

The structure of the core domain of the arginine repressor protein from M. tuberculosis has been determined with (1.85 Å resolution) and without (2.15 Å resolution) the arginine corepressor bound. Three additional arginine molecules have been found to bind to the core domain hexamer at high (0.2 M) arginine concentration.
Keywords: Mycobacterium tuberculosis, arginine repressor proteins, oligomerization domain, core hexamer, arginine-binding sites
Abstract
The Mycobacterium tuberculosis (Mtb) gene product encoded by open reading frame Rv1657 is an arginine repressor (ArgR). All genes involved in the l-arginine (hereafter arginine) biosynthetic pathway are essential for optimal growth of the Mtb pathogen, thus making MtbArgR a potential target for drug design. The C-terminal domains of arginine repressors (CArgR) participate in oligomerization and arginine binding. Several crystal forms of CArgR from Mtb (MtbCArgR) have been obtained. The X-ray crystal structures of MtbCArgR were determined at 1.85 Å resolution with bound arginine and at 2.15 Å resolution in the unliganded form. These structures show that six molecules of MtbCArgR are arranged into a hexamer having approximate 32 point symmetry that is formed from two trimers. The trimers rotate relative to each other by about 11° upon binding arginine. All residues in MtbCArgR deemed to be important for hexamer formation and for arginine binding have been identified from the experimentally determined structures presented. The hexamer contains six regular sites in which the arginine molecules have one common binding mode and three sites in which the arginine molecules have two overlapping binding modes. The latter sites only bind the ligand at high (200 mM) arginine concentrations.
1. Introduction
More than 1.7 billion individuals worldwide are infected with the Mycobacterium tuberculosis (Mtb) pathogen (Raviglione, 2003 ▶) and approximately two million lives are lost to TB annually (http://www.who.int/mediacentre/factsheets/fs104/en/). TB is becoming more difficult to combat owing to the appearance of drug-resistant strains (Coker, 2004 ▶). In order to understand the physiology of this bacillus on a molecular level, the M. tuberculosis Structural Genomics Consortium (TBSGC) was founded with the goal of determining the three-dimensional structures and functions of the proteins from the TB genome (Terwilliger et al., 2003 ▶). Our laboratory has adopted the strategy of targeting the proteins from several Mtb metabolic pathways, in particular the proteins involved in arginine biosynthesis. Recently, the structures of the enzymes encoded by open reading frames Rv1652 (ArgC) and Rv1656 (ArgF) that are involved in this pathway have been determined (Cherney et al., 2007 ▶; Sankaranarayanan et al., 2008 ▶). Rv1657 has been annotated as encoding an arginine repressor (ArgR) that regulates the transcription of genes of the arginine-biosynthetic pathway. The ArgR monomer is composed of two domains: the N-terminal domain is responsible for DNA binding and the C-terminal or core domain is responsible for oligomerization and arginine binding (Tian & Maas, 1994 ▶). In the presence of elevated arginine concentrations, ArgR binds to DNA operators within the promoter regions of the arginine-biosynthetic and catabolism genes (Larsen et al., 2005 ▶; Caldara et al., 2006 ▶), thereby shutting down arginine metabolism. All of the genes involved in the arginine-biosynthetic pathway are essential for optimal growth of the Mtb pathogen (Sassetti et al., 2003 ▶), thus making MtbArgR a potential target for antibiotic drug design.
To date, full structures of the apo arginine repressor from Bacillus stearothermophilus (Bst) and the apo arginine repressor/activator (AhrC) from B. subtilis (Bs) have been reported (Ni et al., 1999 ▶; Dennis et al., 2002 ▶). Structures have also been determined of isolated C-terminal and N-terminal domains of apo ArgR from Escherichia coli (Ec; Van Duyne et al., 1996 ▶; Sunnerhagen et al., 1997 ▶) and of isolated N-terminal domains of BsAhrC that are not bound to DNA (Garnett et al., 2007a ▶) and of the same domains bound to an 18 bp DNA-operator site (Garnett et al., 2008 ▶). In the case of arginine-bound proteins, only the structures of their C-terminal domains (EcCArgR, BstCArgR and BsCAhrC) have been determined (Van Duyne et al., 1996 ▶; Ni et al., 1999 ▶; Garnett et al., 2007b ▶). It was found that these proteins consist of six monomers and form a dimer of trimers with approximate 32 symmetry. The C-terminal domains of the monomers make up a compact core of the hexamers (the core hexamer). The N-terminal domains lie around the core hexamer in loosely associated pairs. The trimers rotate about each other upon arginine binding, which presumably allows the correct matching between the N-terminal domains and the DNA operators. The arginine complexes of EcCArgR, BstCArgR and BsCAhrC have six ligand molecules bound to the core hexamer.
Here, we report several crystal structures of MtbCArgR in the apo form as well as with six and nine arginine molecules bound to the core hexamer. Based on these results, we have been able to study the arginine-binding sites in MtbArgR and to analyze the structural effects resulting from arginine binding.
2. Materials and methods
2.1. Protein preparation, crystallization and data collection
Rv1657 was purified as a GST-fusion protein and then cleaved by restricted proteolysis with TEV proteinase to produce the targeted protein. However, the protein was likely to have undergone additional cleavage by endogenous proteases and the resulting preparation had one half of the expected molecular weight, as described in detail elsewhere (Lu et al., 2007 ▶). The likely cleavage site is the peptide bond following Arg88 of the linker between the N- and C-terminal domains. The C-terminal domain (starting approximately at residues 89–92 and ending at residue 170) had crystallized. Three different crystal forms of MtbCArgR have been grown. Crystallization of the apo form I (Table 1 ▶) from 20% PEG 10 000, 0.1 M HEPES buffer pH 7.5 has been described previously (Lu et al., 2007 ▶). This form belonged to the triclinic space group P1 with six protein subunits in the unit cell (a crystallographic hexamer). Arginine-bound forms were grown under the following crystallization conditions. Equal volumes (1–2 µl) of protein solution (at 10 mg ml−1 in 5 mM Tris–HCl pH 7.4 and 100 mM NaCl) and the crystallization buffer were combined and equilibrated against 1 ml crystallization buffer by vapor diffusion in hanging drops. Crystal form II (Table 1 ▶) resulted from 20% PEG 10 000, 0.1 M Tris–HCl pH 8.5, 0.1 M guanidine–HCl and 5 mM arginine. Crystal form III (Table 1 ▶) grew from slightly different conditions: 12% PEG 10 000, 0.1 M HEPES buffer pH 7.0, 10% glycerol and 0.2 M arginine. Forms II and III of arginine-bound MtbCArgR belonged to the orthorhombic space group P212121 with a hexamer in the asymmetric unit. The crystals chosen for data collection were rinsed in a cryoprotectant containing 30% glycerol in mother liquor and flash-cooled in liquid nitrogen. Data sets were collected either on beamline 7-1 at the Stanford Synchrotron Radiation Laboratory (SSRL) or on beamline 8.3.1 at the Advanced Light Source (ALS) at the Lawrence Berkeley National Laboratory (Table 1 ▶). Raw intensity data were reduced, merged and scaled using the HKL-2000 suite (Otwinowski & Minor, 1997 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the high-resolution shell.
| Crystal | I | II | III |
|---|---|---|---|
| Ligands per hexamer | 0 | 6 | 9 |
| PDB code | 3bue | 2zfz | 3cag |
| Space group | P1 | P212121 | P212121 |
| Unit-cell parameters | |||
| a () | 53.22 | 57.67 | 57.76 |
| b () | 57.24 | 75.95 | 75.65 |
| c () | 57.33 | 107.10 | 108.02 |
| () | 66.2 | 90.0 | 90.0 |
| () | 62.2 | 90.0 | 90.0 |
| () | 82.0 | 90.0 | 90.0 |
| Z | 6 | 24 | 24 |
| Solvent content (%) | 57.8 | 49.2 | 49.5 |
| Data collection | |||
| Temperature (K) | 100 | 100 | 100 |
| Detector | ADSC Q315 | MAR Mosaic 325 | MAR Mosaic 325 |
| Wavelength () | 0.97848 | 0.97947 | 1.10552 |
| Resolution () | 50.002.15 (2.232.15) | 50.001.85 (1.921.85) | 50.001.90 (1.971.90) |
| No. of unique reflections | 28194 (2498) | 41086 (4025) | 36125 (3141) |
| Multiplicity | 1.8 (1.7) | 4.8 (4.5) | 6.9 (6.8) |
| I/(I) | 11.8 (2.3) | 17.8 (2.0) | 15.7 (4.2) |
| Completeness (%) | 99.8 (99.7) | 99.8 (99.2) | 95.6 (84.6) |
| R merge † | 0.061 (0.252) | 0.094 (0.800) | 0.132 (0.422) |
| Refinement | |||
| Refinement resolution | 50.02.15 | 50.01.85 | 50.01.90 |
| R working/R free ‡ | 0.17/0.23 | 0.18/0.23 | 0.17/0.22 |
| No. of atoms | |||
| Protein | 3400 | 3402 | 3466 |
| Water | 361 | 407 | 357 |
| L-Arginine in regular sites | 72 | 72 | |
| L-Arginine in additional sites | 36 | ||
| Guanidine | 8 | ||
| Average B factors (2) | |||
| Protein | 24.6 | 21.0 | 22.25 |
| Water | 35.5 | 33.5 | 35.6 |
| L-Arginine in regular sites | 13.8 | 15.6 | |
| L-Arginine in additional sites | 36.9 | ||
| Guanidine | 31.3 | ||
| R.m.s. deviations from ideality | |||
| Bond lengths () | 0.020 | 0.015 | 0.015 |
| Bond angles () | 1.82 | 1.57 | 1.48 |
| Ramachandran plot | |||
| Highly favored (%) | 94.6 | 93.3 | 94.8 |
| Allowed (%) | 5.1 | 6.7 | 5.2 |
| Generously allowed (%) | 0.3 | 0.0 | 0.0 |
| Disallowed (%) | 0.0 | 0.0 | 0.0 |
R
merge = 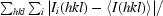
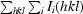 , where I
i(hkl) is the ith intensity measurement and I(hkl) is the mean of all measurements of I(hkl).
, where I
i(hkl) is the ith intensity measurement and I(hkl) is the mean of all measurements of I(hkl).
R
working and R
free = 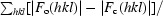
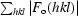 for reflections in the working and test sets. R
free was calculated using 5% of data.
for reflections in the working and test sets. R
free was calculated using 5% of data.
2.2. Structure determination, refinement and analysis
Structure solution and refinement were carried out using programs from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The crystal structure of MtbCArgR was initially solved in the orthorhombic crystal form II by molecular replacement using MOLREP (Vagin & Teplyakov, 1997 ▶). An alanine model was constructed from the trimer of C-terminal domains of BstArgR (PDB code 1b4b; 39% identity) in which all non-identical residues were replaced by alanine residues while identical residues were not changed. This trimer was used as the search model for molecular replacement using data in the resolution range 20–4 Å. Once the correct solution had been achieved, further building of the polypeptide chains was accomplished using ARP/wARP (Perrakis et al., 1999 ▶); structure solutions for the other crystal forms were carried out using MOLREP. Manual fitting of side chains was performed using XtalView/Xfit (McRee, 1999 ▶) and the structures were all refined using REFMAC 5.0 (Murshudov et al., 1997 ▶). The final structures were evaluated using PROCHECK (Laskowski et al., 1993 ▶). Table 1 ▶ summarizes the crystal parameters, data-collection and processing and refinement statistics. The programs ClustalW (Thompson et al., 1994 ▶) and ALIGN (Cohen, 1997 ▶) were used for sequence alignments and structural comparisons, respectively. Quaternary structure analyses were carried out with the EMBL–EBI PISA server (Krissinel & Henrick, 2005 ▶, 2007 ▶). Figures were generated using PyMOL (DeLano, 2002 ▶).
3. Results and discussion
3.1. Overall structure
Well defined electron densities were observed for all hexamer subunits in both the triclinic and orthorhombic crystal forms. The overall structures of the holo MtbCArgR hexamer and the superposed apo MtbCArgR hexamer are shown in Fig. 1 ▶(a). Both hexamers are made up of two trimers and exhibit noncrystallographic 32 symmetry. The bottom trimers were used to perform the superposition while the top trimers retained their relative positions. Overall, the structures of all the trimers are very similar, with a maximum r.m.s.d. of 0.61 Å for 900 main-chain atom pairs between trimers from the arginine-free and arginine-bound hexamers. In the arginine-free hexamer each subunit from the top trimer is located directly above the adjacent subunit from the bottom trimer. However, in the arginine-bound hexamer the top trimer is rotated with respect to the bottom trimer by 11° about the threefold axis relating the subunits in the hexamer (clockwise when viewed from the top) compared with the arginine-free hexamer (Figs. 1 ▶ a and 1 ▶ b). This rotation is less than the equivalent rotation of 15° observed for other arginine repressors (Ni et al., 1999 ▶; Garnett et al., 2007b ▶).
Figure 1.

Overall structure of MtbCArgR. All subunits are shown as ribbon drawings and colored separately. (a) Superposition of apo (darker shade) and holo (lighter shade) hexamers viewed along a dyad symmetry axis. Only subunits D, E and F were used for structural alignment. The holo hexamer has six arginine molecules bound (two of them in this view are labeled RA and RD); these molecules are depicted by sticks with the C atoms in the color of the nearest subunit and with N and O atoms in blue and red, respectively. (b) Superposition as in (a) except viewed along the threefold symmetry axis from the top trimers with the bottom trimers removed for clarity. (c) The subunit of MtbCArgR has α/β topology (αββαββα).
All subunits within the hexamers have well conserved structures, with a maximum r.m.s.d. of 0.3 Å for approximately 300 main-chain atom pairs. Each subunit of the C-terminal domain of the MtbArgR polypeptide chain has an α/β topology in which the three α-helices alternate with pairs of strands in a four-stranded β-sheet (αββαββα; Fig. 1 ▶ c). The β-sheets from the three different subunits associate around the threefold axis to form the trimer. On the other hand, only small regions of helices α1 and α2 are present at the interface between the trimers in the hexamer. As a result, much weaker interactions are expected between trimers than between the individual subunits of each trimer. Indeed, there are 1301 and 1320 Å2 of solvent-accessible surface area buried in the interfaces between subunits in the top and bottom apo trimers, respectively, and 1131 Å2 in the interface between trimers in the hexamer. The EMBL–EBI PISA server gives a change in solvation energy of −45.6 and −46.0 kJ mol−1 upon formation of the interfaces between the subunits in the top and bottom apo trimers, respectively, but only −30.1 kJ mol−1 upon their association into a hexamer. There are also significantly higher total numbers of hydrogen bonds (HB) and salt bridges (SB) at the interfaces between subunits in each apo trimer (21 HB and ten SB for the top trimer and 20 HB and 12 SB for the bottom trimer) than at the interface between trimers (eight HB and nine SB). Thus, the MtbCArgR hexamer is more correctly described as a dimer of trimers than as a hexamer. We shall discuss the interactions between subunits in more detail in §3.4.
3.2. Regular arginine-binding sites
There are a total of six clefts located between the subunits in the top and bottom trimers; these clefts extend roughly parallel to the threefold axis of the dimer of trimers (Fig. 1 ▶ a). Well defined electron density for the arginine ligands is observed in all the clefts. Arginine molecules lying in two adjacent clefts on the top and bottom trimers have their side chains facing each other according to the 32 symmetry of the hexamer. It is worth noting that each arginine-binding site binds the ligand molecule in a unique conformation. Such a binding mode is similar to the pattern observed in the known structures of other arginine repressors (Van Duyne et al., 1996 ▶; Ni et al., 1999 ▶; Garnett et al., 2007b ▶); we will refer to these binding sites as the ‘regular’ binding sites. In this way, at moderate arginine concentrations the MtbCArgR hexamer binds up to six arginine molecules, one in each binding cleft.
Fig. 2 ▶(a) shows the well ordered binding site for arginine molecule RA located in the cleft between subunits A and C (the other regular binding sites look similar). All ligand atoms fit well in the OMIT electron-density map at the 3.0σ contour level (Fig. 2 ▶ a), in agreement with the low values of their atomic B factors. The α-carboxyl O atoms of molecule RA form three hydrogen bonds to the main-chain N atoms of residues Asp146 and Asp147 of the C subunit and to Ala144 of the A subunit (Fig. 2 ▶ a). The α-amino group of RA also forms three hydrogen bonds: a hydrogen bond to a side-chain O atom of Asp132 of the A subunit, a bifurcated hydrogen bond with the main-chain and side-chain O atoms of Thr142 of the A subunit and a bifurcated hydrogen bond with the side-chain O atoms of Asp147 and Thr148 of the C subunit. The other O atom of the Asp132 side chain and the water molecule W bound in the cleft (Fig. 2 ▶ a) accept protons from the side-chain O atoms of Thr142 and Thr148, allowing the bifurcated hydrogen bonds to form. Two N atoms Nη1 and Nη2 of the guanidinium group of the bound arginine make hydrogen bonds and salt bridges to the side-chain O atoms of Asp146 of the F subunit. Finally, there is a hydrogen bond between one of these two arginine N atoms and the unprotonated Nδ1 atom of His125 of the A subunit. An electrostatic surface-potential calculation showed strong negative charge in the binding clefts. The surface of the two adjacent clefts with bound arginine ligands RA and RD is presented in Fig. 2 ▶(b).
Figure 2.

(a) The regular binding site is located in the cleft between subunits A and C; ligand molecule RA is bound to this site. Also shown is the ligand molecule RD bound in the adjacent cleft. The OMIT |F o| − |F c| electron-density map for RA and RD is contoured at the 3.0σ level with the final refined models of RA and RD superposed. Binding residues are represented by sticks with C atoms in the color of the subunit to which the residue belongs. Hydrogen bonds are drawn as black broken lines. The two hydrogen bonds involving the α-amino group of the bound arginine are bifurcated. In their formation, residue Asp132 also plays the role of proton acceptor for Thr142 as does the water molecule W for Thr148. The guanidinum group of each ligand forms an ion pair with the β-carboxyl group of aspartic acid residue Asp146 that belongs to the opposite trimer. (b) Electrostatic surface representation of the two adjacent binding clefts with arginine ligands RA and RD. (c) Superposition of the arginine-binding sites of MtbCArgR (green), EcArgR (magenta), BstArgR (yellow) and BsAhrC (orange). Residues involved in arginine binding are shown as sticks and the MtbArgR numbering is used; also shown are the two bound arginine molecules RA and RD.
There are also hydrophobic contacts between the ligand molecule RA and residues Gly145 of the C subunit and Ala128, Ser129 and Thr142 of the A subunit. However, these residues do not form a deep binding cavity. Superposition of the MtbCArgR hexamer structure with the core-hexamer structures of other arginine repressors (EcArgR, BstArgR and BsAhrC) shows that their known arginine-binding sites are similar to the regular binding sites in MtbArgR (Fig. 2 ▶ c). The binding residues described above are fully conserved, with the exceptions of His125 and Ala144, which are substituted by a glutamine (in EcArgR and BsAhrC) and a cysteine (in BstArgR and BsAhrC), respectively.
3.3. Additional arginine-binding sites
We have found that the MtbCArgR hexamer can bind three additional arginine molecules at 200 mM arginine concentration. Each molecule is distributed between two mutually exclusive binding modes in the gap between trimers. Fig. 3 ▶ shows all nine ligand molecules as viewed along the threefold axis from the top trimer and Fig. 4 ▶(a) demonstrates a closing of the gap with the additional ligands labeled RAE, RBD and RCF. These ligand molecules at 50% occupancy for each binding mode are well defined in the OMIT electron-density map contoured at the 2.0σ level (Fig. 4 ▶ a). The average B factors of the atoms of additional arginine molecules are slightly higher (35–40 Å2) than those of the atoms of the rest of the protein molecules and the six regular arginine molecules (Table 1 ▶). Figs. 3 ▶ and Fig. 4 ▶(a) show that binding of three ligand molecules in the gap between trimers occurs in accordance with the approximate 32 symmetry of the hexamer. In particular, the three twofold axes relate the positions of the two possible mutually exclusive modes of the ligand molecules to each other.
Figure 3.

The MtbCArgR hexamer is bound with six arginine molecules in the regular sites; three disordered arginine molecules are bound in the gap between the trimers. The view is along the threefold-symmetry axis from the top trimer, perpendicular to the view in Fig. 1 ▶(a). The three additional arginine molecules are labeled RAE, RBD and RCF. Each of them adopts two binding positions each at half occupancy. The OMIT |F o| − |F c| electron-density map for the additional ligand molecules is contoured at the 2.0σ level with their final refined models superposed.
Figure 4.

(a) A close-up of the gap between the trimers of the MtbCArgR hexamer bound with the three additional arginine molecules viewed approximately as in Fig. 3 ▶. The molecule RCF stretches between subunits C and F. The molecules RAE and RBD are stretched between subunits A and E and between subunits B and D, respectively. (b) The binding site for the ligand molecule RAE. Binding residues are represented by sticks. Residue His125 adopts two positions that differ in the orientation of the imidazole group. This group must be located far from the ligand if the latter adopts the position with the α-amino and α-carboxyl groups located near the corresponding His125. The imidazole group of the His125 in the other subunit can adopt both positions in this case. Hydrogen bonds are shown as black broken lines. There are also ion pairs between the ligand guanidinum group and the β-carboxyl groups of the aspartic acid residues Asp146.
Fig. 4 ▶(b) shows the additional binding site for the arginine molecule RAE stretched between subunits A and E. The binding sites for RBD and RCF look similar. The two β-carboxyl groups of the neighboring Asp146 residues that play important roles in binding the regular arginine molecules are also largely responsible for binding one additional ligand. The binding is primarily by hydrogen bonding and electrostatic interactions with the guanidinium group of the ligand. As a result, the rest of the ligand could be placed in one of the opposite directions with equal probability. There are three pairs of neighboring β-carboxyl groups of Asp146 in the hexamer, resulting in the binding of three additional arginine molecules, each of which is distributed into two possible binding modes. When the α-amino and α-carboxyl groups of an additional arginine molecule are positioned close to His125 of one subunit, the guanidinium group of the ligand causes His125 of the other subunit to move closer to it to form a hydrogen bond. As a result, the imidazole group of each His125 adopts two conformations.
The α-amino group of arginine RAE in the position shown in green (Fig. 4 ▶ b) forms hydrogen bonds to the main-chain O atom of Gly145 and to the Nδ1 atom of His125 (subunit A). In order for the latter hydrogen bond to form, the imidazole ring of His125 must occupy a position away from the arginine. The α-carboxylate O atom of the bound RAE receives a hydrogen bond from the main-chain N atom of His125. In addition, the α-carboxyl group of arginine is located near to the N-terminus of helix α2 on subunit A. Thus, there is a stabilizing electrostatic interaction from the helix α2 dipole moment and the negative charge of the α-carboxylate of the bound arginine.
The ligand RAE in this position also interacts with residues His125 and Asp146 of subunit E. The N atoms N∊ and Nη2 of the guanidinum group of RAE donate hydrogen bonds (and ion-pair interactions) to the side-chain Oδ2 of Asp146 and to the N∊2 atom of His125 in the position shown in Fig. 4(b) ▶. Thus, interactions that involve the guanidinum group of the bound arginine are likely to play the most important role in binding. The 2|F o| − |F c| electron-density map for the guanidinum group of the additional arginine molecules is better defined than the regions involving the other atoms of the arginine molecules. The same interactions discussed above are also present for the twofold-related RAE molecule (Fig. 4b ▶, cyan) with subunits E and A, respectively. It is worth noting that the residues of the additional binding sites in MtbArgR are conserved in EcArgR, BstArgR and BsAhrC. After superposition of their structures with the MtbCArgR structure, the corresponding residues are well aligned.
The binding of three additional arginine molecules at extreme arginine concentrations shows the capacity of MtbCArgR to bind arginines. The regular arginines bind in the clefts between subunits and the additional arginines bind in the gap between the trimers. The physiological meaning of this finding is not clear.
3.4. Structural effects of arginine binding
The C atoms of residues Leu104, Gly122, Ala123 and Tyr126 participate in hydrophobic contacts at the interface between the trimers. There are also hydrogen bonds and ion pairs at this interface, most of which are relatively weak (Table 2 ▶). Rotation of the trimers with respect to each other upon arginine binding does not change the residues that are responsible for the hydrophobic contacts between the trimers; however, it does change the hydrogen bonds and the ion pairs that occur between them. In the holo hexamer, six new hydrogen bonds are formed between the O and N atoms of residues Gly122 and Tyr126, respectively (one residue from each subunit of one trimer and the other residue from the adjacent subunit of the other trimer). At the same time, most hydrogen bonds and ion pairs (between residues Arg99 and Glu103) present at the interface between the trimers in the apo hexamer are lost as a result of the conformational change in the holo hexamer (Table 2 ▶). The trimers can also interact through hydrogen bonds and salt bridges involving regular and additional ligand molecules (Figs. 2a ▶ and 4b ▶). The major conformational change in each subunit upon binding the three additional arginines occurs in the His125 residues: their imidazole rings adopt two alternative positions in order to accommodate the additional ligands and make hydrogen-bonding interactions with them.
Table 2. Hydrogen-bonding interactions at the interface between trimers in MtbCArgR hexamers.
Residues in italics also form ion pairs between the guanidinum and carboxyl groups of their side chains.
| Apo form (3bue) | Distance () | Holo form with six ligands (2zfz) | Distance () | Holo form with nine ligands (3cag) | Distance () | |||
|---|---|---|---|---|---|---|---|---|
| Subunit A | Subunit F | Subunit A | Subunit F | Subunit A | Subunit F | |||
| Arg99 N1 | Glu103 O1 | 3.3 | Arg99 N1 | Glu103 O1 | 3.6 | Gly122 O | Tyr126 N | 3.5 |
| Glu103 O2 | Arg99 N2 | 3.7 | Glu103 O1 | Tyr126 O | 2.5 | Tyr126 N | Gly122 O | 3.5 |
| His125 N2 | Asp146 O1 | 3.2 | Gly122 O | Tyr126N | 3.5 | |||
| Tyr126 N | Gly122 O | 3.7 | ||||||
| Subunit B | Subunit E | Subunit B | Subunit E | Subunit B | Subunit E | |||
| Glu103 O1 | Arg99 N2 | 3.8 | Arg99 N1 | Glu103 O2 | 3.1 | Gly122 O | Tyr126 N | 3.6 |
| Arg133 N2 | Pro121 O | 3.0 | Arg99 N2 | Glu103 O1 | 3.1 | Tyr126 N | Gly122 O | 3.4 |
| Gly122 O | Tyr126 N | 3.3 | ||||||
| Tyr126 N | Gly122 O | 3.5 | ||||||
| Tyr126 O | Glu103 O1 | 2.4 | ||||||
| Subunit C | Subunit D | Subunit C | Subunit D | Subunit C | Subunit D | |||
| Arg99 N1 | Glu103 O1 | 3.8 | Gly122 O | Tyr126 N | 3.6 | Glu103 O2 | Arg99 N2 | 3.2 |
| Glu103 O2 | Arg99 N1 | 3.7 | Tyr126 N | Gly122 O | 3.5 | Gly122 O | Tyr126 N | 3.5 |
| Glu103 O2 | Arg99 N2 | 3.8 | Tyr126 N | Gly122 O | 3.4 | |||
Here, we have shown that MtbCArgR forms a dimer of trimers that is similar to the other known structures of arginine repressor proteins. Also, the trimers rotate relative to each other in a similar fashion by 11° upon arginine binding. The interactions between the guanidinium groups of the bound arginines with the β-carboxyl groups of Asp146 of the opposite trimer are thought to be the cause of this rotation. The rotation of the two C-terminal trimers about one another enables the two neighboring N-terminal domains to rearrange their positions so that they can properly fit into two major grooves of the DNA operator and as a result block arginine biosynthesis (Ni et al., 1999 ▶; Garnett et al., 2008 ▶).
It has been shown that l-canavanine, which is an arginine mimic, can also interact with ArgR; however, it is not an effective corepressor (Schwartz & Maas, 1960 ▶; Van Duyne et al., 1996 ▶). The binding site of l-canavanine on the C-terminal domain of ArgR is unknown. The oxygen substitution for the Cδ in l-canavanine dramatically decreases the pK of the guanidinium group from 12 in arginine to 6–7 in l-canavanine. Finding an arginine-mimicking compound that is a strong corepressor could produce a new type of drug against TB.
Supplementary Material
PDB reference: C-terminal domain of arginine repressor protein, 3bue, r3buesf
PDB reference: arginine complexes, 2zfz, r2zfzsf
PDB reference: 3cag, r3cagsf
Acknowledgments
X-ray diffraction data were collected on beamline 8.3.1 of the Advanced Light Source (ALS) at Lawrence Berkeley Laboratory under an agreement with the Alberta Synchrotron Institute (ASI). The ALS is operated by the Department of Energy and supported by the National Institutes of Health. Beamline 8.3.1 was funded by the National Science Foundation, the University of California and Henry Wheeler. The ASI synchrotron-access program is supported by grants from the Alberta Science and Research Authority (ASRA), the Alberta Heritage Foundation for Medical Research (AHFMR) and Western Economic Diversification of the Canadian Government. MNGJ is supported by the Canadian Institutes of Health Research and the Alberta Heritage Foundation for Medical Research and is a Canada Institutes of Health Research (CIHR) Canada Research Chair.
References
- Caldara, M., Charlier, D. & Cunin, R. (2006). Microbiology, 152, 3343–3354. [DOI] [PubMed]
- Cherney, L. T., Cherney, M. M., Garen, C. R., Niu, C., Moradian, F. & James, M. N. G. (2007). J. Mol. Biol. 367, 1357–1369. [DOI] [PubMed]
- Cohen, G. E. (1997). J. Appl. Cryst. 30, 1160–1161.
- Coker, R. J. (2004). Trop. Med. Int. Health, 9, 25–40. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, Palo Alto, USA.
- Dennis, C. A., Glykos, N. M., Parsons, M. R. & Phillips, S. E. V. (2002). Acta Cryst. D58, 421–430. [DOI] [PubMed]
- Garnett, J. A., Baumberg, S., Stockley, P. G. & Phillips, S. E. V. (2007a). Acta Cryst. F63, 914–917. [DOI] [PMC free article] [PubMed]
- Garnett, J. A., Baumberg, S., Stockley, P. G. & Phillips, S. E. V. (2007b). Acta Cryst. F63, 918–921. [DOI] [PMC free article] [PubMed]
- Garnett, J. A., Marincs, F., Baumberg, S., Stockley, P. G. & Phillips, S. E. V. (2008). J. Mol. Biol. 379, 284–298. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2005). CompLife 2005, edited by M. R. Berthold, R. Glen, K. Diederichs, O. Kohlbacher & I. Fischer, pp. 163–174. Berlin/Heidelberg: Springer-Verlag.
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Larsen, R., Kok, J. & Kuipers, O. P. (2005). J. Biol. Chem. 280, 19319–19330. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lu, G. J., Garen, C. R., Cherney, M. M., Cherney, L. T., Lee, C. & James, M. N. G. (2007). Acta Cryst. F63, 936–939. [DOI] [PMC free article] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Ni, J., Sakanyan, V., Charlier, D., Glansdorff, N. & Van Duyne, G. D. (1999). Nature Struct. Biol. 6, 427–432. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Perrakis, A., Morris, R. J. & Lamzin, V. S. (1999). Nature Struct. Biol. 6, 458–463. [DOI] [PubMed]
- Raviglione, M. C. (2003). Tuberculosis (Edinb.), 83, 4–14. [DOI] [PubMed]
- Sankaranarayanan, R., Cherney, M. M., Cherney, L. T., Garen, C. R., Moradian, F. & James, M. N. G. (2008). J. Mol. Biol. 375, 1052–1063. [DOI] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Mol. Microbiol. 48, 77–84. [DOI] [PubMed]
- Schwartz, J. H. & Maas, W. K. (1960). J. Bacteriol. 79, 794–799. [DOI] [PMC free article] [PubMed]
- Sunnerhagen, M., Nilges, M., Otting, G. & Carey, J. (1997). Nature Struct. Biol. 4, 819–826. [DOI] [PubMed]
- Terwilliger, T. C. et al. (2003). Tuberculosis (Edinb.), 83, 223–249. [DOI] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Tian, G. & Maas, W. K. (1994). Mol. Microbiol. 13, 599–608. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst. 30, 1022–1025.
- Van Duyne, G. D., Ghosh, G., Maas, W. K. & Sigler, P. B. (1996). J. Mol. Biol. 256, 377–391. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: C-terminal domain of arginine repressor protein, 3bue, r3buesf
PDB reference: arginine complexes, 2zfz, r2zfzsf
PDB reference: 3cag, r3cagsf