

A water network stabilizes the structure of cellular retionic acid binding protein II.
Keywords: CRABPII, retinoic acid, water-mediated interactions, structural integrity
Abstract
The structural integrity of cellular retinoic acid-binding protein II (CRABPII) has been investigated using the crystal structures of CRABPII mutants. The overall fold was well maintained by these CRABPII mutants, each of which carried multiple different mutations. A water-mediated network is found to be present across the large binding cavity, extending from Arg111 deep inside the cavity to the α2 helix at its entrance. This chain of interactions acts as a ‘pillar’ that maintains the integrity of the protein. The disruption of the water network upon loss of Arg111 leads to decreased structural integrity of the protein. A water-mediated network can be re-established by introducing the hydrophilic Glu121 inside the cavity, which results in a rigid protein with the α2 helix adopting an altered conformation compared with wild-type CRABPII.
1. Introduction
Retinoic acid (RA) is the principal metabolite of vitamin A (retinol), which plays important roles in cells such as modulation of cell growth, differentiation and morphogenesis by regulating the transcription of numerous target genes (Wang et al., 1997 ▶; Budhu & Noy, 2002 ▶; Delva et al., 1999 ▶). Administration of RA prevents the symptoms of vitamin A deficiency in animals (Gorry et al., 1994 ▶). Insufficient or excess RA causes malformations or toxicity in embryos and results in severe malfunctions of different cell types in a variety of adult organs (Maden, 1994 ▶; Napoli, 1996 ▶). Therefore, to ensure proper cell growth and function the amount of free RA in the cells must be highly regulated. Two homologous cytoplasmic proteins, cellular retinoic acid-binding protein (CRABP) types I and II (Sani & Hill, 1974 ▶; Ong & Chytil, 1975 ▶; Bailey & Siu, 1988 ▶), bind specifically to RA and regulate its effective concentration by binding to excess RA and/or metabolizing excess RA in the cell through interaction with metabolizing enzymes. CRABPs also solubilize RA and protect it from isomerization (Boylan & Gudas, 1991 ▶, 1992 ▶; Gaub et al., 1998 ▶; Napoli, 1996 ▶, 1999 ▶).
CRABPs are small (molecular weight of ∼16 kDa) soluble proteins that belong to the intracellular lipid-binding proteins (iLBPs) and are found in all vertebrates that need vitamin A (Gorry et al., 1994 ▶). Although both CRABPI and CRABPII are widely expressed in embryos, they do not co-express in the same cells (Maden, 1994 ▶).
CRABPI and CRABPII are highly homologous in humans (74% identity) and in other species. However, there is a higher sequence identity for the same isoform between species than there is for the two isoforms from the same species, which indicates these two homologues should have distinct functions. For example, CRABPI and CRABPII are 99.3 and 93.5% identical, respectively, between human and mouse (Budhu et al., 2001 ▶; Budhu & Noy, 2002 ▶). Although both CRABPs are believed to solubilize and transfer their ligands to the nucleus, experiments have shown that CRABPII enhances the transcriptional activity of RA (Jing et al., 1997 ▶; Bastie et al., 2001 ▶; Budhu et al., 2001 ▶; Budhu & Noy, 2002 ▶; Dong et al., 1999 ▶), while CRABPI enhances the activity of enzymes that catalyze RA degradation and therefore depresses RA efficacy in the cell (Boylan & Gudas, 1991 ▶, 1992 ▶).
Recently, we reported the first crystal structure of apo wild-type CRABPII (WT-CRABPII; Vaezeslami et al., 2006 ▶). The crystal structure of WT-CRABPII–RA has been reported previously by other groups (Kleywegt et al., 1994 ▶); however, a higher resolution structure of this complex determined at 1.48 Å is now available (Vaezeslami et al., 2006 ▶). In the latter work, using three different data sets collected from apo WT-CRABPII, we showed that apo and holo CRABPII have very similar structures. Binding of RA appeared to increase the overall rigidity of the structure, although the induced structural changes were not as pronounced as previously thought (Chen et al., 1998 ▶). The enhanced structural rigidity may be an important determinant for the enhanced nuclear localization of the RA-bound protein. Interestingly, water-mediated interactions are observed in the cavity of apo WT-CRABPII molecule A, connecting Arg111 deep inside the cavity through Arg132 to Ala36 and Val33, both of which are located on the α2 helix. The water-mediated network is not observed in molecule B, probably owing to the crystal packing. The difference in crystal-packing environments between molecules A and B in the α2 helix region is significant, which is apparent from the number of intermolecular interactions that each α2 helix makes with the neighboring molecules: molecule A has no hydrogen bonds and eight van der Waals contacts, and molecule B has seven hydrogen bonds and 43 van der Waals contacts.
Similar water-mediated networks have been observed in other members of the iLBP family, such as CRABPI (Thompson et al., 1995 ▶) and intestinal fatty acid-binding protein (Scapin et al., 1992 ▶), and have been suggested to play important roles in maintaining the stability of these proteins as well. In the rat intestinal fatty acid-binding protein (FABP) structure (PDB code 1ifc; 1.2 Å resolution), the water-mediated network connects Arg106 (corresponding to the conserved residue Arg111 in CRABPII) through Arg126 (corresponding to the conserved residue Arg132 in CRABPII) to Asp34 and Ala32 on the α2 helix at the portal. In the CRABPI structure (PDB code 1cbi), the network is observed in part, but the lower resolution of the structure (2.7 Å) probably leads to an incomplete observation of all the ordered water molecules in the cavity. In the high-resolution structure of cellular retinol-binding protein II (CRBPII, 1.2 Å resolution; PDB code 2rcq), which is also an iLBP-family member, the water-mediated network is again observed and again resembles that in apo WT-CRABPII. In the apo CRBPII structure, the network starts at Glu108 (corresponding to the conserved residue Arg111 in CRABPII based on similarity) and extends to Ala36 and Val33 (both conserved in CRABPII based on identity). These observations further emphasize the structural importance of these water-mediated networks for the iLBP-family proteins.
Here, we report the high-resolution crystal structures of several mutants of CRABPII. These structures show how mutations of the residues that interact with the carboxylate group of RA reposition RA inside the pocket. These structures also demonstrate the importance of certain residues in the structural integrity of CRABPII. Comparison of the structure of apo WT-CARBPII and those of the apo CRABPII mutants reported here shows that mutation of Arg111 causes structural changes in the molecule, indicating the importance of this highly conserved residue of CRABPII. Consistent with previous observations, solvent-mediated interactions are clearly present between residues that are buried deep inside the binding cavity and residues at the portal of the protein. These networks appear to be a determining factor in the structural integrity of this protein.
2. Materials and methods
The structure of CRABPII-R132K:Y134F–RA has been reported in previous work (Vasileiou et al., 2007 ▶). Here, we report the preparation, crystallization, data collection and structure determination of apo R132K:Y134F, apo R132K:R111L:L121E and apo and holo R132K:Y134F:R111L:L121E:T54V CRABPII mutants.
2.1. Site-directed mutagenesis
Site-directed mutagenesis was performed using the CRABPII-pET17b plasmid following the QuikChange protocol (Stratagene). The primers and procedure are described in the supporting information of our previously published data (Crist et al., 2006 ▶; Vasileiou et al., 2007 ▶).
2.2. Protein expression and purification of CRABPII-R132K:Y134F:R111L:L121E:T54V
Human recombinant CRABPII was expressed and purified as described elsewhere (Wang et al., 1997 ▶). The target gene (CRABPII-pET-17b), as isolated from JM109 using the Maxi Prep DNA-isolation kit (Qiagen), was transformed into Escherichia coli strain BL21(DE3)pLysS cells (Stratagene) according to standard protocols. The transformed cells were grown at 310 K on an LB agar plate containing both ampicillin (100 µg ml−1) and chloramphenicol (25 µg ml−1). A single colony was inoculated into 100 ml LB containing the same amounts of the two antibiotics and grown overnight with shaking at 250 rev min−1 at 310 K. This culture was then transferred to 1 l LB with the same antibiotics. The culture was incubated at 310 K until the A 600 was between 0.6 and 0.8. Expression was induced by the addition of 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Cell growth was continued for another 4–5 h at 303 K. The cells were harvested by centrifugation (6000 rev min−1, 30 min) and frozen at 193 K. The frozen cells obtained from a 2 l growth were thawed on ice and resuspended in 100 ml 10 mM Tris–HCl pH 8.0. The suspended cells were lysed by five 45 s bursts of sonication (probe sonicator, Biologics Inc., 60% power) on ice and centrifuged for 15 min at 277 K and 5000 rev min−1. The supernatant was subjected to Fast-Q (Q-Sepharose Fast Flow Resin) column chromatography (80 ml bed volume; Amersham Biosciences), which was pre-equilibrated with 10 mM Tris–HCl pH 8.0 buffer. The column was washed using the same buffer and proteins were eluted using a NaCl gradient of 0–200 mM. CRABPII was eluted in 100–150 mM NaCl. The fractions were analyzed by 20%(w/v) SDS–PAGE and those of highest purity (∼95%) were combined and desalted using a Vivaspin concentrator (Vivascience) with a 5000 Da molecular-weight cutoff (10 000 Da molecular-weight cutoff concentrators showed some protein leakage through the membrane). The desalted protein solution was further purified on a Bio-Rad system (BioLogics Duo Flow, Bio-Rad) using a Source-15Q (Amersham Biosciences) anion-exchange column. Fractions were monitored at A 280 and analyzed by 20% SDS–PAGE. All purification steps were performed at 277 K. The most pure fractions were combined, concentrated and buffer-exchanged (100 mM Tris–HCl pH 8.0) in Vivaspin concentrators (Vivascience) to a concentration of ∼20 mg ml−1, as determined by measuring the A 280. The pure concentrated protein solution was divided into 100 µl aliquots, flash-frozen and stored at 193 K.
2.3. Crystallization and data collection
2.3.1. Apo CRABPII-R132K:Y134F
1.5 µl of the mutant (∼30 mg ml−1) was mixed with 1.5 µl reservoir solution. The protein was crystallized using the same conditions as used for the best diffracting crystals of apo WT-CRABPII [0.2 M sodium acetate, 0.1 M Tris–HCl pH 8.5, 30%(w/v) PEG 4000] and at room temperature. Data were collected on the COM-CAT beamline (32-ID) at the APS synchrotron facility. 360 frames were collected with 1° oscillation at a wavelength of 1.00 Å. Data-collection statistics are reported in Table 1 ▶.
Table 1. X-ray data-collection statistics.
Values in parentheses are for the last resolution shell. KF, R132K:Y134F; KFLEV, R132K:Y134F:R111L:L121E:T54V; KLE, R132K:R111L:L121E.
| Apo KF | Apo KFLEV | KFLEVRA | Apo KLE | |
|---|---|---|---|---|
| Space group | P1 | P1 | P212121 | P1 |
| Z (molecules per ASU) | 2 | 2 | 1 | 2 |
| Unit-cell parameters | ||||
| a () | 34.6 | 34.6 | 45.8 | 34.6 |
| b () | 37.2 | 36.9 | 44.9 | 37.2 |
| c () | 58.8 | 57.8 | 77.9 | 61.1 |
| () | 73.9 | 73.3 | 90 | 73.4 |
| () | 74.8 | 75.9 | 90 | 73.7 |
| () | 87.9 | 87.8 | 90 | 89.7 |
| Wavelength () | 1.00 | 1.00 | 1.00 | 1.00 |
| Resolution range () | 50.01.7 (1.761.70) | 40.01.2 (1.241.20) | 39.51.6 (1.661.60) | 55.91.5 (1.551.50) |
| Average I/(I) | 20.4 (1.6) | 18.2 (2.6) | 27.3 (9.0) | 8.3 (2.4) |
| Total reflections | 254023 | 539004 | 394601 | 112496 |
| Unique reflections | 29672 | 82503 | 21863 | 42752 |
| Completeness | 89.7 (48.9) | 93.8 (89.9) | 99.8 (100) | 96.1 (93.0) |
| R merge (%) | 5.2 (24.0) | 6.6 (27.1) | 5.0 (15.7) | 4.0 (29.8) |
2.3.2. Apo CRABPII-R132K:Y134F:R111L:L121E:T54V
The mutant (∼20 mg ml−1) was crystallized in 0.1 M sodium acetate pH 5.8, 20% PEG 6000 and 5% ethanol at room temperature. The ethanol proved to be necessary as an additive to form well diffracting crystals. Crystals obtained without ethanol were not single and formed clusters. Diffraction data were collected on the COM-CAT beamline (32-ID) at the APS synchrotron facility. 300 frames were collected with 1° oscillation at a wavelength of 1.00 Å and a crystal-to-detector distance of 80.0 mm. Data-collection statistics are reported in Table 1 ▶.
Streaking of the drop with seeds consisting of crushed crystals from the same drop produced larger and well diffracting crystals. The smaller crystals proved to diffract better than the larger ones. In an effort to produce retinal-bound crystals of this mutant, the crystals were soaked for 10 h in a solution prepared by mixing 90–95% of the reservoir with 5–10% of saturated retinal stock solution (∼5.5 × 10−2 M in ethanol) at 277 K. The soaked crystals diffracted to an even higher resolution of 1.2 Å, although our attempt to produce retinal-bound crystals was unsuccessful.
2.3.3. CRABPII-R132K:Y134F:R111L:L121E:T54V–RA
RA is a light-sensitive molecule; all manipulations and crystallizations of RA-bound CRABPII were therefore performed in the dark and under red light.
The penta mutant (∼20 mg ml−1) bound to RA was crystallized in 0.1 M MES pH 6.5, 30%(w/v) PEG 5000, 0.2 M ammonium sulfate at room temperature. The crystallization boxes were wrapped in aluminium foil and kept in dark cabinets. Crystals appeared in 3 d and reached their maximum dimensions in a week. They were quick-soaked in a cryoprotectant solution consisting of the reservoir solution plus 30% glycerol and immediately frozen in liquid nitrogen. Data were collected on the COM-CAT beamline (32-ID) at the APS synchrotron facility. 200 frames were collected with 1° oscillation at a wavelength of 1.00 Å. The crystal-to-detector distance was set to 108.0 mm. Data-collection statistics are reported in Table 1 ▶.
2.3.4. Apo CRABPII-R132K:R111L:L121E
The triple mutant (∼27 mg ml−1) was crystallized in 0.1 M Bis-Tris propane pH 6.5 and 30% PEG 4000 at room temperature (identical to the conditions used for apo WT-CRABPII). Data were collected on the COM-CAT beamline (32-ID) at the APS synchrotron facility. 300 frames were collected with 1° oscillation at a wavelength of 1.00 Å. Data-collection statistics are reported in Table 1 ▶.
In all cases, two data sets were collected: one with minimal exposure to avoid intensity overload of the low-resolution reflections and one with longer exposure times to better measure the weaker high-resolution reflections. Data sets were integrated using DENZO and scaled and merged using SCALEPACK from the HKL package (Otwinowski & Minor, 1997 ▶).
2.4. Structure determination
The structure-determination and refinement calculations were performed using the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
2.4.1. Apo mutants
The structures of the apo mutants were determined using rigid-body refinement in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The coordinates of both of the molecules of the apo WT-CRABPII structure (PDB code 2fs6) were used as the original model. The model was visualized and manually rebuilt using the program TURBO-FRODO (Roussel & Cambillau, 1989 ▶). REFMAC v.5.2.0005 was used to refine the structure against 90–95% of the data (Murshudov et al., 1997 ▶), while 5–10% of the data were chosen randomly for cross-validation using R free (Brünger, 1993 ▶). All three apo CRABPII mutant structures were refined using anisotropic refinement. Alternative conformations of 18 amino-acid side chains and 26 waters were observed for apo CRABPII-R132K:Y134F (molecules A and B combined). In the apo CRABPII-R132K:Y134F:R111L:L121E:T54V structure, 14 amino acids and 22 water molecules were identified as having alternative conformations. In the apo R132K:R111L:L121E mutant, four amino acids and 14 waters were observed with alternative conformations. The refinement statistics are reported in Table 2 ▶.
Table 2. Structure-refinement statistics.
KF, R132K:Y134F; KFLEV, R132K:Y134F:R111L:L121E:T54V; KLE, R132K:R111L:L121E.
| Apo KF | Apo KFLEV | KFLEVRA | Apo KLE | |
|---|---|---|---|---|
| Average B factor (2) | 23.2 | 18.1 | 14.1 | 24.5 |
| Wilson B factor (2) | 21.8 | 16.6 | 24.9 | 19.6 |
| R work (%) | 15.4 | 13.8 | 12.0 | 15.4 |
| R free (%) | 22.2 | 17.6 | 16.8 | 22.7 |
| No. of water molecules | 362 | 399 | 221 | 347 |
| Total reflections used | 25713 | 75296 | 21611 | 41632 |
| RMSDs from ideality | ||||
| Bond lengths () | 0.020 | 0.018 | 0.020 | 0.028 |
| Bond angles () | 1.78 | 1.69 | 1.85 | 2.32 |
| Ramachandran plot | ||||
| Most favored (%) | 93.5 | 92.7 | 94.4 | 94.0 |
| Allowed (%) | 5.6 | 6.9 | 4.8 | 5.2 |
| Generously allowed (%) | 0.4 | 0 | 0 | 0 |
| Disallowed (%) | 0.4 | 0.4 | 0.8 | 0.8 |
| Clashscore (MOLPROBITY) | 9.34 | 4.55 | 6.50 | 12.00 |
| PDB code | 3d96 | 3d95 | 3cwk | 3d97 |
2.4.2. CRABPII-R132K:Y134F:R111L:L121E:T54V–RA
The structure was determined, as described for the apo mutants, using rigid-body refinement. The previously published structure of WT-CRABPII–RA (PDB code 1cbs; Kleywegt et al., 1994 ▶), with RA removed, was used as the starting model. Anisotropic refinement in REFMAC5 resulted in improved R factors and is justified by the thermal ellipsoid plot (not shown). 18 amino acids and 17 waters were observed with alternative conformations. The refinement statistics are reported in Table 2 ▶.
3. Results and discussion
3.1. The structure of the WT-CRABPII–RA complex
All-trans RA binds inside the pocket with its hydrophilic end buried deep inside the cavity and its ionone ring partially solvent-exposed at the portal of the pocket (Fig. 1 ▶ a; Wang & Yan, 1998 ▶; Kleywegt et al., 1994 ▶). RA is mostly surrounded by hydrophobic residues, except for the carboxylate-group region, which makes direct and tight hydrogen bonds with Arg132 and Tyr134 from one side and water-mediated hydrogen bonds with Arg111 and Thr54 from the other side of the pocket (Fig. 1 ▶ b). These tight hydrogen bonds make RA a good ligand for the protein (K d ≃ 2 nM).
Figure 1.

(a) RA in the pocket of WT-CRABPII; (b) hydrogen-bonding interactions of the carboxylate group of RA with the residues inside the pocket. The distances are in angstroms.
Consistent with the structural data, a novel competitive binding assay was developed by Yan and coworkers to measure the relative dissociation constants of site-directed mutants of CRABPs. Arg111 and Arg132 of CRABPII were replaced with Met by site-directed mutagenesis. The relative dissociation constants of the R111M and R132M mutants [K d(R111M)/K d(WT) and K d(R132M)/K d(WT)] were determined to be 40–45 and 6–8, respectively (Wang et al., 1997 ▶). These data indicate that Arg132 and Arg111 are important residues in the binding of RA to CRABPII. Recently, we have also shown the importance of these and several other residues for RA binding in CRABPII (Vasileiou et al., 2008 ▶).
Here, we mostly focus on the structural aspects of each of these mutants to show the importance of the mutated residues on the structural integrity of CRABPII. The crystal structures of these CRABPII mutants clearly show how different binding environments inside the cavity lead to differences in RA binding.
3.2. The structure of apo CRABPII-R132K:Y134F
Arg132 is a fully conserved residue in CRABPII and is strongly conserved as either an Arg or an Asp in the entire iLBP family. Tyr134 is also a highly conserved aromatic residue in this family. Lys and Phe are conservative mutations for Arg and Tyr, respectively. To understand whether these conserved mutations affect the structural integrity of CRABPII, we successfully crystallized both the apo and RA-bound R132K:Y134F mutant and determined their structures. The structure of the apo R132K:Y134F mutant was refined at 1.70 Å resolution with R factors R work = 15.4% and R free = 22.2%. The structure was solved in the same P1 space group and with the same crystal packing as the apo WT-CRABPII structure (Vaezeslami et al., 2006 ▶). The result was a structure that was virtually identical to that of the wild-type protein. The RMSDs between molecules A and molecules B of the two structures are 0.17 and 0.34 Å, respectively, which indicate that these two mutations do not change the overall fold of the structure. The Cα-deviation plot between the apo R132K:Y134F mutant and apo WT-CRABPII is shown in Fig. 2 ▶. Fig. 3 ▶ shows the superimposed structures of molecules A and molecules B of the apo R132K:Y134F:R111L:L121E:T54V mutant and apo WT-CRABPII. Since the overall fold of the apo R132K:Y134F mutant is similar to that of apo WT-CRABPII as well as that of the apo R132K:Y134F:R111L:L121E:T54V mutant structure, the overall structure of the double mutant is almost identical to the structures shown in Fig. 3 ▶.
Figure 2.

Cα-deviation plots between different CRABPII mutants and apo WT-CRABPII. (a) Deviation between molecules A; (b) deviation between molecules B. Green, apo R132K:Y134F; pink, RA-bound R132K:Y134F; yellow, apo R132K:Y134F:R111L:L121E:T54V; blue, RA-bound R132K:Y134F:R111L:L121E:T54V; red, apo R132K:R111L:L121E.
Figure 3.

The overall fold of these mutants (except for molecule A of apo CRABPII-R132K:R111L:L121E) is very well conserved compared with the apo WT-CRABPII structure, as represented by the superimposed structures of (a) molecules A of apo CRABPII-R132K:Y134F:R111L:L121E:T54V (pink) and apo WT-CRABPII (green) and (b) molecules B of apo CRABPII-R132K:Y134F:R111L:L121E:T54V (blue) and apo WT-CRABPII (hot pink).
In both the apo WT-CRABPII and WT-CRABPII–RA structures previously reported, similar solvent-mediated interactions exist which connect Arg111 deep inside the cavity to Ala36 and Val33 located on the α2 helix and the loop that links α2 and βB. Comparison of the apo WT-CRABPII structure with that of the apo R111M mutant (PDB code 1xca) shows that the solvent-mediated interaction is disrupted in the R111M mutant and that the α2 helix is unwound and disrupted. Therefore, these networks were assumed to play an important role in the integrity of the CRABPII structure (Vaezeslami et al., 2006 ▶), which is supported by the fact that similar water-mediated networks have been observed in other members of the iLBP family and suggested to play essential roles in maintaining the structural integrity (Scapin et al., 1992 ▶; Thompson et al., 1995 ▶).
In fact, a similar solvent-mediated interaction bridges Lys132 with the carbonyl groups of Val33 and Ala36 across the binding cavity, as was observed in the structure of apo WT-CRABPII. The networks are shown in Fig. 4 ▶(a) and include (i) guanidino group of Arg111→water 121→water 225→water 290→amino group of Lys132→water 202→water 83→water 44→carbonyl of Val33 (there is also a direct hydrogen bond between the amino group of Lys132 and water 83, but it is a weak interaction with distance 3.4 Å) and (ii) guanidino group of Arg111→water 121→water 225→water 290→amino group of Lys132→water 202→carbonyl of Ala36. The F o − F c OMIT map (calculated at 2.5σ) for the corresponding water molecules is shown in Fig. 4 ▶(b). The B factors, occupancies and number of hydrogen bonds of each water molecule that is involved in the network are listed in Table 3 ▶. The presence of these water molecules is also supported by their low temperature factors as well as the number of hydrogen bonds that the specific water molecules make. Water 290 is the only one that shows a relatively high B factor. However, the appearance of a water molecule at the same position in the apo R132K:Y134F:R111L:L121E:T54V mutant suggests the correct assignment of this water. If we overlay the water-mediated interactions in molecule A of the apo R132K:Y134F mutant with those in molecule A of apo WT-CRABPII, we can see that four of the six waters in the apo R132K:Y134F mutant are conserved between the two structures. Water 121 that interacts with Arg111 moves about 0.2 Å between the two structures. Water 44 that interacts with the Val33 carbonyl O atom moves by 0.6 Å. Water 225 resides at the same position as an acetate O atom in apo WT-CRABPII (0.3 Å difference). Water 202 occupies the position taken by one of the terminal N atoms of Arg132 in apo WT-CRABPII (0.3 Å difference). Waters 83 and 290 do not have equivalents in apo WT-CRABPII owing to the R132K mutation. Waters 83 and 290 compensate for the reduced side-chain volume that results from the R132K mutation, which is key for re-establishing a continuous hydrogen-bonding network.
Figure 4.
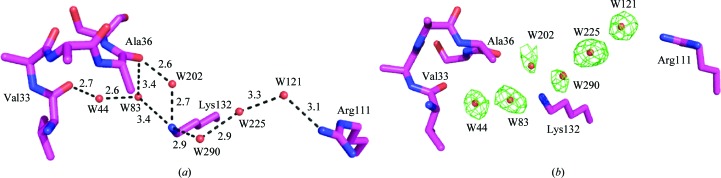
(a) The water-mediated interaction between Arg111 and Ala36 and Val33 in apo CRABPII-R132K:Y134F (the distances are in angstroms); (b) F o − F c OMIT map of the water molecules calculated at 2.5σ.
Table 3. B factors, occupancies and number of hydrogen bonds associated with the ordered water molecules that are involved in the water-mediated interactions in molecules A of the CRABPII mutants.
| No. of hydrogen bonds | ||||||
|---|---|---|---|---|---|---|
| Mutant | Overall B factor (2) | Water | B factor (2) | Occupancy | To protein | To solvent |
| Apo R132K:Y134F | 23.2 | 44 | 32.5 | 1 | 1 | 2 |
| 83 | 36.9 | 1 | 3 | 2 | ||
| 202 | 36.7 | 1 | 2 | 1 | ||
| 290 | 56.8 | 1 | 1 | 1 | ||
| 225 | 34.3 | 1 | 0 | 2 | ||
| 121 | 28.9 | 1 | 2 | 2 | ||
| R132K:Y134FRA | 21.3 | 134 | 42.8 | 1 | 3 | 1 |
| 11 | 25.8 | 1 | 3 | 1 | ||
| 10 | 24.1 | 1 | 2 | 1 | ||
| Apo R132K:Y134F:R111L:L121E:T54V | 18.1 | 230 | 18.0 | 0.5 | 1 | 3 |
| 89 | 27.4 | 1 | 1 | 3 | ||
| 83 | 28.8 | 1 | 2 | 1 | ||
| 391 | 40.2 | 1 | 1 | 1 | ||
| 392A | 26.3 | 0.5 | 0 | 2 | ||
| 392B | 19.7 | 0.5 | 2 | 2 | ||
| 393 | 36.6 | 1 | 2 | 1 | ||
| R132K:Y134F:R111L:L121E:T54VRA | 14.1 | 65 | 33.4 | 1 | 1 | 1 |
| 62 | 24.0 | 1 | 2 | 2 | ||
| 187 | 28.6 | 1 | 1 | 1 | ||
| Apo R132K:R111L:L121E | 24.5 | 18 | 22.9 | 1 | 2 | 2 |
| 212 | 32.8 | 1 | 0 | 3 | ||
| 102 | 31.6 | 1 | 2 | 2 | ||
| 13 | 25.7 | 1 | 1 | 2 | ||
This elegant water-mediated interaction was not observed in molecule B of this mutant, similar to molecule B of apo WT-CRABPII. Lys132 has two evenly occupied conformations in molecule B. Although one of these two conformers interacts with Arg111 through two water molecules, it does not interact with the loop region. This structure shows that the crystal-packing interactions in the loop region of molecule B can dictate the interactions inside the pocket, similar to what has been observed in molecule B of apo WT-CRABPII. Molecule B of this mutant is almost identical to molecule B of apo WT-CRABPII (RMSD = 0.34 Å) and makes similar intermolecular interactions with neighboring symmetry-related molecules (Figs. 2 ▶ and 3 ▶). For more details, see the discussion on the comparison of molecules A and B of apo WT-CRABPII in Vaezeslami et al. (2006 ▶).
More importantly, this structure shows that a similar structure to apo WT-CRABPII, with similar solvent-mediated interactions in the binding cavity, is obtained when Arg111 is intact and other residues in the binding cavity are conservatively mutated.
3.3. The structure of the CRABPII-R132K:Y134F–RA complex
As reported previously (Vasileiou et al., 2007 ▶), the structure of the R132K:Y134F mutant bound to RA (CRABPII-R132K:Y134F–RA) has been determined and refined at 1.70 Å resolution with good crystallographic R factors R work = 14.5% and R free = 20.3%. This structure is superimposed on the structure of WT-CRABPII–RA in Fig. 5 ▶(a), which shows that the structure of CRABPII-R132K:Y134F–RA is almost identical to that of WT-CRABPII–RA (RMSD = 0.18 Å). This similarity further indicates that these two mutations do not change the integrity of the structure.
Figure 5.

(a) Overlaid structures of R132K:Y134F–RA (brown) and WT CRABPII–RA (yellow); (b) water-mediated interaction between Arg111 deep inside the cavity and Ala36 located on the loop connecting α2 to βB in R132K:Y134F–RA. The distances are in ansgtroms.
As shown in Fig. 5 ▶(b), a similar water-mediated network that connects Arg111 to Ala36 is observed. This further suggests the importance of Arg111 and water-mediated interactions in the structural integrity of CRABPII. The path of the water-mediated network is as follows: guanidino group of Arg111→water 11→carboxylate group of RA→amino group of Lys132→carbonyl of Ala36. The B factors, occupancies and number of hydrogen bonds of each water molecule that is involved in the network are listed in Table 3 ▶. Both waters 11 and 134 are conserved compared with apo WT-CRABPII and apo CRABPII-R132K:Y134F. Water 10 does not have an equivalent counterpart in apo WT-CRABPII, probably because it is not critical in constructing the interaction network. Unlike in the WT-CRABPII–RA and apo WT-CRABPII structures, Arg111 does not have a water-mediated interaction with Val33 on the α2 helix. This suggests that the interaction with Ala36 is more of a determinant for the structure.
3.4. The structure of apo CRABPII-R132K:Y134F:R111L:L121E:T54V
The structure of the apo R132K:Y134F:R111L:L121E:T54V mutant (the penta mutant) was determined at a high resolution of 1.20 Å with crystallographic R factors R work = 13.8% and R free = 17.6%. The superimposed structures of molecules A and molecules B of the penta mutant and apo WT-CRABPII are shown in Fig. 3 ▶. The RMSDs between molecules A and molecules B of the two structures are 0.26 and 0.53 Å, respectively. Comparing the two structures indicates that these five mutations do not change the overall fold of the structure. However, during refinement we realised that although the α2 helix has an identical conformation to that in apo WT-CRABPII, it also has an additional conformation.
To generate an unbiased map of the loop and α2 helix regions, residues 26–40, which are located in the α2 helix and the loop connecting this helix to βB, were deleted from map calculations. An F o − F c OMIT electron-density map was calculated for this region, which clearly showed one conformation for the helix (Fig. 6 ▶). This conformation was identical to that in apo WT-CRABPII. After the helix and loop had been modeled in the OMIT map, some extra positive electron density was generated. However, the extra density was not definitive enough to build a second conformation. The B factors of the atoms in this region (residues 26–40) were ∼20 Å2, which are close to the average B factor of this structure (18.1 Å2). This observation indicates that although Arg111 is mutated to a hydrophobic residue, the helix and loop are not as severely destabilized as was observed in the R111M mutant structure. In addition, calculation of the secondary structure using the DSSP program confirms that, similar to apo WT-CRABPII, residues 26–35 comprise the second helix and afford three full helical turns (Kabsch & Sander, 1983 ▶).
Figure 6.

F o − F c OMIT map calculated at 2.5σ after deleting residues 26–40 from molecule A of apo CRABPII-R132K:Y134F:R111L:L121E:T54V. Only one conformation is observed for this helix.
Leu121 is not a conserved residue in CRABPII and is therefore not expected to be critical to the structural integrity of the protein. We believe that either the R111L or the T54V mutation (or both at the same time) is responsible for partially destabilizing the helix and the loop regions. Thr54 is a conserved residue (80%) and also engages in a solvent-mediated interaction with the helix–loop region, similar to that between Arg111 and the helix–loop in the apo WT-CRABPII and the apo R132K:Y134F mutant structures.
In order to understand why the R111M structure has a destabilized α2 helix (B factor of ∼75 Å2) and yet the penta mutant has a well defined α2 helix, we investigated the interaction of the residues inside the binding pocket of the latter structure. Interestingly, according to the overlay of the apo WT-CRABPII structure and that of the penta mutant, the carboxylate O atoms of Glu121 occupy a position right between the amino group of Lys132 and the guanidino group of Arg111 (mutated to Leu in the penta mutant). This particular arrangement allows the penta mutant to establish a chain of water-mediated interactions connecting Glu121 and the α2 helix–loop region in the absence of Arg at position 111. This water-mediated network is similar to that between Arg111 and the α2 helix in the apo and holo R132K:Y134F mutant structures. Specifically, there are water-mediated interactions between Glu121 and Val33/Ala36. The paths of the two water networks are shown in Fig. 7 ▶(a) and include (i) carboxylate of Glu121→water 393→water 392 (in two closely located conformations)→water 391→amino group of Lys132→water 83→carbonyl of Ala36 and (ii) carboxylate of Glu121→water 393→water 392→water 391→amino group of Lys132→water 83→water 89→water 230→carbonyl of Val33. The F o − F c OMIT map is shown in Fig. 7 ▶(b). The B factors, occupancies and numbers of hydrogen bonds of these water molecules are listed in Table 3 ▶. Interestingly, five of the six water molecules involved in the network in the apo penta mutant molecule A are highly conserved when compared with those in the apo R132K:Y134F mutant molecule A. Within the five conserved water molecules, the largest distance between the two structures is only 0.64 Å. The only nonconserved water is water 393, which interacts with Glu121. This is predictable since the R111L and L121E mutations completely switch the hydrophilicities of these two residues deep inside the cavity and would require a change of the hydrogen-bonding interactions associated with them.
Figure 7.

(a) The water-mediated interaction between Glu121 and Val33 and Ala36 in apo CRABPII-R132K:Y134F:R111L:L121E:T54V (the distances are in angstroms); (b) F o − F c OMIT map calculated at 2.5σ for the corresponding water molecules.
Similar to the apo CRABPII-R132K:Y134F structure discussed earlier, the water-mediated interaction was not observed in molecule B of this penta mutant. Glu121 interacts with Lys132 through a water molecule; however, Lys132 does not make any interaction with the second helix and loop region.
3.5. The structure of the CRABPII-R132K:Y134F:R111L:L121E:T54V–RA complex
The structure of the R132K:Y134F:R111L:L121E:T54V mutant bound to RA (CRABPII-R132K:Y134F:R111L:T54V:L121E–RA) was determined and refined at 1.60 Å resolution with crystallographic R factors R work = 12.1% and R free = 16.8%. The structure belongs to space group P212121 and has one molecule in the asymmetric unit. The data-collection and refinement statistics are reported in Tables 1 ▶ and 2 ▶, respectively. The overall structure is almost identical to those of WT-CRABPII–RA (RMSD = 0.49 Å) and CRABPII-R132K:Y134F–RA (RMSD = 0.32 Å) (see Fig. 5 ▶ a for a representation of the structure).
As shown in Fig. 8 ▶(a), RA occupies a similar position in the penta mutant as in the wild-type protein. The ionone ring and most of the backbone of the chromophore occupy identical positions; however, the carboxylate group moves toward Glu121, tilting the backbone by 5.9° and moving the carboxylate C atom by 1.3 Å (Fig. 8 ▶ a). This motion is the consequence of different interactions between the protein and ligand in this mutant relative to the wild type. In the latter, RA forms hydrogen bonds to Arg111, Lys132 and Tyr134; however, in the penta mutant these three residues are all mutated to hydrophobic residues.
Figure 8.

(a) RA and the neighboring residues of its carboxylate group in the pocket of the penta mutant (magenta) and WT-CRABPII–RA (yellow) (only the residues of the penta mutant are labeled). (b) The binding pocket of CRABPII-R132K:Y134F:R111L:T54V:L121E–RA. RA and Glu121 make tight dicarboxylate interactions with each other, indicating that both groups are protonated. (c) Water-mediated interactions connect Glu121 to the residues on the α2 helix region. The distances are in angstroms.
Interestingly, the crystal structure of the RA-bound penta mutant showed that RA binds to this protein by forming a tight dicarboxylic acid interaction with Glu121 (Fig. 8 ▶ b). This observation could explain the increased affinity of RA for the penta mutant (K d = 250 ± 19 nM) compared with the tetra mutant R132K:Y134F:R111L:T54V (K d = 900 ± 64 nM). As shown in Fig. 8 ▶(b), the carboxylate group of Glu121 has two conformations (each are 50% occupied). The O atoms of the two carboxylate groups are ∼2.6 Å away from each other at their shortest distances. This tight dicarboxylic acid interaction indicates that both RA and Glu121 must be protonated. This is not surprising because the pocket of the penta mutant is highly hydrophobic. Considering the fact that a hydrophobic environment cannot tolerate charges, it is expected that both carboxylate groups become protonated in this pocket. Previous reports have shown that within a hydrophobic cavity the pK a of a Glu residue can increase by as much as five units (Fitch et al., 2002 ▶). A comprehensive study of the retinoic acid-binding affinities of various CRABPII mutants is ongoing (Vasileiou et al., 2008 ▶).
As shown in Fig. 8 ▶(b), the hydrogen-bond interaction observed between Arg111 and RA is broken upon the R111L mutation. A 50% occupied water molecule forms a hydrogen bond to the carboxylate group of RA. This water cannot be fully occupied because it is positioned too close to one of the conformations of the carboxylate group of Glu121. The 2.1 Å shown in red does not represent the true distance between water 1 and Glu121. Water 1 will be disordered when the carboxylate moves toward it. In this structure, unlike other structures, Lys132 has moved towards RA and Glu121 and makes direct hydrogen bonds with both of them. It may also be neutral owing to deprotonation.
Comparing the structures of the apo mutants of CRABPII with that of the R111M mutant (PDB code 1xca; Chen et al., 1998 ▶), in which Arg111 is mutated to a hydrophobic residue, shows that this mutation destabilizes the structure, particularly at the α2 helix and the loop connecting this helix to βB. However, although in the structure of the RA-bound penta mutant Arg111 is mutated to Leu, the structure is almost identical to that of RA-bound WT-CRABPII. A similar water-mediated interaction is made between the designed Glu121 and two residues at the portal, Ala36 (the first residue on the loop connecting α2 to βB) and Val33 (on α2), as observed in other structures between Arg111 and the two latter residues. The water-mediated interactions are carboxylate of Glu121→carboxylate of RA→amino group of Lys132→water 187→water 62→carbonyl of Ala36 and carboxylate of Glu121→carboxylate of RA→amino group of Lys132→water 187→water 62→water 65→carbonyl of Val33 (Fig. 8 ▶ c). Again, the B factors, occupancies and numbers of hydrogen bonds of these water molecules are listed in Table 3 ▶. Only three waters were identified in this chain of interactions. Two of them, waters 65 and 62, are conserved and are involved in hydrogen bonding to Ala36 and Val 33, respectively. Water 65 is 1.19 Å away from its counterpart in apo WT-CRABPII and water 62 is 1.09 Å away from the terminal N atom of Arg111 in apo WT-CRABPII. Lys132 moves more towards Glu121 in the RA-bound penta mutant compared with the apo penta mutant. This movement is accompanied by the appearance of the nonconserved water 187 to establish the continuous interaction network. The interaction between Glu121 and Lys132 is now mediated by the O atom of the ligand carboxylate group and is no longer mediated by water.
3.6. The structure of apo CRABPII-R132K:R111L:L121E
The structure of the apo R132K:R111L:L121E mutant was determined at 1.50 Å resolution and refined with R factors R work = 15.4% and R free = 22.7%. The RMSDs between molecules A and molecules B of this mutant and apo WT-CRABPII are 1.16 and 0.46 Å, respectively.
A buffer molecule, Bis-Tris propane (BTP), from the crystalization conditions is present inside the binding pocket of molecule A of this apo CRABPII-R132K:R111L:L121E structure (Fig. 9 ▶ a) but not in molecule B. The average B factor for BTP is 31.0 Å2, which is higher than the overall B factor (24.5 Å2). BTP contains a total of six hydroxyl groups, with three at each end of the molecule. These hydroxyl groups make over ten hydrogen-bonding interactions with nearby residues or ordered water molecules inside molecule A of apo CRABPII-R132K:R111L:L121E, creating a network interconnecting Glu73, Glu121 and Lys132 as shown in Fig. 9 ▶(b) which is similar to the water-mediated network observed in the previously discussed structures (apo and holo R132K:Y134F and apo and holo R132K:Y134F:R111L:L121E:T54V mutants). The interactions are as follows: carboxylate of Glu73→O8, O12 hydroxy groups of BTP, then O10 of BTP→carboxylate of Glu121→N20, O19 of BTP→amino group of Lys132→water 13; the network then branches off at water 13: water 13→water 102→OH of Ser37, water 13→water 102→water 18→carbonyls of Ala32 and Ala34 and water 13→water 212→water 18→carbonyls of Ala32 and Ala34. The B factors, occupancies and numbers of hydrogen bonds of these water molecules are listed in Table 3 ▶. The interaction between Glu121 and Lys132 is now mediated by a hydroxy group of BTP. The water molecules here are all involved in mediating the interaction going from Lys132 to the helix portal. However, owing to the partial unwinding of the C-terminus of the α2 helix, these water molecules are not at the conserved positions and constitute a new pattern of hydrogen-bonding interactions in this area.
Figure 9.

(a) 2F o − F c map for BTP calculated at 1σ. (b) The hydrogen-bonding network involving both the BTP and ordered waters linking Glu121 and the α2 helix and the loop connecting this helix to βB in molecule A of apo CRABPII-R132K:R111L:L121E (the distances are in angstroms). (c) Superimposed structures of molecule A of apo CRABPII-R132K:R111L:L121E (purple) and apo WT-CRABPII (yellow).
In this mutant the last helical turn of the α2 helix of molecule A is completely unwound at its C-terminus (residues ∼34–40) and the βC–βD hairpin loop (residues ∼54–61) moves concurrently (Figs. 2 ▶ and 9 ▶ c). The differences between molecules A of this mutant and apo WT-CRABPII become noticeable at the N-terminus of α1 and reach a maximum divergence at Ala36, the first residue on the loop connecting α2 to βB. The secondary-structure calculation of this mutant using DSSP shows that the α2 helix has lost its last helical turn in this structure and unlike in apo WT-CRABPII, where residues 26–35 comprise the α2 helix, here only residues 26–33 form the helix. This unwinding is probably not a consequence of the binding of BTP as the same unwinding was observed in other apo CRABPII mutant structures when no BTP was present in the cavity (data not shown). However, the altered water network within the protein does result in the loss of a water-mediated interaction between Lys132 and the main-chain carbonyl of Ala36. In the wild-type structure there is a direct interaction between Arg132 and the main-chain carbonyl of Ala36, whereas in all of the Lys132-containing structures the water-mediated interaction between Lys132 and Ala36 replaces this interaction. Only in the structure of the triple mutant is the water network altered such that this water molecule is not present, probably leading to further destabilization of the helical conformation of the α2 helix.
Since R132K is a conservative mutation and Leu121 is not a conserved residue, this structure further suggests the importance of Arg111 in defining the structural integrity of CRABPII and in particular the α2 helix. Although α2 and the loop in the apo R132K:R111L:L121E mutant have different conformations compared with WT-CRABPII, they are well defined. The B factors of the atoms in these regions (residues 26–40) are 19.8 Å2 for molecule A and 18.3 Å2 for molecule B, both of which are lower than the average B factor of the whole structure without BTP (19.8 Å2 for molecule A and 24.8 Å2 for molecule B). This observation is contrary to what was observed in the structure of the R111M mutant determined by Chen et al. (1998 ▶). In the R111M mutant, although Arg132 interacts with Ala36 it makes no interaction with the other residues. The helix in this structure is highly destabilized and has a high B factor of ∼75 Å2. As reported by the authors, the electron-density map of the helix region in molecule A was built only with great difficulty, especially for residues 33–37.
In the apo R132K:R111L:L121E mutant (and our other structures discussed here) the electron-density map of these residues was well defined from the beginning of refinement, which indicates that the region is stable despite the R111L mutation. It appears that Glu121 stabilizes the helix in the triple mutant. Therefore, although the R111L mutation led to a different conformation for the helix and loop, they are not destabilized. In other words, in this structure Glu121 mimics the role of Arg111 in apo WT-CRABPII, but through a different water-mediated interaction, which leads to a different conformation in the loop and helix region.
Our attempts to crystallize holo CRABPII-R132K:Y134F:R111L only resulted in apo crystals, most probably owing to the low binding affinity of this mutant for RA [K d(R132K:R111L:L121E) = 1000 ± 28 nM, K d(R132K:Y134F) = 100 ± 7 nM, K d(R132K:Y134F:R111L:L121E:T54V) = 250 ± 19 nM].
4. Conclusion
Together, the mutant CRABPII structures described here elaborate on a theme that is becoming clear for CRABPII. The structural integrity of the protein in its apo unbound form depends on a series of water-mediated interactions that extend from the top of the binding cavity at the α2 helix to the bottom of the binding cavity at the Arg111 residue. This chain of interactions acts as a ‘pillar’ that supports the structure in the absence of ligand. Disruption of this concerted interaction by loss of Arg111 results in the partial collapse of this chain of water-mediated interactions and lower structural integrity of the protein. We have further seen that loss of Arg111 can be partially compensated by the addition of another hydrophilic residue, in this case Glu121, which can then recapitulate the chain of interactions. However, the result is not identical to the wild type and results in yet another conformation of the α2 helix. This rather surprising chain of water-mediated interactions extending over 12 Å may be a necessary consequence of the relatively large internal cavity within the CRABPII structure. Such a large void is not common in protein structures as small as CRABPII (only 16 kDa) and may require such a chain of interactions to preserve the integrity of the structure when the cavity is not filled by ligand.
Supplementary Material
PDB reference: CRABPII, apo R132K:Y134F, 3d96, r3d96sf
PDB reference: apo R132K:Y134F:R111L:L121E:T54V, 3d95, r3d95sf
PDB reference: R132K:Y134F:R111L:L121E:T54V–RA, 3cwk, r3cwksf
PDB reference: apo R132K:R111L:L121E, 2g7b, r2g7bsf
Acknowledgments
We are grateful to the beamline staff at COM-CAT sector 32-ID at the APS, Argonne National Laboratory. Use of the APS was supported by the US Department of Energy, Office of Basic Energy Sciences under contract No. W-31-109-ENG-38. This research was supported by the NIH (GM84563 to JHG and GM82800 to BB).
References
- Bailey, J. S. & Siu, C. H. (1988). J. Biol. Chem. 263, 9326–9332. [PubMed]
- Bastie, J. N., Despouy, G., Balitrand, N., Rochette-Egly, C., Chomienne, C. & Delva, L. (2001). FEBS Lett. 507, 67–73. [DOI] [PubMed]
- Boylan, J. F. & Gudas, L. J. (1991). J. Cell Biol. 112, 965–979. [DOI] [PMC free article] [PubMed]
- Boylan, J. F. & Gudas, L. J. (1992). J. Biol. Chem. 267, 21486–21491. [PubMed]
- Brünger, A. T. (1993). Acta Cryst. D49, 24–36. [DOI] [PubMed]
- Budhu, A., Gillilan, R. & Noy, N. (2001). J. Mol. Biol. 305, 939–949. [DOI] [PubMed]
- Budhu, A. S. & Noy, N. (2002). Mol. Cell. Biol. 22, 2632–2641. [DOI] [PMC free article] [PubMed]
- Chen, X., Tordova, M., Gilliland, G. L., Wang, L. C., Li, Y., Yan, H. G. & Ji, X. H. (1998). J. Mol. Biol. 278, 641–653. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Crist, R. M., Vasileiou, C., Rabago-Smith, M., Geiger, J. H. & Borhan, B. (2006). J. Am. Chem. Soc. 128, 4522–4523. [DOI] [PubMed]
- Delva, L., Bastie, J. N., Rochette-Egly, C., Kraiba, R., Balitrand, N., Despouy, G., Chambon, P. & Chomienne, C. (1999). Mol. Cell. Biol. 19, 7158–7167. [DOI] [PMC free article] [PubMed]
- Dong, D., Ruuska, S. E., Levinthal, D. J. & Noy, N. (1999). J. Biol. Chem. 274, 23695–23698. [DOI] [PubMed]
- Fitch, C. A., Karp, D. A., Lee, K. K., Stites, W. E., Lattman, E. E. & Garcia-Moreno, B. (2002). Biophys. J. 82, 3289–3304. [DOI] [PMC free article] [PubMed]
- Gaub, M. P., Lutz, Y., Ghyselinck, N. B., Scheuer, I., Pfister, V., Chambon, P. & Rochette-Egly, C. (1998). J. Histochem. Cytochem. 46, 1103–1111. [DOI] [PubMed]
- Gorry, P., Lufkin, T., Dierich, A., Rochette-Egly, C., Décimo, D., Dollé, P., Mark, M., Durand, B. & Chambon, P. (1994). Proc. Natl Acad. Sci. USA, 91, 9032–9036. [DOI] [PMC free article] [PubMed]
- Jing, Y. K., Waxman, S. & Mira-y-Lopez, R. (1997). Cancer. Res. 57, 1668–1672. [PubMed]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed]
- Kleywegt, G. J., Bergfors, T., Senn, H., Lemotte, P., Gsell, B., Shudo, K. & Jones, T. A. (1994). Structure, 2, 1241–1258. [DOI] [PubMed]
- Maden, M. (1994). Vitamin A in Health and Disease, edited by R. Blomhoff, pp. 289–322. New York: Marcel Dekker.
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Napoli, J. L. (1996). FASEB J. 10, 993–1001. [DOI] [PubMed]
- Napoli, J. L. (1999). Biochim. Biophys. Acta, 1440, 139–162. [DOI] [PubMed]
- Ong, D. E. & Chytil, F. (1975). J. Biol. Chem. 250, 6113–6117. [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Roussel, A. & Cambillau, C. (1989). Silicon Graphics Geometry Partners Directory. Mountain View: Silicon Graphics.
- Sani, B. P. & Hill, D. L. (1974). Biochem. Biophys. Res. Commun. 61, 1276–1282. [DOI] [PubMed]
- Scapin, G., Gordon, J. I. & Sacchettini, J. C. (1992). J. Biol. Chem. 267, 4253–4269. [DOI] [PubMed]
- Thompson, J. R., Bratt, J. M. & Banaszak, L. J. (1995). J. Mol. Biol. 252, 433–446. [DOI] [PubMed]
- Vaezeslami, S., Mathes, E., Vasileiou, C., Borhan, B. & Geiger, J. H. (2006). J. Mol. Biol. 363, 687–701. [DOI] [PubMed]
- Vasileiou, C., Vaezeslami, S., Crist, R. M., Rabago-Smith, M., Geiger, J. H. & Borhan, B. (2007). J. Am. Chem. Soc. 129, 6140–6148. [DOI] [PubMed]
- Vasileiou, C., Vaezeslami, S., Lee, K. S. S., Crist, R. M., Goins, S. M., Geiger, J. H. & Borhan, B. (2008). In the press.
- Wang, L. C., Li, Y. & Yan, H. G. (1997). J. Biol. Chem. 272, 1541–1547. [DOI] [PubMed]
- Wang, L. C. & Yan, H. G. (1998). Biochemistry, 37, 13021–13032. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CRABPII, apo R132K:Y134F, 3d96, r3d96sf
PDB reference: apo R132K:Y134F:R111L:L121E:T54V, 3d95, r3d95sf
PDB reference: R132K:Y134F:R111L:L121E:T54V–RA, 3cwk, r3cwksf
PDB reference: apo R132K:R111L:L121E, 2g7b, r2g7bsf