

The X-ray crystallographic structure of a dimer variant of fructose-1,6-bisphosphate aldolase demonstrates a stable oligomer that mirrors half of the native tetramer. The presence of product demonstrates that this is an active form.
Keywords: dimeric aldolase, hemolytic anemia, D128V, oligomerization
Abstract
Fructose-1,6-bisphosphate aldolase (aldolase) is an essential enzyme in glycolysis and gluconeogenesis. In addition to this primary function, aldolase is also known to bind to a variety of other proteins, a property that may allow it to perform ‘moonlighting’ roles in the cell. Although monomeric and dimeric aldolases possess full catalytic activity, the enzyme occurs as an unusually stable tetramer, suggesting a possible link between the oligomeric state and these noncatalytic cellular roles. Here, the first high-resolution X-ray crystal structure of rabbit muscle D128V aldolase, a dimeric form of aldolase mimicking the clinically important D128G mutation in humans associated with hemolytic anemia, is presented. The structure of the dimer was determined to 1.7 Å resolution with the product DHAP bound in the active site. The turnover of substrate to produce the product ligand demonstrates the retention of catalytic activity by the dimeric aldolase. The D128V mutation causes aldolase to lose intermolecular contacts with the neighboring subunit at one of the two interfaces of the tetramer. The tertiary structure of the dimer does not significantly differ from the structure of half of the tetramer. Analytical ultracentrifugation confirms the occurrence of the enzyme as a dimer in solution. The highly stable structure of aldolase with an independent active site is consistent with a model in which aldolase has evolved as a multimeric scaffold to perform other noncatalytic functions.
1. Introduction
Fructose-1,6-bisphosphate aldolase (aldolase; EC 4.1.2.13) is a ubiquitously expressed glycolytic enzyme that catalyzes the reversible cleavage of fructose 1,6-bisphosphate (Fru 1,6-P2) to dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P) in glycolysis and the reverse reaction in gluconeogenesis. In addition, aldolase is involved in the metabolism of fructose, converting fructose 1-phosphate (Fru 1-P) to DHAP and glyceraldehyde (Horecker et al., 1975 ▶).
There are two classes of aldolases based on their chemical mechanism: class I enzymes are cofactor-independent and catalyze C—C bond cleavage via the formation of a Schiff-base intermediate with the sugar, whereas class II enzymes require a divalent metal ion for catalysis. The class I aldolases of vertebrates are further subdivided into three isozymes that have different activities and vary in their tissue distribution (Rutter, 1964 ▶). The isoforms aldolase A (predominantly present in muscle) and aldolase C (in brain) have higher activity towards Fru 1,6-P2 than towards Fru 1-P. Aldolase B (in liver) has equal turnover numbers for both Fru 1,6-P2 and Fru 1-P, corresponding to its role in fructose metabolism in the liver (Penhoet & Rutter, 1971 ▶). The amino-acid sequences of these isozymes are highly conserved, predominantly at the subunit interface and the active site (Rottmann et al., 1984 ▶, 1987 ▶; Lai et al., 1974 ▶; Deng & Smith, 1999 ▶), with 81, 70 and 70% overall sequence identity between A and C, between A and B and between B and C, respectively.
Biochemical studies supported by crystal structures have shown that class I aldolases are invariably present as homotetramers (Penhoet et al., 1967 ▶). Each subunit has an independent active site that resides in the center of the barrel in a (α/β)8-barrel fold (Sygusch et al., 1987 ▶). Although the quaternary structure is not required for enzymatic activity (Beernink & Tolan, 1996 ▶), the dissociation constant is unusually low, of the order of 10−26 M 3 for the monomer–tetramer equilibrium (Tolan et al., 2003 ▶). This dissociation constant is lower than those of other glycolytic enzymes and raises the question of why aldolase has evolved to possess such a very tight interaction at the protein–protein interfaces. Steady-state kinetic studies of aldolase isozymes have shown no cooperativity among the subunits, thus aldolase does not display allosteric regulation (Penhoet & Rutter, 1971 ▶). Perhaps linked to these findings, there is considerable evidence that aldolase participates in nonmetabolic processes in addition to its enzymatic role. Aldolase is involved in binding interactions with several other proteins, but the functional role of aldolase in these interactions has not been fully characterized. Aldolases A and C have been found to bind to the neurofilament light-chain mRNA in neurons (Cañete-Soler et al., 2005 ▶). Aldolase A associates with the vacuolar H+-ATPase (Lu et al., 2001 ▶) and is thought to mediate its assembly, expression, and activity as a proton pump (Lu et al., 2004 ▶). Aldolase also plays a role in the control of proteins involved in endocytosis (Lundmark & Carlsson, 2004 ▶). There are other binding partners for which no functional role has been established, including γ-tubulin (Volker & Knull, 1997 ▶), heparin (Ta et al., 1999 ▶), phospholipase D (Kim et al., 2002 ▶) and the glucose transporter GLUT4 (Kao et al., 1999 ▶). Aldolase A has been proposed to play a structural role in the assembly of the actin cytoskeleton in vivo owing to its well established ability to bind F-actin (Arnold & Pette, 1968 ▶; Arnold et al., 1971 ▶; O’Reilly & Clarke, 1993 ▶). Further studies have shown that tetrameric aldolase can cross-link F-actin filaments into a gel or rafts, which is consistent with a potential role for this enzyme in the regulation of cytoskeleton structure (Pagliaro & Taylor, 1988 ▶; Wang et al., 1996 ▶; Sukow & DeRosier, 1998 ▶). The ability to cross-link actin points to the multimeric state of aldolase as a prerequisite for its ‘moonlighting’ functions, i.e. a single protein–protein interface can thus provide multiple interactions.
Some of the activities of aldolase appear to link catalytic activity to quaternary structure. Aldolase A deficiency is an autosomal recessive trait and has been associated with nonspherocytic hemolytic anemia in humans (Beutler et al., 1973 ▶). One of the most well studied alleles, D128G (Kishi et al., 1987 ▶), results in a thermally unstable protein. The source of this instability has been studied further using this and other Asp128 substitutions (Beernink & Tolan, 1994 ▶) and showed that the varying degrees of thermolability correlated with varying degrees of dissociation into dimers. The largest shift in the equilibrium towards the dimer is caused by the substitution of Asp128 by Val (Beernink & Tolan, 1994 ▶). The structure of the aldolase tetramer and the position of Asp128 are shown in Fig. 1 ▶.
Figure 1.

Rabbit muscle fructose-1,6-bisphophate aldolase tetramer (Blom & Sygusch, 1997 ▶; PDB code 1ado). Each monomer is colored separately, with the A and B interfaces indicated by arrows as described previously (Beernink & Tolan, 1994 ▶). The sites of the D128V substitutions are indicated by red spheres on interface B and the C- and N-termini are labeled for monomer A.
Here, using D128V aldolase, a high-resolution crystal structure of the dimeric form was explored. The data provided a structural basis for understanding the loss of quaternary structure and showed that the protein was stable in the dimeric form. Remarkably, the dimer did not differ significantly from the corresponding half-tetramer in native aldolases. Since the asymmetric unit contained a single monomer, analysis of D128V aldolase was performed via analytical ultracentrifugation to support the assignment of the crystallographically generated dimer as the biological unit. The product DHAP was bound to the active site after crystallization with the substrate Fru 1,6-P2, which showed that D128V aldolase was catalytically competent. The crystal structure of the dimer will provide a structural basis for studies of aldolase in its role as a binding partner in cellular processes.
2. Materials and methods
2.1. Expression and purification of recombinant aldolase
D128V aldolase was purified as previously described (Beernink & Tolan, 1994 ▶), with the exception that in the last step of purification the CM-Sepharose Fast Flow (GE Healthcare) column was washed with 50 mM TAPS–glycine–KOH pH 8.3, 1 mM dithiothreitol (DTT) and D128V aldolase was eluted with the same buffer containing 2 mM Fru 1,6-P2. Activity was determined by following NADH oxidation at 340 nm in a coupled assay with triosephosphate isomerase and glycerol-3-phosphate dehydrogenase (Racker, 1947 ▶). The active fractions were concentrated and exchanged with crystallization buffer comprised of 50 mM Tris–HCl pH 7.0, 50 mM NaCl, 25 mM ammonium acetate using an Ultrafree centrifugal concentrator (Millipore) with a 10 kDa molecular-weight cutoff membrane. The protein was >95% pure as assessed by SDS–PAGE.
2.2. Crystallization, data collection and processing
D128V aldolase crystals were grown by the hanging-drop vapor-diffusion method using Hampton Research Crystal Screens I and II to screen initial crystallization conditions. The final optimized condition used a 1:1 ratio of protein in crystallization buffer to reservoir solution (0.2 M ammonium sulfate, 25% PEG 2K monomethyl ether (PEG 2K MME), 100 mM sodium acetate pH 5.1) with a final volume of 2 µl, which was equilibrated against 0.5 ml reservoir solution at 291 K. The initial condition was optimized by the addition of osmolytes (naturally occurring compounds that affect protein stability; Bolen & Baskakov, 2001 ▶) to favor ordering in the C-terminal region. The addition of 100 mM sarcosine to the crystallization condition yielded pyramidal crystals with dimensions 0.1 × 0.05 × 0.05 mm within 3 d. X-ray diffraction data from D128V aldolase crystals were collected on beamline X29 at Brookhaven National Laboratory. The crystals were cryoprotected by passing them through Paratone-N prior to flash-freezing in a stream of gaseous N2 at 100 K. The programs DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶) were used to integrate and scale the data to a resolution of 1.70 Å. D128V aldolase crystallized in space group P21212, with unit-cell parameters a = 53.32, b = 137.90, c = 50.15 Å. The data-collection statistics are summarized in Table 1 ▶.
Table 1. Crystallographic data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data-collection statistics | |
| Unit-cell parameters (Å) | a = 53.32, b = 137.90, c = 51.15 |
| Space group | P21212 |
| Wavelength (Å) | 1.10 |
| Resolution range (Å) | 100–1.70 |
| Total reflections | 267879 (24004) |
| Total unique reflections | 42325 (4185) |
| Completeness (%) | 99.0 (100.0) |
| I/σ(I) | 44.4 (7.3) |
| Rmerge† | 0.09 (0.4) |
| Redundancy | 6.3 (5.7) |
| Refinement statistics | |
| No. of protein atoms per ASU | 3029 |
| No. of reflections (working set/free set) | 40471/4071 |
| Rwork‡/Rfree§ (%) | 0.189/0.209 |
| Average B factors (Å2) | |
| Overall | 15.8 |
| Main-chain atoms | 14.6 |
| Side-chain atoms | 15.6 |
| DHAP | 26.6 |
| Active-site sulfate | 45.1 |
| Surface sulfate | 65.9 |
| Acetate | 47.0 |
| Water | 31.2 |
| R.m.s. deviation from ideal | |
| Bond lengths (Å) | 0.004 |
| Angles (°) | 1.3 |
| Dihedrals (°) | 21.5 |
| Impropers (°) | 0.92 |
R
merge = 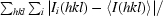
 , where 〈I(hkl)〉 is the mean intensity of the multiple Ii(hkl) observations of symmetry-related reflections.
, where 〈I(hkl)〉 is the mean intensity of the multiple Ii(hkl) observations of symmetry-related reflections.
R
work = 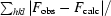
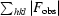 .
.
R
free = 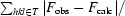 , where the test set T includes 10% of the data.
, where the test set T includes 10% of the data.
2.3. Structural solution and refinement
The structure was solved by molecular replacement with the program AMoRe (Navaza, 1994 ▶) using a monomer from the structure of wild-type human muscle aldolase A (PDB code 1ald; Gamblin et al., 1991 ▶) as the initial search model. The orientation of the monomer obtained by molecular replacement was improved by rigid-body refinement using the Crystallography & NMR System (CNS) program suite (Brünger et al., 1998 ▶). σA-weighted electron-density maps with coefficients 2F o − F c calculated from the model after rigid-body refinement in the program CNS showed good electron density for all areas of the monomer with the exception of the N-terminal (1–4) and C-terminal regions (345–363), which failed to appear even after subsequent rounds of refinement. To minimize model bias in the electron-density maps, the side chain of residue 128 was modeled as an alanine. The density was clearly that of a valine in the 2F o − F c map and was modeled as such in subsequent rounds of refinement. Iterative rounds of model building using Coot (Emsley & Cowtan, 2004 ▶) were performed alternately with rounds of positional, group temperature-factor and simulated-annealing refinement in the program CNS. A test set consisting of 10% of the data was used for R free calculations. In the final rounds of refinement water molecules were included using a 3σ cutoff. A total of 655 water molecules were added to the model as well as six sulfates which were bound at the surface of the protein. In the final round of refinement, a composite-omit electron-density map was used to fit the models of DHAP and sulfate to the active site. The final structure had a working R factor of 0.189 and a free R factor of 0.209. Analysis of the Ramachandran plot defined by PROCHECK (Laskowski et al., 1993 ▶) showed good statistics for the model, with 91.8% of residues in the most favorable regions and 8.2% of residues in additionally allowed regions for D128V aldolase.
2.4. Analytical ultracentrifugation
Purified D128V aldolase was dialyzed against 0.2 M ammonium sulfate, 100 mM sodium acetate and 0.1 mM DTT. The buffer was kept consistent with the crystallization buffer but with the exclusion of 25% PEG 2K MME, which forms a highly viscous solution that slows the equilibration process. Analytical ultracentrifugation (Beckman XLA-90, Beckman Coulter) was performed to determine the oligomerization state of D128V aldolase using 0.2, 0.5 and 1 mg ml−1 aldolase at 6400, 8000 and 12 000 rev min−1 at 291 K. The equilibrium state was verified at each speed and protein concentration was monitored at 280 nm. A two-compartment pharmacokinetic model was fitted to individual data by means of the program WINNONLIN v.2.1 (Metzler, 1989 ▶).
2.5. Analysis of domain movement
The change in intersubunit orientation of the dimer in comparison to the tetramer was measured using the program DynDom (Hayward et al., 1997 ▶). The buried surface area between monomers was calculated for the structures of aldolase (PDB code 1ado) and D128V aldolase using CNS with a probe radius of 1.4 Å (Lee & Richards, 1971 ▶).
3. Results
3.1. Overall structure
The asymmetric unit contained a single monomer of D128V aldolase, with residues 5–344 of the subunit modeled into electron density. The structure was refined to a resolution of 1.7 Å with a final R factor of 18.9% and an R free of 21.0%. The first four N-terminal residues and the 19 C-terminal residues (345–363) were disordered with no observable electron density, which is consistent with previously reported crystal structures (Sygusch et al., 1987 ▶; Choi et al., 1999 ▶). Crystallographic data-collection and refinement statistics are reported in Table 1 ▶. The structure of the monomer is not significantly different from that of wild-type aldolase, with an r.m.s. deviation of 0.50 Å. The density at the site of substitution was well accounted for, reflecting a change in the side chain with no molecular disorder in the region (Fig. 2 ▶).
Figure 2.

Stereoview of the site of substitution in D128V aldolase. The residues flanking the site of substitution are shown with the corresponding 2F o − F c electron-density map (blue cages) contoured at 1σ.
3.2. Crystal-packing analysis
The crystallographic asymmetric unit of the aldolase structure contained one monomer. Upon building the symmetry mates, the monomer formed a tightly packed dimer with its counterpart in the neighboring asymmetric unit. However, there were no counterparts of this symmetry dimer to form the native tetramer. Thus, the structure demonstrated a dimeric oligomerization state. The structural differences between the D128V aldolase dimer and the wild-type tetramer (PDB code 1ado) were not significant. An overlay of the main-chain Cα atoms of the dimer with those of the wild-type aldolase A dimer (subunits B and C) gave an overall r.m.s. deviation of 0.81 Å (Fig. 3 ▶). The 19 residues in the C-terminal region (residues 345–363) were not included in the calculations owing to their lack of ordered structure. The difference in the relative orientations of the monomers within the D128V aldolase compared with half of the tetramer was analyzed using the program DynDom (Hayward et al., 1997 ▶). The conformational difference was measured by overlaying a single monomer from the D128V aldolase on one monomer of the aldolase tetramer and measuring the displacement of the second monomer, yielding a 4.1° rotation at the dimer interface. The result was largely a rigid-body movement of the monomer with the A interface of the dimer acting as a hinge axis.
Figure 3.

Overlay of D128V aldolase (cyan) and wild-type aldolase (yellow). The Cα traces are oriented as in Fig. 1 ▶.
3.3. Analytical ultracentrifugation
Sedimentation-equilibrium analytical ultracentrifugation was employed to show that D128V aldolase is dimeric under the same conditions as those used for crystallization for X-ray crystallography. The data were a good fit to a monodisperse model with a molecular weight equal to that of the aldolase dimer, 79 580 ± 350 Da (Fig. 4 ▶), which is nearly the same as the calculated molecular weight of 78 388 Da. The data were not consistent when a monomer or tetramer was used for fitting (fit not shown). These data confirm that D128V aldolase forms a dimer in solution.
Figure 4.

Sedimentation-equilibrium analytical ultracentrifugation of D128V aldolase under crystallization conditions. The sedimentation-equilibrium trace using 20 µM protein at 291 K and 8000 rev min−1 (monitored by absorbance at 280 nm) is shown in the lower panel. The solid line depicts the best fit to the data (open circles) performed as described in §2.4. Residual errors are presented in the upper panel.
3.4. Subunit interface
The native aldolase is a homotetramer with two unique interfaces described as interface A and interface B (see Fig. 1 ▶). In contrast to the A interface, which is comprised of helix-packing interactions, the B interface exhibits loop–loop interactions formed by ten buried hydrogen bonds (Table 2 ▶). At each B interface, six of the hydrogen bonds involve the carboxylate O atoms of Asp128 interacting with the backbone amides of three consecutive residues (Gly126, Leu127 and Asp128) of the neighboring subunit (Fig. 5 ▶). The hydrophobic side chain of valine cannot form hydrogen bonds. Thus, D128V aldolase retains only the A interface, consistent with a crystallographic dimer. The loop region flanking the substituted Asp128 residue shows no major displacement arising from the loss of these conserved hydrogen bonds.
Table 2. Interactions at interface B .
| Monomer A | Monomer B | Distance (Å) |
|---|---|---|
| Gln125 CB | Gly129 N | 3.0 |
| Gly126 N | Asp128 OD1 | 2.8 |
| Leu127 N | Asp128 OD2 | 3.2 |
| Asp128 N | Asp128 OD2 | 3.1 |
| Asp128 OD1 | Gly126 N | 2.9 |
| Asp128 OD2 | Asp128 N | 2.9 |
| Asp128 OD2 | Leu127 N | 3.2 |
| Gly129 N | Gln125 OE1 | 3.0 |
| Glu165 OE1 | Asn168 ND2 | 3.0 |
| Asn168 ND2 | Glu165 OE1 | 3.0 |
Figure 5.

The B interface of wild-type aldolase A. Depiction of the hydrogen bonds (dashed lines) made with Asp128 at the B interface. The coloring scheme is identical to that of Fig. 1 ▶ for the ribbons, with a ball-and-stick representation in yellow for one monomer and in orange for the second monomer.
3.5. Calculation of the buried surface area
Calculation of the buried surface area for the tetramer (PDB code 1ado) showed extensive interactions between the monomer interfaces. The dimer interface buries 869 Å2 of surface area, while the tetramer buries 3460 Å2. The buried surface area of D128V aldolase did not significantly differ from the same dimer interface of the tetramer (851 Å2). If the crystal contacts from adjacent dimers were included, a total value of 2146 Å2 was obtained. There was a net loss of 1314 Å2 surface area for the D128V aldolase dimer when compared with the tetramer. Thus, the crystal contacts do not compensate for the loss in buried surface area of the B interface. It is curious that D128V aldolase did not pack into the wild-type tetramer during crystallization. Certainly, there are no steric constraints to prevent this. In contrast, the dimeric aldolase B A149P variant (Malay et al., 2002 ▶) did form the tetramer in the crystalline state (Malay et al., 2005 ▶).
3.6. Active site
Numerous crystal structures of aldolase A bound to substrates or products have been determined (Blom & Sygusch, 1997 ▶; Choi et al., 1999 ▶, 2001 ▶) and have provided insight into aldolase substrate recognition and catalysis. The active site of rabbit muscle aldolase A is located in the center of the barrel and includes a number of conserved charged residues (Arg42, Lys107, Lys146, Glu187 and Arg303) that are in close proximity to the Schiff base-forming Lys229 (Lai et al., 1965 ▶). A molecule of the product DHAP and a sulfate ion were found liganded to the active site of the dimeric D128V aldolase (Fig. 6 ▶ a). The presence of the product was a consequence of the cleavage of Fru 1,6-P2 (retained from the purification process, which included elution with substrate). The C1 phosphate was bound by hydrogen bonds to the ∊-amino group of Lys229 and the main-chain N atoms of Ser271 and Gly272 as is found in other aldolase–DHAP structures (Blom & Sygusch, 1997 ▶; Choi et al., 2001 ▶; St-Jean et al., 2007 ▶). The C2 carbonyl and the C3 hydroxyl make hydrogen bonds to the Arg303 main-chain N atom and guanidino group, respectively. The sulfate ion binds to a site comprised of residues Arg303, Lys41 and Arg42, all of which are known to bind the C6 phosphate of Fru 1,6-P2 (Choi et al., 1999 ▶; St-Jean et al., 2005 ▶). There was some displacement of residues Lys41, Arg42 and Arg303 in the D128V aldolase structure when compared with previously solved DHAP-liganded structures (Fig. 6 ▶ b) owing to their binding interaction with the sulfate ion.
Figure 6.

Stereoview of the active site of fructose 1,6-bisphosphate. (a) DHAP and a sulfate ion depicted in ball-and-stick representation along with conserved active-site residues in D128V aldolase. A 1σ electron-density map is shown (violet cages). (b) Superposition of the active-site residues of D128V aldolase in red with DHAP and sulfate ion in yellow and the residues of the wild-type DHAP-bound structure (Blom & Sygusch, 1997 ▶; PDB code 1ado) in green. Hydrogen bonds are shown in black for D128V aldolase and in green for the wild-type structure.
4. Discussion
Fructose-1,6-bisphosphate aldolase has a highly conserved tetrameric structure with an atypical, low dissociation constant (Tolan et al., 2003 ▶). A possible explanation for this unusual stability is that it may have evolved as a tether molecule for roles in the cell other than those as a catalyst. The two interfaces in the quaternary structure (A and B) are highly conserved amongst aldolases from varied sources (Beernink & Tolan, 1994 ▶, 1996 ▶). The A interface is relatively hydrophobic, while the B interface is hydrophilic in nature and has buried hydrogen bonds making loop–loop interactions. It has been shown previously that substitution at one of these interfaces leads to the dissociation of aldolase into dimers (Beernink & Tolan, 1994 ▶). Monomers have been formed by making single mutations at each of the two interfaces. Both the monomeric and dimeric forms are fully active (Beernink & Tolan, 1996 ▶). Dissociation of the wild-type enzyme can only be accomplished under harsh conditions (Hsu & Neet, 1973 ▶, 1975 ▶; Lebherz, 1972 ▶; Rudolph et al., 1976 ▶; Gerschitz et al., 1977 ▶; Engelhard et al., 1976 ▶). Remarkably, previous studies of aldolase unfolding show that in 4 M urea, although most of the backbone is unfolded, three short segments located at the subunit interface remain intact, maintaining the protein in a tetrameric state (Deng & Smith, 1999 ▶).
Mutations that lead to inborn errors in metabolism are often associated with the subunit interfaces. The human mutations D128G, E206K and G346S are associated with nonspherocytic hemolytic anemia and myopathy (Kishi et al., 1987 ▶; Kreuder et al., 1996 ▶; Esposito et al., 2004 ▶). Biochemical studies on the D128G substitution have shown that the consequence of substitution at position Asp128 is an active dimer that is thermolabile. Other substitutions at Asp128 showed that the tetramer–dimer dissociation is shifted further to the right by the substitution of Asp128 by Val (Beernink & Tolan, 1994 ▶) than by substitution by Gly or Ala.
The structure of D128V aldolase showed that this substitution yielded a stable dimer. There was a negligible perturbation in the tertiary structure of the monomers. Moreover, the A interface of the dimer was retained and did not differ significantly from that of the tetramer, indicating that the interactions were strong enough to maintain the stable dimer. The most compelling evidence was that at 291 K the crystal-packing forces did not reform the tetramer although significant buried surface area was lost. The enzyme was stable enough to form a dimer in solution as observed via analytical ultracentrifugation. The strength of this interface is consistent with a potential role of the quaternary structure of aldolase as a tether molecule.
Finally, the presence of the product DHAP trapped in the active site after crystallization with Fru 1,6-P2 was an indication that the enzyme remains catalytically active in the absence of its native tetrameric structure. Together, these results strongly suggest that the quaternary structure of native aldolase is crucial to the performance of its many other cellular roles.
Supplementary Material
PDB reference: fructose-1,6-bisphosphate aldolase A dimer variant, 3bv4, r3bv4sf
Acknowledgments
This work was supported by grants GM060616 (to DRT and KNA) and DK065089 (to DRT) from the National Institutes of Health. Data for this study were measured on beamline X29 of the National Synchrotron Light Source. Financial support for NSLS comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy and from the National Center for Research Resources of the National Institutes of Health. We thank Dr Nicholas Silvaggi for careful reading of this manuscript.
References
- Arnold, H., Henning, R. & Pette, D. (1971). Eur. J. Biochem.22, 121–126. [DOI] [PubMed]
- Arnold, H. & Pette, D. (1968). Eur. J. Biochem.6, 163–171. [DOI] [PubMed]
- Beernink, P. T. & Tolan, D. R. (1994). Protein Sci.3, 1383–1391. [DOI] [PMC free article] [PubMed]
- Beernink, P. T. & Tolan, D. R. (1996). Proc. Natl Acad. Sci. USA, 93, 5374–5379. [DOI] [PMC free article] [PubMed]
- Beutler, E., Scott, S., Bishop, A., Margolis, N., Matsumoto, F. & Kuhl, W. (1973). Trans. Assoc. Am. Physicians, 86, 154–166. [PubMed]
- Blom, N. & Sygusch, J. (1997). Nature Struct. Biol.4, 36–39. [DOI] [PubMed]
- Bolen, D. W. & Baskakov, I. V. (2001). J. Mol. Biol.310, 955–963. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Cañete-Soler, R., Reddy, K. S., Tolan, D. R. & Zhai, J. (2005). J. Neurosci.25, 4353–4364. [DOI] [PMC free article] [PubMed]
- Choi, K. H., Mazurkie, A. S., Morris, A. J., Utheza, D., Tolan, D. R. & Allen, K. N. (1999). Biochemistry, 38, 12655–12664. [DOI] [PubMed]
- Choi, K. H., Shi, J., Hopkins, C. E., Tolan, D. R. & Allen, K. N. (2001). Biochemistry, 40, 13868–13875. [DOI] [PubMed]
- Deng, Y. & Smith, D. L. (1999). J. Mol. Biol.294, 247–258. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Engelhard, M., Rudolph, R. & Jaenicke, R. (1976). Eur. J. Biochem.67, 447–453. [DOI] [PubMed]
- Esposito, G., Vitagliano, L., Costanzo, P., Borrelli, L., Barone, R., Pavone, L., Izzo, P., Zagari, A. & Salvatore, F. (2004). Biochem. J.380, 51–56. [DOI] [PMC free article] [PubMed]
- Gamblin, S. J., Davies, G. J., Grimes, J. M., Jackson, R. M., Littlechild, J. A. & Watson, H. C. (1991). J. Mol. Biol.219, 573–576. [DOI] [PubMed]
- Gerschitz, J., Rudolph, R. & Jaenicke, R. (1977). Biophys. Struct. Mech.3, 291–302. [DOI] [PubMed]
- Hayward, S., Kitao, A. & Berendsen, H. J. (1997). Proteins, 27, 425–437. [DOI] [PubMed]
- Horecker, B. L., Tsolas, O. & Lai, C. Y. (1975). The Enzymes, edited by P. D. Boyer, Vol. 7, pp. 213–258. New York, London: Academic Press.
- Hsu, L.-S. & Neet, K. E. (1973). Biochemistry, 12, 586–595. [DOI] [PubMed]
- Hsu, L.-S. & Neet, K. E. (1975). J. Mol. Biol.97, 351–357. [DOI] [PubMed]
- Kao, A. W., Noda, Y., Johnson, J. H., Pessin, J. E. & Saltiel, A. R. (1999). J. Biol. Chem.274, 17742–17747. [DOI] [PubMed]
- Kim, J. H., Lee, S., Lee, T. G., Hirata, M., Suh, P. G. & Ryu, S. H. (2002). Biochemistry, 41, 3414–3421. [DOI] [PubMed]
- Kishi, H., Mukai, T., Hirono, A., Fujii, H., Miwa, S. & Hori, K. (1987). Proc. Natl Acad. Sci. USA, 84, 8623–8627. [DOI] [PMC free article] [PubMed]
- Kreuder, J., Borkhardt, A., Repp, R., Pekrun, A., Gottsche, B., Gottschalk, U., Reichmann, H., Schachenmayr, W., Schlegel, K. & Lampert, F. (1996). N. Engl. J. Med.334, 1100–1104. [DOI] [PubMed]
- Lai, C. Y., Hoffee, P. & Horecker, B. L. (1965). Arch. Biochem. Biophys.112, 567–579. [DOI] [PubMed]
- Lai, C. Y., Nakai, N. & Chang, D. (1974). Science, 183, 1204–1206. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lebherz, H. (1972). Biochemistry, 11, 2243–2250. [DOI] [PubMed]
- Lee, B. & Richards, F. M. (1971). J. Mol. Biol.55, 379–400. [DOI] [PubMed]
- Lu, M., Holliday, S., Zhang, L., Dunn, W. A. Jr & Gluck, S. L. (2001). J. Biol. Chem.276, 30407–30413. [DOI] [PubMed]
- Lu, M., Sautin, Y. Y., Holliday, L. S. & Gluck, S. L. (2004). J. Biol. Chem.279, 8732–8739. [DOI] [PubMed]
- Lundmark, R. & Carlsson, S. R. (2004). J. Biol. Chem.279, 42694–42702. [DOI] [PubMed]
- Malay, A. D., Allen, K. N. & Tolan, D. R. (2005). J. Mol. Biol.347, 135–144. [DOI] [PubMed]
- Malay, A. D., Procious, S. L. & Tolan, D. R. (2002). Arch. Biochem. Biophys.408, 295–304. [DOI] [PubMed]
- Metzler, M. (1989). J. Chromatogr.489, 11–21. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- O’Reilly, G. & Clarke, F. (1993). FEBS Lett.321, 69–72. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pagliaro, L. & Taylor, D. L. (1988). J. Cell Biol.107, 981–991. [DOI] [PMC free article] [PubMed]
- Penhoet, E. E., Kochman, M., Valentine, R. & Rutter, W. J. (1967). Biochemistry, 6, 2940–2949. [DOI] [PubMed]
- Penhoet, E. E. & Rutter, W. J. (1971). J. Biol. Chem.246, 318–323. [PubMed]
- Racker, E. (1947). J. Biol. Chem.167, 843–854. [PubMed]
- Rottmann, W. H., Deselms, K. R., Niclas, J., Camerato, T., Holman, P. S., Green, C. J. & Tolan, D. R. (1987). Biochimie, 69, 137–145. [DOI] [PubMed]
- Rottmann, W. H., Tolan, D. R. & Penhoet, E. E. (1984). Proc. Natl Acad. Sci. USA, 81, 2734–2742. [DOI] [PMC free article] [PubMed]
- Rudolph, R., Engelhard, M. & Jaenicke, R. (1976). Eur. J. Biochem.67, 455–462. [DOI] [PubMed]
- Rutter, W. J. (1964). Fed. Proc.23, 1248–1257. [PubMed]
- St-Jean, M., Izard, T. & Sygusch, J. (2007). J. Biol. Chem.282, 14309–14315. [DOI] [PubMed]
- St-Jean, M., Lafrance-Vanasse, J., Liotard, B. & Sygusch, J. (2005). J. Biol. Chem.280, 27262–27270. [DOI] [PubMed]
- Sukow, C. & DeRosier, D. (1998). J. Mol. Biol.284, 1039–1050. [DOI] [PubMed]
- Sygusch, J., Beaudry, D. & Allaire, M. (1987). Proc. Natl Acad. Sci. USA, 84, 7846–7850. [DOI] [PMC free article] [PubMed]
- Ta, T. V., Takano, R., Kamei, K., Xu, X. Y., Kariya, Y., Yoshida, K. & Hara, S. (1999). J. Biochem. (Tokyo), 125, 554–559. [DOI] [PubMed]
- Tolan, D. R., Schuler, B., Beernink, P. T. & Jaenicke, R. (2003). Biol. Chem.384, 1463–1471. [DOI] [PubMed]
- Volker, K. W. & Knull, H. (1997). Arch. Biochem. Biophys.338, 237–243. [DOI] [PubMed]
- Wang, J., Morris, A. J., Tolan, D. R. & Pagliaro, L. (1996). J. Biol. Chem.271, 6861–6865. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: fructose-1,6-bisphosphate aldolase A dimer variant, 3bv4, r3bv4sf