signal due to pH noted above and cannot be interpreted quantitatively.
Steady State GTPase Activity of MtbFtsZ
The GTPase activity of MtbFtsZ was much lower than that of EcFtsZ at all pH values. Because the GTPase is zero below the critical concentration, we calculated the hydrolysis rate (GDP FtsZ−1 min−1, Table 1) relative to the FtsZ concentration above the critical concentration (12). Note that these rates were determined at room temperature. Because our previous GTPase assays were mostly at 37 °C, we compared the rates at the two temperatures for a single preparation of EcFtsZ. The GTPase of EcFtsZ was 2- or 3-fold lower at 22 °C than at 37 °C. This is similar to the previous results of de Boer et al. (21), who reported that the GTPase of EcFtsZ is 2-fold higher at 37 °C than at 30 °C.
TABLE 1. Summary of GDP-induced disassembly, subunit turnover, and GTPase at steady state.
All measurements were done at room temperature, so the GTPase of EcFtsZ is 2−3 times lower than the values we reported previously at 37 °C (12).
| pH | GDP disassembly t½ | Turnover t½ | GTPase | 1/GTPase |
|---|---|---|---|---|
| s | s | GTP FtsZ−1 min−1 | s | |
| E. coli | ||||
| 6.5 | 6 | 8 | 4 ± 2 | 15 |
| 7.7 |
4 |
5.5 |
7 ± 1 |
8.6 |
| M. tuberculosis | ||||
| 6.5 | 131 | (163)a | (0.08)a ± 0.02 | (750)a |
| 6.9 | 51 | (80)a | (0.26 ± 0.05)a | (230)a |
| 7.7 | 28 | 42 | 0.8 ± 0.2 | 75 |
Turnover numbers for MtbFtsZ at pH 6.5 and 6.9 are in parentheses because turnover was limited in extent; 1/GTPase numbers are exaggerated for these cases because they are based on total FtsZ, including the subunits that are not turning over.
MtbFtsZ hydrolyzed 0.8 and 0.08 GTP/FtsZ/min at pH 7.7 and 6.5 (Fig. 3d, Table 1). In our previous study of EcFtsZ mutants we considered a value of 0.13 to be the noise from contaminating GTPases (12). However, we believe the 0.08 value measured here is valid because the His tag provided an extra step of purification, eliminating most of the contaminating GTPases. Previous studies of MtbFtsZ at pH 6.5 have reported values of 0.04 ((8) (a much lower value was quoted in the text, but we calculated this value from the 13 μm curve in their Fig. 8) and 0.13 (10), both in good agreement with our measurement. The data in Table 1 show that MtbFtsZ hydrolyzes GTP 50 and 9 times slower than does EcFtsZ at pH 6.5 and 7.7.
Kinetics Analysis: MtbFtsZ Assembly Is Cooperative with a Weak Dimer Nucleus
We measured assembly kinetics over a range of protein concentration at both pH 6.5 and 7.7 (Fig. 4). Assembly at both pH 6.5 and 7.7 could be fit with the same kinetic model that fit EcFtsZ. This comprised three steps: monomer activation, formation of a weak dimer nucleus, and elongation (see Scheme 1 under “Experimental Procedures”). The model and fitting procedure are described in detail in our previous papers (1, 4). The main difference between EcFtsZ and MtbFtsZ (at pH 7.7) is that MtbFtsZ has a much slower rate constant for elongation. Table 2 lists the kinetic parameters derived from the fitting and compares them to the parameters determined previously for EcFtsZ.
FIGURE 4. Assembly kinetics and fitting results at different concentrations of MtbFtsZ in MMK buffer (a) and HMK buffer (b).
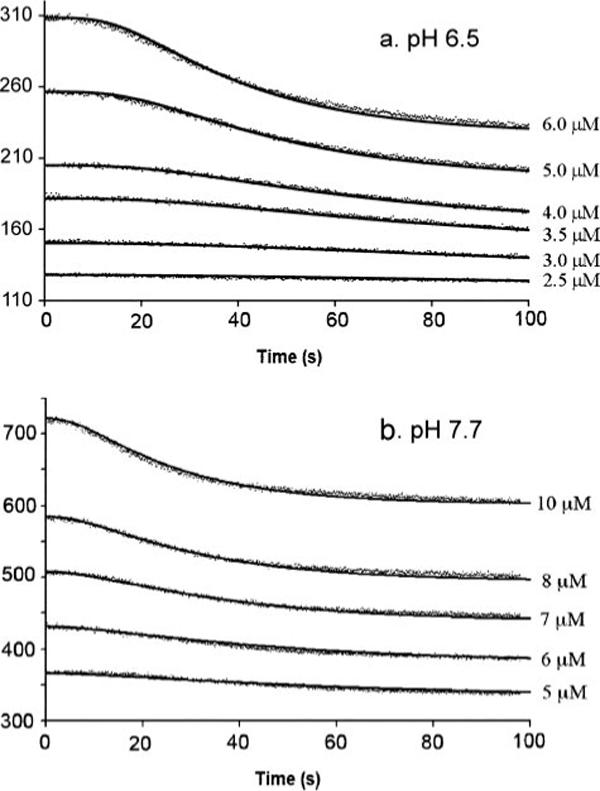
The measured donor fluorescence is represented by closely spaced dots. A solid line through each set of data is the computer fit based on the nucleation model. The values of the kinetic parameters giving the best fit are shown in Table 2.
TABLE 2.
Comparison of the kinetic parameters, critical concentration (Cc), and GTPase activity of MtbFtsZ and EcFtsZ
| Parameters | MtbFtsZ |
EcFtsZ-F268Ca | |
|---|---|---|---|
| MMK (pH 6.5) | HMK (pH 7.7) | HMK (pH 7.7) | |
| k1 (s−1) | 0.10 | 0.21 | 0.38 |
| k2 (μm−1 s−1) | 0.50 | 0.62 | 0.72 |
| k−2 (s−1) | 2347 | 352.0 | 199.6 |
| ke (μm−1 s−1) | 0.95 | 0.20 | 6.63 |
| k−e (s−1) | 1.17 (1.9)b | 0.41 (1.3)b | 4.0 (8.0)b |
| k−2/k2 (μm) | 4694 | 567.7 | 277.2 |
| k−e/ke (μm) | 1.22 | 2.05 | 0.60 |
| Cc (μm) | 2.0 | 3.2 | 1.2 |
Data are taken from our previous study (1).
Calculated from k−e = ke Cc.
Protofilament Dynamics at Steady State
A particular advantage of the FRET assay is that it can measure assembly dynamics and subunit turnover at steady state. We did two experiments to measure the assembly dynamics of MtbFtsZ at steady state. First, we induced disassembly by adding a 20-fold excess of GDP, which blocks the reassembly reaction and lets one observe the rate of disassembly. During the depolymerization processes, FRET is lost and donor fluorescence increases. The depolymerization of MtbFtsZ fit a single exponential decay (Fig. 5a), F(t) = Feq +ΔF·e−t/τ (where F(t) is the fluorescence at time t, Feq is the final equilibrium fluorescence, ΔF is the change in fluorescence from time zero to equilibrium, and τ is the characteristic decay time; the half-time of the reaction is t½ = τ·ln2). The decay half-time was 28 s at pH 7.7 and increased to 131 s at pH 6.5 (Fig. 5a). In a similar experiment EcFtsZ showed a half-time of 4 s (Fig. 5A, bottom panel).
FIGURE 5. GDP-induced disassembly (a) and the rate of subunit turnover measured by FRET techniques (b).
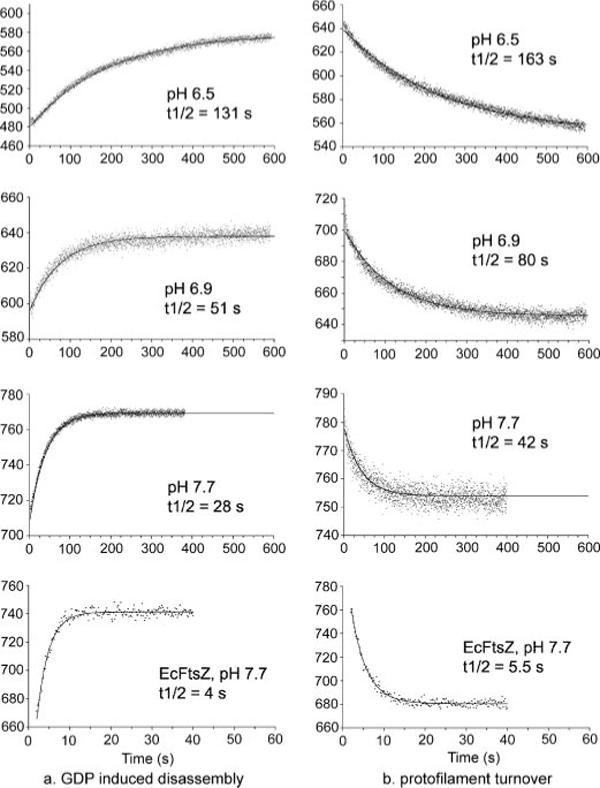
a, 10 μm MtbFtsZ at different pH was assembled to steady state with 100 μm GTP, and disassembly was induced by adding 2 mm GDP. Disassembly caused loss of FRET, indicated here by the increase in donor fluorescence. The solid lines are the single-exponential fit. b, separate preassembled protofilaments of 10 μm MtbFtsZ-fluorescein and MtbFtsZ-TMR were mixed at time zero. The decrease in donor fluorescence is because of the FRET signal that develops when protofilaments disassemble and reassemble into mixed filaments. The solid lines are the single-exponential fit.
The second experiment directly measured the subunit turnover at steady state by mixing separate pools of preassembled donor and acceptor protofilaments. Initially there was no FRET (except for some homo-FRET by fluorescein) because the donors and acceptors were on separate protofilaments. FRET developed as the protofilaments disassembled and reassembled into mixed polymers. Fig. 5b shows the change in the FRET signal over time in three different pH buffers: pH 6.5, 6.9, and 7.7. These curves were all fit well by single-exponentials. The half-time of MtbFtsZ turnover was about 42 s at pH 7.7 and 163 s at pH 6.5. The turnover of EcFtsZ was much faster with a half-time of 5 s at pH 7.7 (Fig. 5b, bottom panel). Also, we found that at pH 6.5 only about half of the MtbFtsZ subunits exchanged. Thus, in addition to the slow turnover, half of the subunits remained stably in their original two-stranded filaments and did not exchange.
We repeated these experiments with different total concentrations of protein. We expected turnover to be independent of the total FtsZ concentration, because once the assembly has reached steady state all protofilaments will reassemble from the pool of subunits at the steady state concentration. We tested both the GDP-induced disassembly and the subunit turnover at 7, 10, and 15 μm MtbFtsZ. The rates were essentially identical at the three concentrations (at pH 7.7 and 7, 10, and 15 μm MtbFtsZ, the half-times for GDP-induced disassembly were 28, 28, and 25 s and turnover was 42, 42, and 41 s, respectively).
In Vivo Dynamics of the Z-ring in M. smegmatis
We used FRAP to examine the dynamics of MtbFtsZ polymers in vivo. As we were not able to culture M. tuberculosis strains directly for this study, instead we performed FRAP on two strains of M. smegmatis. Both strains express M. smegmatis FtsZ (MsmFtsZ) from the genome. The first strain, mC2-79, also expresses MsmFtsZ fused to green fluorescent protein (GFP) for detection purposes, and the second strain, mC2-11, expresses MtbFtsZ fused to GFP. When induced by acet-amide, the FtsZ-GFP level was higher than that from genomic expression (17). In the present study we did not induce with acetamide but relied on the leakiness of the promoter. In this case the FtsZ-GFP is about half the level of the genomic MsmFtsZ (22).>3
One representative FRAP time series for MsmFtsZ is shown in Fig. 6a. As in our previous FRAP studies, we attempted to bleach half of a Z-ring in each case, and we observed a range of recovery half-times (see Fig. 6, b and c). The average half-time for strain mC2-79 (MsmFtsZ-GFP) was 34 s (n = 9) and for strain mC2-11 (MsmFtsZ plus MtbFtsZ-GFP) was 25 s (n = 9). In both cases there was a large spread in the half-times, which is shown in the histograms in Fig. 6, b and c. These are not experimental errors but represent real variations from one bacterium to another. We should also note that ∼20% of the FRAP series obtained showed very little recovery (less than 10%), and these were not included in the averages given above. Thus the average half-times we report here are likely underestimates.
FIGURE 6. In vivo FRAP of MsmFtsZ and MtbFtsZ Z-rings in M. smegmatis.
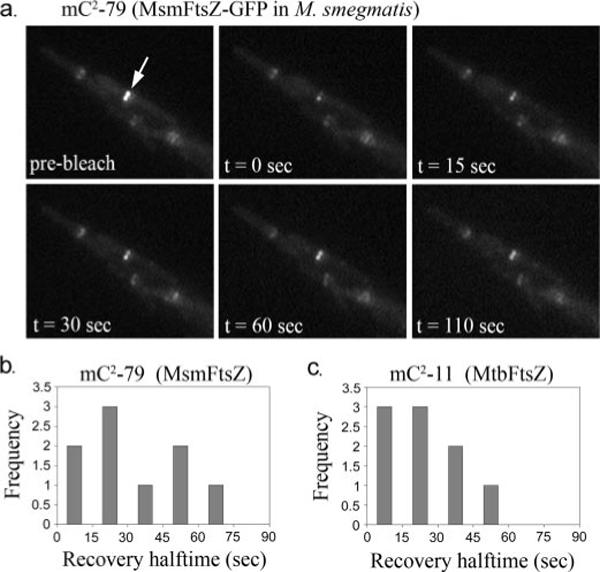
a, representative FRAP time series for strain mC2-79 (expressing MsmFtsZ-GFP). The white arrow indicates the half of the ring about to be bleached. b, histogram of FRAP recovery half-times obtained for MsmFtsZ-GFP in M. smegmatis. c, histogram of FRAP recovery times for MtbFtsZ-GFP in M. smegmatis.
DISCUSSION
The Effect of pH on Assembly of MtbFtsZ: One- and Two-stranded Filaments
The previous studies of MtbFtsZ were done only at pH 6.5 (8, 10). We have confirmed most of these observations; assembly of MtbFtsZ is about 10 times slower than EcFtsZ, and produces almost exclusively long, two-stranded filaments at pH 6.5. In the present study we also tested MtbFtsZ at pH 7.7 and two intermediate pH values. At pH 7.7 MtbFtsZ produced short, one-stranded protofilaments similar to those of EcFtsZ.
Using a light scattering assay, White et al. (8) report a strong assembly of MtbFtsZ at pH 6.5, but found that the signal dropped to about one-tenth at pH 7.0 and disappeared completely at pH 7.5. We believe this illustrates the problem inherent in the light scattering assay, namely that the scattering signal is strongly dependent on the size of the polymers. Thus the long, two-stranded filaments at pH 6.5 scatter very strongly, whereas the same mass of short, one-stranded filaments at pH 7.7 scatters very weakly. The FRET assay avoids this problem and gives a signal proportional to the mass of polymer and independent of filament length. We confirmed, both by the FRET assay and by electron microscopy, that MtbFtsZ assembles quite well at pH 7.2 and 7.7.
Bacteria maintain a fairly constant internal pH when growing in a range of external pH. In optimal growth conditions, E. coli maintains a cytoplasmic pH near 7.7 (23, 24). Mycobacterium species maintain their internal pH in a range of 6.5 to 7 (25, 26). At the pH 6.6 of our growth medium, M. smegmatis maintains an internal pH of 6.9−7.0 (25, 26). In this physiological range MtbFtsZ formed two-stranded filaments, as well as some one-stranded filaments. We found that higher potassium, which is present at 300+ mm in bacterial cytoplasm (27), also favored small bundles of filaments. This suggests that a mixture of one- and two-stranded filaments and small bundles may be the physiologically relevant FtsZ polymer in Mycobacterium.
The average t½ of 25 s that we measured for turnover in vivo is significantly faster than the 80 s measured in vitro at pH 6.9. This discrepancy is in contrast to the case for E. coli, where the turnover rates were very close in vivo and in vitro (1). We noted that the FRAP data for M. smegmatis were much more scattered than for E. coli, and we excluded 20% of the measurements that showed minimal recovery; thus the actual turnover in vivo is likely slower than the 25 s average half-time.
Turnover in vivo measured by FRAP might be faster or slower than turnover in vitro measured by FRAP. If turnover in vivo involved dissociation and reassociation of entire protofilaments, it could be faster than the turnover of single subunits measured by FRET. On the other hand, if subunits disassembled from the Z-ring and quickly reassembled into the Z-ring without exchanging with the cytoplasmic pool, turnover in vivo would appear slower than in vitro. We have previously discussed in detail how the in vivo FRAP correlates with in vitro assembly and turnover for EcFtsZ (1, 3). For MtbFtsZ the analysis is complicated by the unknown mixture of one- and two-stranded filaments, so we prefer not to speculate further on the modest discrepancy of turnover in vivo and in vitro.
Kinetics Analysis: Both One- and Two-stranded Filaments Show Cooperative Assembly with a Dimer Nucleus
We had expected that the assembly mechanism for the two-stranded polymers at pH 6.5 might be very different from that of the one-stranded protofilaments at pH 7.7. In particular, a two-stranded filament could provide a rationale for cooperative assembly and a dimer nucleus, which remain an enigma for a one-stranded filament (4). However the kinetics of initial assembly were fit by the same kinetic model at both pH 6.5 and 7.7: monomer activation, a dimer nucleus, and elongation. The numerical values of the kinetic parameters were different. Elongation was five times slower at pH 7.7 compared with pH 6.5, but nucleation was eight times more favorable, leading to a similar time course for initial assembly. Because nucleation was actually less favorable for the two-stranded polymer, the two-stranded structure apparently does not play a role in nucleation. We suggest that the basic assembly mechanism at both pH 7.7 and 6.5 is the formation of one-stranded protofilaments. The formation of two-stranded filaments at pH 6.5 may be a secondary association event with little effect on kinetics.
We caution that the dimer nucleus is a hypothetical intermediate inferred from the kinetic modeling. Because the protofilaments are one-stranded it is difficult to understand the structure of this nucleus. Another potential problem is that, although the dissociation constant (k−2/k2, Table 2) of the dimer is approximately the same for EcFtsZ and MtbFtsZ at pH 7.7 (277 and 567 μm), it is 10 times higher for MtbFtsZ at pH 6.5 (4,700 μm). This much slower nucleation is balanced by a five times faster elongation to give a total assembly time approximately the same for the two pH values. This coincidence suggests caution in the interpretation of the kinetics model. At present the dimer nucleus is a hypothesis that provides a good fit to the kinetics, but we do not even know that it exists as a structure.
Correlations among Steady State Turnover, GTP Hydrolysis, and GDP-induced Disassembly
The steady state assembly dynamics appear to be related to GTP hydrolysis. The half-time for subunit turnover is about half the time for GTP turnover (1/GTPase, Table 1) for EcFtsZ at pH 6.5 and 7.7 and for MtbFtsZ at pH 7.7. (The two-stranded filaments of MtbFtsZ at pH 6.5 and 6.9 are complicated by having only a partial turnover, which we will ignore for the present.) This correlation suggests the following mechanism. Following GTP hydrolysis the GDP is trapped in the protofilament interface, and the subunit has to dissociate in order to exchange the GDP for GTP. Once a subunit dissociates and the exchange has occurred, the subunit is activated for a new round of assembly and hydrolysis. This mechanism is well established for tubulin, and our data suggest that it may apply also to FtsZ.
Steady state subunit turnover also correlates well with the GDP-induced disassembly. For all five cases in Table 1, the half-time for GDP-induced disassembly was about 65−75% that for subunit turnover measured by FRET. This is consistent with the model above and with our assumption that the excess of GDP binds to free subunits and blocks their ability to reassemble. The protofilaments then disassemble at their natural rate. Although fit by a single exponential, this disassembly reaction is likely complex and involves two mechanisms. One is the natural disassembly from the protofilament end, given by the kinetic constant k−e. The second mechanism involves GTP hydrolysis, where we assume that each hydrolysis event is followed by dissociation of the subunit and exchange of GDP for GTP.
The steady state subunit turnover measured by FRET is only slightly slower than this disassembly rate. This suggests that during normal turnover protofilaments are disassembling almost as quickly as when reassembly is completely blocked by GDP.
Exchange of Nucleotide into Protofilaments
Some previous studies have proposed that GTP can exchange directly into assembled protofilaments (28-32). In the most recent study (28) the earliest time point examined was 10 s, the time at which exchange was found to be complete (at pH 7.5, 500 mm KCl). However, our work shows that the half-time for subunit turnover for EcFtsZ was 5.5 s under comparable conditions (Table 1), so that most or all of this exchange could take place on free subunits as they are turning over.
Tadros et al. (28) repeated the exchange experiment in 500 mm RbCl and found a slower exchange with a half-time of ∼10 s. The GTPase was greatly reduced in rubidium (0.6 min−1) and was eight times slower than the rate measured for GTP exchange. The authors concluded that nucleotide exchange in rubidium was occurring directly into assembled protofilaments. However, we have repeated these GTPase measurements and compared them directly to subunit exchange. We found that the GTPase at pH 7.7 and room temperature was 2 min−1 in rubidium (about four times faster than reported by Tadros et al. (28)) and was 6−7 min−1 in potassium. The half-time of subunit exchange was 11 s in rubidium and 4 s in potassium. Thus the half-time for subunit exchange was about one-third of the time for GTP hydrolysis in both rubidium and potassium, similar to the ratio of one-half that we found for EcFtsZ at pH 6.5 and 7.7 and for MtbFtsZ at pH 7.7 (Table 1).
In an earlier study our laboratory investigated nucleotide exchange into EcFtsZ polymers stabilized by DEAE-dextran (33). In DEAE-dextran FtsZ-GDP assembled into tubes, with the protofilaments in the helical, curved conformation. FtsZ-GTP formed sheets of straight protofilaments. When polymers were assembled in one nucleotide and an excess of the other was added, they transformed in shape. However, the transformation took 2−4 min, suggesting disassembly of the original, an exchange of nucleotide on the free subunits, and reassembly of the new form. Rapid exchange directly into assembled protofilaments was found under only one condition. When GTP plus EDTA was added to GDP tubes, the tubes transformed into sheets of straight protofilaments in less than 15 s. We concluded that chelation of magnesium permitted the exchange of GTP directly into the curved conformation of GDP protofilaments.
The close correlation of GDP-induced disassembly and subunit turnover in the continued presence of GTP is inconsistent with nucleotide exchange into protofilaments. If GDP could enter the protofilaments, we would expect the GDP-induced disassembly to be much faster. However, in all cases it was only 25−35% faster. If GTP could enter the protofilaments, we would expect it to replace the GDP rapidly following hydrolysis and eliminate the observed correlation of hydrolysis with turnover.
Modeling Details of Protofilament Turnover
In the future, we would like to develop a detailed model for events involved in turnover of protofilaments at steady state. However, at present there are too many unknowns. The site of GTP hydrolysis is one of the major unknowns. Most of the subunits in polymer carry unhydrolyzed GTP (34), so there must be a considerable lag between the time a subunit enters the protofilament and when it hydrolyzes its GTP. What triggers hydrolysis is unknown. One possibility is that these subunits hydrolyze GTP at random and that the protofilament then fragments at the site of the newly formed GDP. A very different model would propose that spontaneous hydrolysis of GTP occurs rarely, but when it does happen additional hydrolysis is accelerated vectorially on the adjacent subunits. This type of coupled or vectorial hydrolysis has been proposed to explain how the GTP cap generates microtubule dynamic instability (35).
Acknowledgment
We thank Gina Briscoe for help with protein purification.
Footnotes
This work was supported by National Institutes of Health Grants GM66014 (to H. P. E.) and AI48417 and AI64502 (to M. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: EcFtsZ, FtsZ from Escherichia coli; MtbFtsZ, FtsZ from Mycobacterium tuberculosis; FRET, fluorescence resonance energy transfer; TMR, tetramethylrhodamine; FRAP, fluorescence recovery after photobleaching; GFP, green fluorescent protein; MES, 4-morpholineethanesulfonic acid; MOPS, 4-morpholinepropanesulfonic acid.
M. Rajagopalan, unpublished data.
REFERENCES
- 1.Chen Y, Erickson HP. J. Biol. Chem. 2005;280:22549–22554. doi: 10.1074/jbc.M500895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stricker J, Maddox P, Salmon ED, Erickson HP. Proc. Natl. Acad. Sci. U. S. A. 2002;99:3171–3175. doi: 10.1073/pnas.052595099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson DE, Gueiros-Filho FJ, Erickson HP. J. Bacteriol. 2004;186:5775–5781. doi: 10.1128/JB.186.17.5775-5781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Bjornson K, Redick SD, Erickson HP. Biophys. J. 2005;88:505–514. doi: 10.1529/biophysj.104.044149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchison TJ, Kirschner MW, RayChaudhuri D. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11821–11826. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Q, Kirikae F, Kirikae T, Pepe A, Amin A, Respicio L, Slayden RA, Tonge PJ, Ojima I. J. Med. Chem. 2006;49:463–466. doi: 10.1021/jm050920y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Galgoci A, Kodali S, Herath KB, Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D, Singh S. J. Biol. Chem. 2003;278:44424–44428. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]
- 8.White EL, Ross LJ, Reynolds RC, Seitz LE, Moore GD, Borhani DW. J. Bacteriol. 2000;182:4028–4034. doi: 10.1128/jb.182.14.4028-4034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung AK, Lucile White E, Ross LJ, Reynolds RC, DeVito JA, Borhani DW. J. Mol. Biol. 2004;342:953–970. doi: 10.1016/j.jmb.2004.07.061. [DOI] [PubMed] [Google Scholar]
- 10.Rajagopalan M, Atkinson MA, Lofton H, Chauhan A, Madiraju MV. Biochem. Biophys. Res. Commun. 2005;331:1171–1177. doi: 10.1016/j.bbrc.2005.03.239. [DOI] [PubMed] [Google Scholar]
- 11.Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. Anal. Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- 12.Redick SD, Stricker J, Briscoe G, Erickson HP. J. Bacteriol. 2005;187:2727–2736. doi: 10.1128/JB.187.8.2727-2736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barshop BA, Wrenn RF, Frieden C. Anal. Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 14.Zimmerle CT, Frieden C. Biochem. J. 1989;258:381–387. doi: 10.1042/bj2580381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frieden C. Methods Enzymol. 1994;240:311–322. doi: 10.1016/s0076-6879(94)40053-9. [DOI] [PubMed] [Google Scholar]
- 16.Dang Q, Frieden C. Trends Biochem. Sci. 1997;22:317. doi: 10.1016/s0968-0004(97)01062-1. [DOI] [PubMed] [Google Scholar]
- 17.Dziadek J, Rutherford SA, Madiraju MV, Atkinson MA, Rajagopalan M. Microbiology. 2003;149:1593–1603. doi: 10.1099/mic.0.26023-0. [DOI] [PubMed] [Google Scholar]
- 18.Dziadek J, Madiraju MV, Rutherford SA, Atkinson MA, Rajagopalan M. Microbiology. 2002;148:961–971. doi: 10.1099/00221287-148-4-961. [DOI] [PubMed] [Google Scholar]
- 19.Maddox PS, Bloom KS, Salmon ED. Nat. Cell Biol. 2000;2:36–41. doi: 10.1038/71357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Biochemistry. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 21.de Boer P, Crossley R, Rothfield L. Nature. 1992;359:254–256. doi: 10.1038/359254a0. [DOI] [PubMed] [Google Scholar]
- 22.Rajagopalan M, Maloney E, Dziadek J, Poplawska M, Lofton H, Chauhan A, Madiraju MV. FEMS Microbiol. Lett. 2005;250:9–17. doi: 10.1016/j.femsle.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 23.Kashket ER. Biochemistry. 1982;21:5534–5538. doi: 10.1021/bi00265a024. [DOI] [PubMed] [Google Scholar]
- 24.Padan E, Zilberstein D, Rottenberg H. Eur. J. Biochem. 1976;63:533–541. doi: 10.1111/j.1432-1033.1976.tb10257.x. [DOI] [PubMed] [Google Scholar]
- 25.Rao M, Streur TL, Aldwell FE, Cook GM. Microbiology. 2001;147:1017–1024. doi: 10.1099/00221287-147-4-1017. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Zhang H, Sun Z. J. Antimicrob. Chemother. 2003;52:56–60. doi: 10.1093/jac/dkg287. [DOI] [PubMed] [Google Scholar]
- 27.Cayley S, Lewis BA, Guttman HJ, Record MT., Jr. J. Mol. Biol. 1991;222:281–300. doi: 10.1016/0022-2836(91)90212-o. [DOI] [PubMed] [Google Scholar]
- 28.Tadros M, Gonzalez JM, Rivas G, Vicente M, Mingorance J. FEBS Lett. 2006;580:4941–4946. doi: 10.1016/j.febslet.2006.07.083. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez JM, Jimenez M, Velez M, Mingorance J, Andreu JM, Vicente M, Rivas G. J. Biol. Chem. 2003;278:37664–37671. doi: 10.1074/jbc.M305230200. [DOI] [PubMed] [Google Scholar]
- 30.Scheffers DJ, de Wit JG, den Blaauwen T, Driessen AJ. Biochemistry. 2002;41:521–529. doi: 10.1021/bi011370i. [DOI] [PubMed] [Google Scholar]
- 31.Mingorance J, Rueda S, Gomez-Puertas P, Valencia A, Vicente M. Mol. Microbiol. 2001;41:83–91. doi: 10.1046/j.1365-2958.2001.02498.x. [DOI] [PubMed] [Google Scholar]
- 32.Oliva MA, Cordell SC, Lowe J. Nat. Struct. Mol. Biol. 2004;11:1243–1250. doi: 10.1038/nsmb855. [DOI] [PubMed] [Google Scholar]
- 33.Lu CL, Erickson HP. Cell Struct. Funct. 1999;24:285–290. doi: 10.1247/csf.24.285. [DOI] [PubMed] [Google Scholar]
- 34.Romberg L, Mitchison TJ. Biochemistry. 2004;43:282–288. doi: 10.1021/bi035465r. [DOI] [PubMed] [Google Scholar]
- 35.Flyvbjerg H, Holy TE, Leibler S. Phys. Rev. A. 1996;54:5538–5560. doi: 10.1103/physreve.54.5538. [DOI] [PubMed] [Google Scholar]