

The crystal structure of M. tuberculosis phosphoribosyl-ATP pyrophosphohydrolase, the second enzyme in the histidine-biosynthetic pathway, is presented. The structural and inferred functional relationships between M. tuberculosis phosphoribosyl-ATP pyrophosphohydrolase and other members of the nucleoside-triphosphate pyrophosphatase-fold family are described.
Keywords: histidine biosynthesis, pyrophosphatase, tuberculosis, actinobacteria, catalytic mechanism
Abstract
Phosphoribosyl-ATP pyrophosphohydrolase is the second enzyme in the histidine-biosynthetic pathway, irreversibly hydrolyzing phosphoribosyl-ATP to phosphoribosyl-AMP and pyrophosphate. It is encoded by the hisE gene, which is present as a separate gene in many bacteria and archaea but is fused to hisI in other bacteria, fungi and plants. Because of its essentiality for growth in vitro, HisE is a potential drug target for tuberculosis. The crystal structures of two native (uncomplexed) forms of HisE from Mycobacterium tuberculosis have been determined to resolutions of 1.25 and 1.79 Å. The structure of the apoenzyme reveals that the protein is composed of five α-helices with connecting loops and is a member of the α-helical nucleoside-triphosphate pyrophosphatase superfamily. The biological unit of the protein is a homodimer, with an active site on each subunit composed of residues exclusively from that subunit. A comparison with the Campylobacter jejuni dUTPase active site allowed the identification of putative metal- and substrate-binding sites in HisE, including four conserved glutamate and glutamine residues in the sequence that are consistent with a motif for pyrophosphohydrolase activity. However, significant differences between family members are observed in the loop region between α-helices H1 and H3. The crystal structure of M. tuberculosis HisE provides insights into possible mechanisms of substrate binding and the diversity of the nucleoside-triphosphate pyrophosphatase superfamily.
1. Introduction
Unlike most mammals, which are dependent on a dietary supply of histidine, many microorganisms, fungi and plants are able to synthesize histidine de novo. The histidine-biosynthesis pathway consists of ten enzymatic steps (Brenner & Ames, 1971 ▶; Winkler, 1996 ▶), starting with the condensation of ATP and phosphoribosyl pyrophosphate (PRPP) to form phosphoribosyl ATP (PRATP). The intermediates in this pathway are also used as precursors for purine biosynthesis, as well as for the synthesis of natural products such as the antitumor agent coformycin (Hanvey et al., 1987 ▶). Thus, the histidine-biosynthesis pathway is an ideal target for the development of inhibitors which could possess desirable antibacterial and/or herbicidal properties with low mammalian toxicity. In fact, all the genes in this pathway have been shown to be essential to the growth of Mycobacterium tuberculosis (Mtb) in vitro (Sassetti et al., 2001 ▶).
HisE is responsible for the second step in the pathway, the hydrolysis of pyrophosphate from PRATP. In Escherichia coli and many other microorganisms, a single gene, hisIE, encodes the enzymes for the second and third steps of the histidine-biosynthesis pathway (Smith & Ames, 1965 ▶). HisIE is a bifunctional enzyme; its amino-terminal and carboxy-terminal domains have phosphoribosyl-AMP cyclohydrolase (HisI) and phosphoribosyl-ATP pyrophosphohydrolase (HisE or PRATP-PH) activity, respectively. In contrast, many archaea and some actinobacteria, including Mtb, have independent genes that encode the pyrophosphohydrolase (hisE) and cyclohydrolase (hisI) activities (Beckler & Reeve, 1986 ▶). In the Mtb genome, hisE, together with hisG, is encoded in a different operon (Rv2121c–Rv2122c) from the rest of the genes of the histidine-biosynthesis pathway (Rv1599–Rv1606).
Through sequence and structural analysis, hisE-encoded PRATP-PH has been assigned to a superfamily of α-helical NTP pyrophosphatases (α-NTP-PPases; Moroz et al., 2005 ▶). The α-NTP-PPase superfamily is composed of structurally related α-helical enzymes that hydrolyze nucleoside triphosphates and share several conserved sequence motifs in their active sites, including a conserved Mg2+-binding EXXE sequence fingerprint (Moroz et al., 2005 ▶). These enzymes also share a highly conserved core composed of two central antiparallel α-helices that form the active site on one face and are also involved in oligomer assembly (Harkiolaki et al., 2004 ▶; Moroz et al., 2004 ▶). Other members of this superfamily, which are generally larger and contain additional α-helices, include homodimeric dUTPases from Trypanosoma cruzi (Harkiolaki et al., 2004 ▶) and Campylobacter jejuni (Moroz et al., 2004 ▶), the MazG NTP-PPase from Sulfolobus solfataricus and several less well characterized proteins such as the mouse protein RS21-C6, which is overexpressed in the thymus (Wu et al., 2007 ▶). These enzymes function by hydrolyzing the α–β phosphodiester bond of nucleoside triphosphates using essential Mg2+ ion(s) to activate a proposed water nucleophile and stabilize the charged intermediates to facilitate catalysis (Moroz et al., 2004 ▶). The α-NTP-PPase enzymes are unrelated to the well known nucleotide-binding enzymes with Rossmann-like and P-loop folds (Cheek et al., 2005 ▶).
In this paper, we describe the high-resolution crystal structures of ATP-phosphoribosyl pyrophosphatase from Mtb in two native (uncomplexed) crystal forms. The structure is found to be mostly α-helical, which is consistent with previous structures from this family. The protein folds into two antiparallel α-helices and dimerizes into a four α-helix bundle, forming two inferred PRATP active sites on the outer faces. While the structures reported here do not contain any bound ligands, conserved residues thought to be important for Mg2+ binding in other NTP-PPases can be identified. However, there are significant differences in the substrate-binding pocket, especially in the loop connecting α-helices H1 and H3, which suggest that HisE might bind PRATP differently to the manner in which nucleotide triphosphates are bound by other family members.
2. Experimental procedures
2.1. Expression and purification of phosphoribosyl-ATP-PH
The hisE (Rv2122c) gene from the Mtb H37Rv genome encoding the PRATP-PH was overexpressed in E. coli by cloning the gene into the pET23a(+) vector from Novagen without an affinity tag. Selenomethionine-labeled protein was produced in E. coli strain B834 using the medium described by LeMaster and coworkers (Davies et al., 2000 ▶). Protein expression was induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside for 4 h at 310 K. After harvesting, the cells were resuspended in 20 mM Tris–HCl pH 8.0 buffer containing protease inhibitors (Boehringer Mannheim), lysozyme (0.5 µg ml−1) and DNaseI (0.2 µg ml−1) and was stirred for 20 min. The cells were then lysed by passage through a French press at 83 MPa. Cell debris was removed by centrifugation at 23 426g. The supernatant was filtered through a 0.45 µm cellulose acetate syringe filter and then loaded onto a fast flow Q-Sepharose column (Amersham Pharmacia Biotech) that had been equilibrated with the same buffer prior to elution with a linear gradient to 250 mM NaCl in the same buffer. Solid ammonium sulfate was added to the pooled protein from the anion-exchange column to a final concentration of 1.0 M. The clear supernatant was then loaded onto a phenyl-Sepharose (Amersham Pharmacia Biotech) column previously equilibrated with 20 mM Tris–HCl pH 8.0 containing 1 M ammonium sulfate. The protein was eluted with a 1 l linear 1.0–0.0 M ammonium sulfate gradient at a flow rate of 1 ml min−1. Pooled fractions from the hydrophobic interaction column were concentrated to 4 ml using a Centriprep-3 (Amicon, Inc.) and then purified on a Superdex-75 HiPrep 16/100 column (Amersham Pharmacia Biotech). The active fractions that were >95% homogeneous, as determined by SDS–PAGE, were pooled and stored at 193 K.
2.2. Crystallization
Hanging-drop vapor diffusion produced two different crystal forms under different conditions. For both forms, the protein concentration was 10 mg ml−1 in 20 mM HEPES pH 7.5. Native and selenomethionine-containing protein crystals (form I) were obtained in 4 d after mixing protein solution and reservoir buffer (100 mM sodium cacodylate pH 6.5, 1.0 M sodium citrate, 0.1 M guanidinium hydrochloride) in a 1:1 ratio. The resulting rectangular crystals are orthorhombic and belong to space group I222. Crystal form II was obtained in 1.0 M sodium formate, 0.1 M sodium iodide, 0.1 mM calcium chloride. Crystal form II is monoclinic and belongs to space group C2.
2.3. Data collection and structure solution
The crystals were flash-frozen in a gaseous nitrogen stream at 100 K. Paratone-N oil (Hampton Research) was used as a cryoprotectant for both form I and II crystals. A multiple-wavelength anomalous dispersion (MAD) data set for the selenomethionine protein in crystal form I was collected to 1.23 Å resolution on the BioCARS 14BMD beamline at the Advanced Photon Source using a Q4 CCD detector (ADSC). The data sets were integrated and scaled with MOSFLM and SCALA from the CCP4 suite of programs (Leslie, 1992 ▶; Collaborative Computational Project, Number 4, 1994 ▶). The data for crystal form II were collected to 1.79 Å resolution from a single crystal at a home source using a Nonius/MacScience/DIP2030 image-plate detector with Osmic optics coupled to a Rigaku X-ray generator with a copper rotating anode (Cu Kα, wavelength 1.54 Å). The data for crystal form II were processed and scaled using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶).
Data-collection and refinement statistics for both form I and form II crystals are summarized in Tables 1 ▶ and 2 ▶. The programs SHELXD and SHELXE (Sheldrick, 2008 ▶) were employed for structure determination of crystal form I. One selenium site was found, corresponding to the Se atom incorporated into Met79. The high-energy remote data set, with anomalous signals merged, was used as the native data for phasing. The initial quality of the phases from SHELXE was quite high, with a figure of merit (FOM) of 0.817. Solvent flattening and histogram matching were applied to further improve the phases using the DM program from the CCP4 package (Cowtan & Main, 1998 ▶). The initial model was built on a Silicon Graphics workstation using the program Xfit from the XtalView program suite (McRee, 1999 ▶). The program REFMAC5 (Murshudov et al., 1997 ▶) was employed for refinement by the maximum-likelihood target function using anisotropic B factors (as afforded by the higher resolution). The model from crystal form I was then used to solve the monoclinic crystal form II by molecular replacement using MOLREP (Vagin & Teplyakov, 1997 ▶). Rigid-body rotation and translation searches identified four subunits in the asymmetric unit. The resulting model for crystal form II was edited in XtalView and refined with REFMAC5 (using isotropic B factors). The quality of the obtained models was validated with the program PROCHECK (Laskowski et al., 1993 ▶) and no residues were in disallowed regions.
Table 1. Crystallographic statistics.
Values in parentheses are for the outer resolution shell (1.231.31 for crystal form I and 1.791.85 for crystal form II).
| Crystal form I | Crystal form II | |||||
|---|---|---|---|---|---|---|
| Data set | Native† | Peak | Inflection | Low-energy remote | High-energy remote | Native (home source) |
| Space group | I222 | C2 | ||||
| Subunits in ASU | 1 | 4 | ||||
| Unit-cell parameters | ||||||
| a () | 37.93 | 117.70 | ||||
| b () | 62.60 | 43.12 | ||||
| c () | 67.05 | 65.55 | ||||
| () | 90.0 | 90.0 | ||||
| () | 90.0 | 96.43 | ||||
| () | 90.0 | 90.0 | ||||
| Wavelength () | 0.9572 | 0.9802 | 0.9800 | 1.0037 | 0.9572 | 1.542 |
| Resolution range () | 23.31.23 | 23.31.23 | 23.31.23 | 23.31.23 | 23.31.23 | 67.421.79 |
| No. of reflections | 317517 | 486815 | 215905 | 299934 | 317517 | 99562 |
| Unique reflections | 24103 | 22983 | 22523 | 21027 | 24103 | 30085 |
| R merge ‡ | 0.049 (0.44) | 0.056 (0.60) | 0.054 (0.58) | 0.045 (0.38) | 0.065 (0.51) | 0.052 (0.68) |
| I/(I) | 8.1 (1.6) | 6.7 (1.2) | 7.5 (1.3) | 8.0 (2.0) | 6.8 (1.4) | 26.0 (1.6) |
| Completeness (%) | 95.3 (88.2) | 96.5 (89.0) | 95.7 (88.2) | 96.1 (88.2) | 95.3 (88.2) | 98.9 (92.1) |
Native data for crystal form I were taken from the high-energy remote data set, with anomalous signals merged.
R
merge = 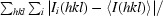
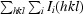 , where I(hkl) is the mean intensity of multiple Ii(hkl) observations of symmetry-related reflections.
, where I(hkl) is the mean intensity of multiple Ii(hkl) observations of symmetry-related reflections.
Table 2. Refinement statistics.
| Crystal form I | Crystal form II | |
|---|---|---|
| Resolution range () | 45.641.23 | 67.421.79 |
| No. of reflections used for refinement | 19181 | 30085 |
| No. of non-H atoms | 828 | 2941 |
| No. of water molecules | 151 | 252 |
| No. of ligand atoms | 0 | 0 |
| R work † (%) | 18.3 | 20.7 |
| R free ‡ (%) | 20.7 | 25.6 |
| Average B factor (2) | 11.05 | 27.45 |
| R.m.s. deviations from ideal geometry | ||
| Bond distance () | 0.006 | 0.01 |
| Bond angles () | 0.98 | 1.15 |
| Ramachandran plot | ||
| Residues in most favored regions (%) | 97.3 | 97.4 |
| Residues in additional allowed regions (%) | 2.7 | 2.6 |
| Residues in disallowed regions (%) | 0 | 0 |
| PDB code | 1y6x | 3c0 |
R factor = 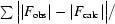
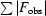 .
.
R free was monitored using 5% of the reflection data excluded from refinement.
3. Results and discussion
3.1. Amino-acid sequence analysis
Although the original annotation of the Mtb H37Rv genome sequence (Cole et al., 1998 ▶) did not list any open reading frames annotated as phosphoribosyl-ATP pyrophosphatase, in the updated annotation (Camus et al., 2002 ▶) this function was assigned to the Rv2122c gene (93 amino acids). Indeed, sequence analysis showed that its product shares 31% sequence identity with the carboxy-terminal domain of the bifunctional hisIE from E. coli. An alignment of related amino-acid sequences of predicted PRATP-PH enzymes from various bacterial and archaeal species showed a total of 11 residues that are conserved in 80–100% of the sequences (Fig. 1 ▶): Arg20, Ser28, Gly37, Lys44, Glu47, Glu48, Glu51, Ala55, Glu67, Leu71 and Val77.
Figure 1.

Multiple alignment of amino-acid sequences of HisE homologs from various bacterial species. The top three rows are from actinobacteria (Act.; Mycobacterium tuberculosis, Streptomyces coelicolor and Corynebacterium glutamicum). The remaining species include Listeria innocua, Bacillus cereus, Clostridium acetobutylicum, Methanococcus jannaschii, Solfolobus solfataricus, Neisseria meningitidis, Chromobacterium violaceum, Pseudomonas aeruginosa and Caulobacter crescentus. A representative MazG sequence from S. solfataricus has also been included for reference (bottom row). The species names in bold indicate proteins for which crystal structures have been solved. The alignment has been adjusted manually to be consistent with known gap placement in these structures. Gray boxes indicate residues that participate in the putative magnesium-binding site. ‘+’ indicates highly conserved residues (identical in at least ten of the 12 HisE sequences).
3.2. Overall structure
The structures obtained from crystal form I (space group I222), solved at 1.25 Å resolution, and crystal form II (space group C2) were essentially identical, except for a few minor differences that are discussed separately below. Both structures were in the native form and lacked density for any ligands other than water molecules in the active site. The crystal structure of Mtb HisE reveals that the protein is built entirely of α-helices and connecting loops (Fig. 2 ▶ a). The structure is composed of five α-helices (H1, residues 10–23; H2, 28–36; H3, 38–58; H4, 60–83; H5, 86–93), with the core of the protein formed primarily by the two long α-helices H3 and H4. The overall dimensions of an individual subunit are approximately 20 × 40 × 20 Å.
Figure 2.

(a) Backbone representation of the fold of Mtb HisE (residues 7–93). Helices H1–H5 are labeled. All molecular graphics in this paper were produced using SPOCK (Christopher, 1998 ▶). (b) Backbone representation of the dimer formed by two subunits packing together to form a four-helix bundle. Helix H5 in the C-terminus of each subunit wraps around the other subunit and contacts helix H1, interlocking the dimer.
The protein forms a dimer, with two PRATP-PH subunits related by a crystallographic twofold axis. Two pairs of long central α-helices, H3 and H4, pack together in an inverted fashion to form a four α-helix bundle (Fig. 2 ▶ b). The extensive interaction between the two subunits of the dimer buries 1960 Å2 of surface area per subunit, which corresponds to 31% of the total subunit surface area, as calculated using SPOCK (Christopher, 1998 ▶). There are a total of 28 residues involved in the dimer interface, with a high content of nonpolar residues (about 80%). The hydrophobic residues participating in protein dimerization include Leu13, Leu45, Leu46, Leu64, Leu71, Leu72, Leu78, Leu84, Leu93, Val38, Val52, Val89, Ile68, Ala49, Ala56, Ala65, Thr9, Thr75, Met79, Phe10, Trp53 and Tyr90. The aromatic residue Phe10 from the N-terminal α-helix H1 packs closely with its symmetry mate, contributing further to dimer stabilization. While Ala65 is the only residue that is perfectly conserved in the multiple sequence alignment of bacterial HisE homologs (see Fig. 1 ▶), many residues in the interface are highly conserved and are generally restricted to hydrophobic substitutions. In addition, there are three charged/polar interactions between subunits on the periphery of the interface which are solvent-exposed: His39–Glu57 (salt bridge), Asp62–Lys92 (salt bridge) and Asn61–Arg82 (reciprocal side-chain/backbone interactions). The amino- and carboxy-termini of each subunit are also involved in the dimer interaction (α-helix H5 from each subunit wraps around the other subunit, interlocking the dimer). The first six residues of each subunit are disordered.
In the form II crystals Mtb HisE crystallized in space group C2, with four molecules in the asymmetric unit, and the structure was solved at 1.79 Å resolution. The molecules appear as two independent dimers, each with an extensive interface identical to the interaction observed in the dimer in the form I crystal structure. With symmetry operations, the full tetramer can be reconstructed with approximately the same packing as observed in other HisE homologs (e.g. Bacillus cereus; PDB code 1yvw), with two subunits forming a tight dimer as a four α-helix bundle and two dimers associating in a looser complex with two α-helix bundles packing at an angle of roughly 45° (burying 791 Å2 of surface area per dimer). The structure of HisE in the form II crystals is largely equivalent to the structure in the form I crystals and there are no large-scale conformational changes. However, there is a small backbone shift in the region of residues 22–30. The Cα root-mean-square distance (r.m.s.d.) in this region is 1.60 Å, whereas the r.m.s.d. in the rest of the protein (residues 7–21 and 31–93) is 0.40 Å. This loop region is involved in a crystal lattice contact (247 Å2 of surface area per monomer is buried by interactions between residues 27–29 and the same residues of a symmetry-related molecule), so the observed structural shift could reflect an artifact of crystal packing. This loop is near the putative substrate-binding region (discussed below) based on superposition with other NTP-PPase superfamily members complexed with substrate analogs. There are three potentially relevant changes in the side-chain conformations of crystal form II compared with crystal form I. Thr29 Cβ moves 2.25 Å away from the putative nucleobase-binding site, Arg20 rotates inward to fill the shallow depression formed by the loop and Glu16 flips outward to solvent as a steric swap with Arg20 (Arg20 and Glu16 can occupy two roughly equivalent positions but not at the same time, so they alternate between the two structures). In the rest of the structure, the conformations of the backbone and side-chain atoms are substantially the same. The importance of crystal form II is that it has different rotamers for two residues in the putative metal-ion-binding site that better demonstrate the expected coordination geometry, as discussed below.
3.3. Comparison with related structures in the NTP-PPase family
Three other HisE (PRATP-PH) structures are available for the enzymes from Streptomyces coelicolor (PDB code 1yxb), B. cereus (1yvw) and Chromobacterium violaceum (2a7w) (all three were solved by the Northeast Structural Genomics Consortium). Despite having limited sequence identity (25–62% to Mtb, see rows 2, 5 and 10 in Fig. 1 ▶), the four enzymes exhibit strong conservation of the overall three-dimensional structure, including the core pair of α-helices. Superposition of the Cα atoms between Mtb HisE and the enzymes from S. coelicolor, B. cereus and C. violaceum gives r.m.s.d.s of 0.85, 1.15 and 1.77 Å (over 81, 83 and 80 residues, respectively). The S. coelicolor enzyme is the most similar to Mtb HisE (62% identity). The B. cereus and C. violaceum enzymes have an additional ∼18-residue C-terminal tail that partially extends α-helix H5 before becoming disordered.
A similarity search using a single subunit of PRATP-PH against the PDB with the program DALI (Holm & Sander, 1997 ▶) revealed structural similarities to a number of other proteins in the α-NTP-PPase superfamily (Moroz et al., 2005 ▶). Despite weak sequence similarity (<19% identity), good structural similarity is observed with C. jejuni dUTPase (PDB code 1w2y; Z score 10.4) and T. cruzi dUTPase (PDB code 1ogk). These enzymes are larger (229 and 283 residues, respectively) and the Mtb HisE structure superposes on a subset of α-helices in the core. Using SSM (Krissinel & Henrick, 2004 ▶), the backbone r.m.s.d.s of 1w2y and 1ogk to HisE were found to be 2.5 and 3.5 Å (over 69 and 74 residues), respectively, primarily reflecting conformational differences in the orientation of α-helix H5 and the loop connecting α-helices H1 and H3. An important difference between the HisE and dUTPase enzymes is that the dUTPases have an insertion of approximately ten residues in the loop connecting α-helices H3 and H4 and this loop reaches over to cover the substrate-binding pocket of an adjacent subunit in the dimer. HisE lacks this extended loop and the substrate-binding pocket is formed from residues in a single subunit.
Another significant match (Z score 7.2) was to S. solfataricus MazG (PDB code 1vmg), which is representative of a subfamily of NTP-PPases (Robinson et al., 2007 ▶). These enzymes are similar in length to PRATP pyrophosphohydrolases. For example, 1vmg is 95 residues in length, similar to the HisE enzyme, and has an almost identical fold (1.71 Å r.m.s.d. over 65 residues using SSM), despite having only 22% amino-acid identity to Mtb HisE (see the last row in Fig. 1 ▶). Mtb HisE also bears similarity to RS21-C6 (PDB codes 2oig and 2oie) from mouse (Wu et al., 2007 ▶) and an integron-associated MazG (PDB codes 2q9l, 2q5z and 2q73) from Vibrio, a marine bacterium (Robinson et al., 2007 ▶), which are also members of the MazG family. Structural similarity was also observed to the recently solved crystal structures of B. subtilis YpjD (PDB code 2gta), a putative pyrophosphatase, and an uncharacterized mouse protein, Mm.29898 (PDB code 2a3q). The primary difference between the MazG structures and the HisE structures is the divergence of the backbone in the H1–H3 loop region. For example, there is 4.35 Å Cα r.m.s.d. between residues 24–36 in Mtb HisE and 39–51 in Vibrio MazG, the significance of which is discussed below.
3.4. Analysis of the active site
With the aim of determining the structure of a complex between PRATP-PH and a substrate analog, we tried direct soaking of crystals in a solution containing 5 mM ATP analogs (NAD, AMP-PNP or AMP-CPP) in the presence of 50 mM Mg2+. However, we failed to find any density in the Fourier difference map for any of the above-mentioned ATP analogs. Cocrystallization also failed to yield a substrate complex. Nonetheless, some inferences about the putative active site can be made based on comparison with other NTP-PPase superfamily members which have been solved in complex with nucleoside triphosphate analogs. The corresponding site in Mtb HisE is on a solvent-exposed face of the helical core (facing away from the dimer axis). The core of the site is lined primarily by residues from α-helices H3 and H4.
In comparison with C. jejuni dUTPase, the active site in HisE is much more open and exposed. In 1w2y the site is formed by α-helices H1, H3 and H4, together with extensive contacts by residues from the additional C-terminal α-helices H9 and H10 which are completely absent in HisE. In addition, as noted above, several residues from the loop between α-helices H3 and H4 in an adjacent subunit (Lys57, His58 and Trp59 in 1w2y) extend as a flap to cover over the site, which is not the case for HisE. However, nucleic acid substrates can still bind to the less well formed active site in HisE without contributions from the additional α-helices of dUTPase, as evidenced by the complex of 5-methyl-dCTP with the mouse RS21-R6 MazG homolog (PDB code 2oig; Wu et al., 2007 ▶). The nucleotide binds in approximately the same position as in 1w2y (complexed with dUpNHp), but interacts with residues from all four N-terminal α-helices (H1–H4) in RS21-C6.
There is considerable difference among the enzymes with respect to the structure of the loop connecting α-helices H1 and H3 (residues 24–37 in Mtb HisE; Fig. 3 ▶). In dUTPase this region does not interact with the ligand, while in RS21-R6 it forms α-helix H2 that interacts extensively with the ligand (ring stacking and hydrogen bonding). In HisE, α-helix H2 moves closer in and α-helix H1 moves further away by several angstroms. In fact, Thr29 in α-helix H2 would conflict sterically with the ligand based on superposition with 2oig and this encroachment by α-helix H2 destroys the concavity of the active site. In the currently available HisE crystal structures, there is no obvious substrate-binding cavity and the binding mode of PRATP, particularly the phosphoribosyl moiety, remains unclear.
Figure 3.

Superposition of the backbones of Mtb HisE (PDB code 1w2y; white), S. solfataricus MazG (1vmg; red), mouse RS21-C6 MazG homolog (2oig; green) and T. cruzi dUTPase (1w2y; yellow; residues 110–229 deleted for clarity). The figure illustrates the high variability of the backbone in the loop region connecting helices H1 and H3 (residues 24–37 in Mtb HisE, including helix H2), in contrast to the good alignment of the central helix pair H3 and H4. Also shown is the 5-methyl-dCTP that was complexed with 2oig, indicating the putative substrate-binding region.
Comparison of the structures of these enzymes also provides insights into the residues that are potentially involved in metal binding and catalysis. The structure of the C. jejuni dUTPase complexed with a substrate analog (dUpNHp, a nonhydrolyzable derivative of deoxyuridine diphosphate) showed three magnesium ions bound per subunit. All three ions were chelated to the side-chain carboxylates of three strictly conserved glutamates (Glu46, Glu49 and Glu74) and one aspartate (Asp77) in an intricate network. The two O atoms in each glutamate bridge a different pair of Mg2+ ions (none are bidentate to a single ion), filling a total of eight coordination positions among the three bound ions; only one oxygen of Asp77 is involved, but one of the O atoms of Glu49 simultaneously fills the coordination spheres of two magnesiums. Two of the Mg2+ ions are in turn complexed with O atoms in the diphosphate tail of the ligand and the coordination system is completed by six water molecules. The divalent cations are thought to stabilize intermediates in the reaction (Moroz et al., 2004 ▶). The glutamates and aspartate are also conserved in the mouse R21-C6 MazG homolog (2oig), clustering in proximity to the triphosphate tail of liganded dCTP, although no Mg2+ ions were observed in the crystal structure and the triphosphate moiety projects into the solvent (Wu et al., 2007 ▶).
Superposition of the native PRATP-PH structure with these complexes reveals that the overall structure of the putative metal-binding site of HisE is quite similar to that of dUTPase (Fig. 4 ▶ a). The Mg2+-binding site occupies a negatively charged pocket that consists of the side chains of three conserved glutamates, Glu48, Glu51 and Glu67, and a glutamine, Gln70, which are positioned in close proximity to one another on the solvent-exposed face of α-helices H3 and H4 (Fig. 4 ▶ b). While the three glutamates are 100% conserved, position 70 is an aspartate in all organisms apart from actinobacteria, where it is uniquely substituted by a glutamine (see Fig. 1 ▶). The importance of the three key residues homologous to Glu48, Glu51 and Glu67 of Mtb PRATP-PH has been examined by mutational studies in T. maritima MazG, in which the mutation of the conserved equivalent residues Glu42, Glu45 and Glu61 in the active site of the enzyme resulted in the abolition of the hydrolysis of GTP as a substrate (Zhang et al., 2003 ▶).
Figure 4.

(a) A superposition of the metal-binding site between Mtb HisE crystal form II (white backbone and side chains) and T. cruzi dUTPase (1w2y; green backbone, gray side chains) complexed with three Mg2+ ions (orange spheres) and a dUDP analog (dUpNHp). The HisE residues (white) are labeled E48, E51, E67 and Q70. The corresponding dUTPase residues (gray) are E46, E49, E74 and D77. This figure illustrates the coordination of all three Mg2+ ions by the side chains in 1w2y (gray), including the central Mg2+ ion, which is coordinated by all four side chains simultaneously, and coordination of two of the Mg2+ ions to the two phosphates in the ligand. Furthermore, the side chains in the HisE structure are in approximately the same conformation, although they appear to be bound only to water molecules, since the HisE structure is uncomplexed. (b) Molecular surface showing the putative substrate-binding site (crystal form I). The substrate-binding pocket is slightly opened (owing to rotation of Gln70 away), but still constitutes a cluster of highly conserved negatively charged residues (E48, E51 and E67). (c) Superposition of the EXXE metal-binding motifs from the form I crystal (red) and form II crystal (blue). The ordered molecule (density peak modeled as a water molecule) in crystal form II is shown in the center (blue sphere) to illustrate the coordination to all four side chains. In contrast, in crystal form I Q70 is flipped away and the carboxylate of E51 is rotated differently.
In crystal form II (space group C2), in which the protein was crystallized in the presence of 2.0 M sodium formate and 0.1 M calcium chloride, the four conserved residues (Glu48, Glu51, Glu67 and Gln70) are found in approximately the same conformation as their catalytic counterparts in the metal-binding site of the C. jejuni dUTPase complex (Glu46, Glu49, Glu74 and Asp77; Fig. 4 ▶ a). A peak in the density exists in the middle of the cluster, coordinated to all four side chains. Although the density is not particularly strong (∼3σ), it appears to have octahedral coordination geometry (73–98° angles between adjacent coordinating side-chain atoms; 148° angle across from Glu51 to Gln70), with another water molecule in the fifth position in two of the four subunits and the sixth position open. The peak is positioned at 2.60, 2.30, 2.50 and 2.27 Å from O atoms in the side chains of residues Glu48, Glu51, Glu67 and Gln70, respectively. These distances are too long for typical oxygen–Mg2+ coordination bonds (2.0–2.1 Å) and are more typical of carboxylate–water distances. Therefore, this peak has preliminarily been modeled as a water molecule (and was refined as such), although we cannot rule out the possibility that it could be a magnesium ion carried along during the purification process or a calcium ion from the calcium chloride in the crystallization buffer. In contrast, in the form I crystal structure the metal-binding site is partially opened by the rotation of two side chains (Gln70 and Glu51) out of the cluster (Fig. 4 ▶ c). Gln70 adopts a different rotamer and is flipped out of the pocket and the carboxylate of Glu51 rotates 90° and is perpendicular to the orientation in crystal form I. However, Glu48 and Glu67 maintain similar positions and also coordinate a water molecule in the form I crystals in the same position as in the form II crystals (distances of 2.37 and 2.65 Å, respectively).
4. Discussion
One of the most interesting distinctions between HisE and other members of the NTP-PPase superfamily is its specificity for PRATP as part of its role in catalyzing the second step in the histidine-biosynthesis pathway. In particular, the binding of the phosphoribosyl (PR) group is important to elucidate because it represents the most significant difference in substrate recognition from the other NTP-PPase family members, which generally bind nucleoside triphosphates. The nucleobase-binding region in the dUTPase and MazG homologs is not large enough to accommodate the phosphoribosyl extension; in fact, this pocket is even more closed and more hydrophobic (with fewer hydrogen-bond donors and acceptors) in HisE.
One hypothesis is that the region between α-helices H1 and H3 (residues 24–37) might undergo a conformational rearrangement (e.g. shifting or unraveling of α-helix H2) to open the site upon substrate binding. Such a rearrangement has also been proposed for the Vibrio integron MazG, which appears to alternate between an open and closed conformation based on a comparison of native and complexed structures (Robinson et al., 2007 ▶). However, all crystal structures to date for HisE have been uncomplexed and no such rearrangement has yet been observed experimentally; it was also not observed between the native and complexed forms of RS21-R6 (2oie versus 2oig; Wu et al., 2007 ▶).
An alternative hypothesis, requiring no major conformational changes, would be that the substrate binds in a different mode, perhaps nestling the phosphoribosyl (PR) end of the ligand into a shallow depression near α-helix H1. The depression is large enough to accommodate an adenine as well as the phosphoribosyl extension. Interestingly, this depression is lined with a number of hydrophobic and hydrophilic residues, including Glu16, Leu17, Arg20, Pro25, Ser28, Thr30 and Tyr73, most of which are highly conserved among PRATP-PH homologs (Fig. 1 ▶). Adenine could π-stack on top of Tyr73, the ribose of PR could hydrogen-bond to several polar side chains and the phosphate could be positioned near the guanidinium group of Arg20.
Yet another possibility is that PRATP binding may involve complex formation with HisI, since HisE and HisI are often fused into a single bifunctional enzyme in many other organisms (Beckler & Reeve, 1986 ▶). Residues from HisI might help to more fully form the substrate-binding pocket and contribute to binding of the phosphoribosyl moiety. Discrimination of these possibilities will have to await further biochemical or crystallographic characterization.
Supplementary Material
PDB reference: phosphoribosyl-ATP pyrophosphohydrolase, space group I222, 1y6x, r1y6xsf
PDB reference: space group C2, 3c90, r3c90sf
Acknowledgments
We thank Dr Michael Galperin for fruitful discussions. This work was supported by grants PO1-AIO68135 and N01-AI-75320 from the National Institutes of Health and by funds from the Robert A. Welch Foundation (to JCS). We thank the scientists at the Advanced Photon Source (APS) of the Argonne National Laboratory (ANL) for their help with data collection. Use of BioCARS Sector 14 was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. DE-AC02-06CH11357.
References
- Beckler, G. S. & Reeve, J. N. (1986). Mol. Gen. Genet. 204, 133–140. [DOI] [PubMed]
- Brenner, M. & Ames, B. N. (1971). Metabolic Regulation, Vol. 5, edited by H. J. Vogel, pp. 349–387. New York: Academic Press.
- Camus, J. C., Pryor, M. J., Medigue, C. & Cole, S. T. (2002). Microbiology, 148, 2967–2973. [DOI] [PubMed]
- Cheek, S., Ginalski, K., Zhang, H. & Grishin, N. V. (2005). BMC Struct. Biol. 5, 6. [DOI] [PMC free article] [PubMed]
- Christopher, J. A. (1998). SPOCK Reference Manual. http://quorum.tamu.edu.
- Cole, S. T. et al. (1998). Nature (London), 393, 537–544.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cowtan, K. & Main, P. (1998). Acta Cryst. D54, 487–493. [DOI] [PubMed]
- Davies, C., Heath, R. J., White, S. W. & Rock, C. O. (2000). Structure, 8, 185–195. [DOI] [PubMed]
- Hanvey, J. C., Hawkins, E. S., Tunac, J. B., Dechter, J. J., Baker, D. C. & Suhadolnik, R. J. (1987). Biochemistry, 26, 5636–5641. [DOI] [PubMed]
- Harkiolaki, M., Dodson, E. J., Bernier-Villamor, V., Turkenburg, J. P., Gonzalez-Pacanowska, D. & Wilson, K. S. (2004). Structure, 12, 41–53. [DOI] [PubMed]
- Holm, L. & Sander, C. (1997). Nucleic Acids Res. 25, 231–234. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Leslie, A. G. W. (1992). Jnt CCP/ESF–EACBM Newsl. Protein Crystallogr. 26
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Moroz, O. V., Harkiolaki, M., Galperin, M. Y., Vagin, A. A., Gonzalez-Pacanowska, D. & Wilson, K. S. (2004). J. Mol. Biol. 342, 1583–1597. [DOI] [PubMed]
- Moroz, O. V., Murzin, A. G., Makarova, K. S., Koonin, E. V., Wilson, K. S. & Galperin, M. Y. (2005). J. Mol. Biol. 347, 243–255. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Robinson, A., Guilfoyle, A. P., Harrop, S. J., Boucher, Y., Stokes, H. W., Curmi, P. M. & Mabbutt, B. C. (2007). Mol. Microbiol. 66, 610–621. [DOI] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2001). Proc. Natl Acad. Sci. USA, 98, 12712–12717. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Smith, D. W. E. & Ames, B. N. (1965). J. Biol. Chem. 240, 3056–3063. [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst. 30, 1022–1025.
- Winkler, M. E. (1996). Escherichia coli and Salmonella: Cellular and Molecular Biology, Vol. 1, edited by F. C. Niedhardt & R. Curtiss, pp. 485–505. Washington: ASM Press.
- Wu, B., Liu, Y., Zhao, Q., Liao, S., Zhang, J., Bartlam, M., Chen, W. & Rao, Z. (2007). J. Mol. Biol. 367, 1405–1412. [DOI] [PubMed]
- Zhang, J., Zhang, Y. & Inouye, M. (2003). J. Biol. Chem. 278, 21408–21414. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: phosphoribosyl-ATP pyrophosphohydrolase, space group I222, 1y6x, r1y6xsf
PDB reference: space group C2, 3c90, r3c90sf