

Crystal structures of M. tuberculosis folylpolyglutamate synthetase complexed with two nucleotides have been determined at 2.0 and 2.3 Å resolution, revealing an active-site loop movement and associated changes that influence substrate binding.
Keywords: Mycobacterium tuberculosis, folate metabolism, nucleotide binding, conformational change, cobalt binding
Abstract
Folate derivatives are essential vitamins for cell growth and replication, primarily because of their central role in reactions of one-carbon metabolism. Folates require polyglutamation to be efficiently retained within the cell and folate-dependent enzymes have a higher affinity for the polyglutamylated forms of this cofactor. Polyglutamylation is dependent on the enzyme folylpolyglutamate synthetase (FPGS), which catalyzes the sequential addition of several glutamates to folate. FPGS is essential for the growth and survival of important bacterial species, including Mycobacterium tuberculosis, and is a potential drug target. Here, the crystal structures of M. tuberculosis FPGS in complex with ADP and AMPPCP are reported at 2.0 and 2.3 Å resolution, respectively. The structures reveal a deeply buried nucleotide-binding site, as in the Escherichia coli and Lactobacillus casei FPGS structures, and a long extended groove for the binding of folate substrates. Differences from the E. coli and L. casei FPGS structures are seen in the binding of a key divalent cation, the carbamylation state of an essential lysine side chain and the adoption of an ‘open’ position by the active-site β5–α6 loop. These changes point to coordinated events that are associated with dihydropteroate/folate binding and the catalysis of the new amide bond with an incoming glutamate residue.
1. Introduction
Folic acid and its derivatives are essential vitamins that are required for cell growth and replication in both prokaryotic and eukaryotic cells. Folic acid serves as a carrier of single-carbon units, typically in the form of methyl and formyl substituents at the N-5 and N-10 positions or as bridging N-5/N-10 methylene groups (Fig. 1 ▶). These folate derivatives are essential for a number of diverse biochemical pathways, including methionine metabolism, the biosynthesis of purine nucleotides and thymidylate synthesis. Because most of the reactions involving folate substrates result in the transfer of single-carbon units, the reactions are collectively known as one-carbon metabolism (Shane, 1989 ▶). Folate coenzymes are of particular importance during pregnancy or infancy and folate deficiency can manifest itself in several ways including placenta-mediated diseases (Ray & Laskin, 1999 ▶), neural tube defects in newborns (Pitkin, 2007 ▶), hyperhomocysteinaemia (Bailey, 1995 ▶; Durand et al., 1998 ▶) and the development of some cancers (Kim, 1999 ▶).
Figure 1.

Schematic representation of the reactions catalyzed by (a) dihydrofolate synthetase (DHFS) and (b) folylpolyglutamate synthetase (FPGS). In both reactions the dihydropteroate or folate substrates have the same co-substrates (ATP and l-glutamate) and the reaction mechanisms are identical, involving (1) activation of the free carboxylate by ATP and (2) nucleophilic attack on the resulting acyl-phosphate intermediate by l-glutamate. The different forms of the one-carbon derivatives of tetrahydrofolate are shown on the lower molecule: (i) 10-methyl, (ii) 5,10-methylene and (iii) 10-formyl.
Folates within the cell are generally found as polyglutamate derivatives. Since the presence of the long negatively charged tail effectively prevents efflux through the cell membrane, it was initially thought that polyglutamylation was simply a way of storing folate and raising the cellular concentration (Shane, 1989 ▶). However, it has since been shown that many folate-dependent enzymes have a higher affinity for its polyglutamylated forms (Lu et al., 1984 ▶; Lowe et al., 1993 ▶; Schirch & Strong, 1989 ▶). Folate antagonists (so-called antifolates), which have been used in cancer chemotherapy and as antibiotics, are also polyglutamylated, which can greatly enhance their ability to inhibit certain enzymes (Synold et al., 1996 ▶).
In organisms that synthesize folates de novo (e.g. plants, bacteria, fungi and protozoa), two enzymes catalyze the addition of glutamic acid residues: dihydrofolate synthetase (DHFS) adds the first glutamic acid residue to dihyropteroate (DHP) to produce dihydrofolate (DHF), after which the ubiquitous enzyme folylpolyglutamate synthetase (FPGS; EC 6.3.2.17) catalyzes folate polyglutamylation. This occurs in all cells, including those that require exogenous folate (e.g. mammalian cells). In some bacteria, DHFS and FPGS activities reside on a single gene (e.g. Escherichia coli and Corynebacterium), whereas in plants and fungi it appears that distinct genes encode the two enzymes (Ravanel et al., 2001 ▶). Both enzymes use a stepwise reaction in which glutamate residues are added by the ATP-dependent formation of an activated acyl-phosphate intermediate, followed by nucleophilic attack on the acyl phosphate by the incoming glutamate molecule to produce the new amide bond (Fig. 1 ▶). The number of glutamate residues added by FPGS appears to be species-dependent; E. coli for example favors only three residues (Osborne et al., 1993 ▶), whereas Lactobacillus casei can add up to nine residues (Toy & Bognar, 1994 ▶).
Crystal structures of FPGS are available from three bacterial species: L. casei (Sun et al., 1998 ▶, 2001 ▶; Sheng et al., 2002 ▶; Smith et al., 2006 ▶), Thermotoga maritima (unpublished work; PDB code 1o5z) and the bifunctional E. coli DHFS/FPGS (Mathieu et al., 2005 ▶). In each case the enzyme comprises two domains: an N-terminal ATPase domain and a C-terminal Rossmann-fold domain, with the active site at the interface between the two domains. The nucleotide-binding site appears to be highly conserved in each case, but different sites approximately 5 Å apart have been found to bind pterin substrates in the L. casei and E. coli enzymes (Sun et al., 2001 ▶; Mathieu et al., 2005 ▶). It has been suggested that the site observed in E. coli DHFS/FPGS may be used to add the first one or two glutamate residues and the second site, observed in L. casei FPGS, may be used to add subsequent residues as the tail elongates (Tan & Carlson, 2005 ▶; Smith et al., 2006 ▶). How the tail is moved through the active site so that the terminal γ-carboxylate is always presented to the ATP and where this growing tail is accommodated in the enzyme structure are yet to be determined, but it is clear that protein dynamics must play a significant part.
As the production of polyglutamylated folates in vivo is of vital importance to all living systems, this emerging evidence of a distinction between dihydropteroate (DHP) and tetrahydrofolate (THF) binding by bacterial FPGS suggests that it may be possible to design inhibitors of bacterial DHFS activity that do not inhibit FPGS activity in humans, thereby selectively inhibiting folate metabolism in bacteria. FPGS in Mycobacterium tuberculosis (MtFPGS) has been shown to be an essential gene for the growth of M. tuberculosis (Sassetti et al., 2003 ▶) and like E. coli FPGS is thought to exhibit both DHFS and FPGS activities. This raises the possibility that the development of inhibitors of MtFPGS could also provide potential leads for chemotherapy against tuberculosis (TB).
Here, we report the structures of M. tuberculosis FPGS in complex with ADP and AMPPCP at resolutions of 2.0 and 2.3 Å, respectively, and demonstrate differences in loop closure, divalent cation binding and the carbamylation of an active-site lysine residue that relate to substrate binding and the reaction mechanism.
2. Experimental procedures
2.1. Cloning, expression and protein purification
The cloning, expression and purification of MtFPGS were carried out as described previously (Young et al., 2006 ▶). Briefly, the open reading frame encoding MtFPGS (Rv2445) was PCR-amplified from M. tuberculosis genomic DNA and cloned into a modified pET42a plasmid (Novagen), pET42a-rTEV, to produce the expression plasmid GST-His6-MtFPGS. Recombinant MtFPGS protein was expressed in BL21 (λDE3) pGROELS chaperone strain cells which were ‘cold-shocked’ before induction with IPTG at 293 K for 16 h. Selenomethionine-labelled MtFPGS (SeMet-MtFPGS) was produced using a modified protocol based on inhibition of methionine biosynthesis, as previously described (Young et al., 2006 ▶).
Bacteria expressing MtFPGS were lysed and recombinant protein was purified from the clarified soluble fraction by IMAC as described previously (Young et al., 2006 ▶). Eluted fractions containing the bulk of the recombinant protein were pooled and dialyzed with a 1:20 ratio of His6-tagged rTEV protease at 277 K for 16 h to cleave the affinity tag from MtFPGS. MtFPGS was separated from the rTEV-His6 protease and cleaved GST-His6 affinity tag by passage through a nickel-chelating column and then further purified by size-exclusion chromatography. SeMet-MtFPGS was purified using the same procedures as used for native MtFPGS.
2.2. Crystallization, data collection and structure solution
MtFPGS was crystallized by a batch method as described previously (Young et al., 2006 ▶). Protein pre-incubated with either 1.5 mM ADP or AMPPCP and 2 mM MgCl2 was mixed with an equal volume of precipitant solution comprising 14%(w/v) PEG 8000, 30%(v/v) MPD, 10 mM CoCl2 and 50 mM sodium acetate pH 5.5 and crystals were grown under paraffin oil. Crystals appeared after 16 h and grew to a maximum size of 100 µm after 96 h. Before flash-freezing in liquid nitrogen, the crystals were soaked for 60 min in a 60:40 mix of cryoprotectant [crystallization buffer + 30%(v/v) glycerol] and protein buffer. Crystals of SeMet-MtFPGS, pre-incubated with 1.5 mM ADP and 2 mM MgCl2, were grown using the same batch method as used for native protein.
Multiwavelength anomalous dispersion (MAD) diffraction data were collected from a single crystal of SeMet-MtFPGS–ADP on beamline BL9-1 at the SSRL. A total of 55 images were collected with an oscillation range of 1° per image, 20 s exposure per image and a crystal-to-detector distance of 240 mm. Diffraction data from native MtFPGS–AMPPCP crystals were collected on BL9-2 at the SSRL. A total of 60 images were collected with an exposure time of 2 s and an oscillation range of 1° per image.
All data were indexed and integrated with MOSFLM (Leslie, 2006 ▶) and reduced with SCALA (Evans, 2006 ▶) from the CCP4 suite (Potterton et al., 2003 ▶; Collaborative Computational Project, Number 4, 1994 ▶). Some data-collection statistics are presented in Table 1 ▶. The crystals proved to be cubic, space group P213, with unit-cell parameters a = b = c = 112.4 Å and one MtFPGS molecule in the asymmetric unit, with an estimated solvent content of 46% (Matthews, 1968 ▶). The structure of MtFPGS–ADP was determined by SAD using autoSHARP (Vonrhein et al., 2006 ▶; de La Fortelle & Bricogne, 1997 ▶) with data collected at the peak of the Se absorption edge (0.979 Å). Eight of the nine possible Se sites were located with SHELXD (Sheldrick, 2008 ▶). Phase refinement was performed with SHARP (de La Fortelle & Bricogne, 1997 ▶; Bricogne et al., 2003 ▶) and the phases were further improved by solvent flattening using SOLOMON and DM (Abrahams & Leslie, 1996 ▶). Automatic tracing using ARP/wARP (Perrakis et al., 2001 ▶) was used to partially build the model and the remainder of the model was built and refined with the programs Coot (Emsley & Cowtan, 2004 ▶) and REFMAC, with the final steps using TLS refinement (Murshudov et al., 1997 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell: 2.11–2.0 Å for MtFPGS–ADP and 2.42–2.3 Å for MtFPGS–AMPPCP.
| MtFPGS–ADP | MtFPGS–AMPPCP | |
|---|---|---|
| Maximum resolution (dmin) (Å) | 2.0 | 2.3 |
| Observed reflections | 221039 | 150909 |
| Unique reflections to dmin | 32271 | 21320 |
| Rmerge† (%) | 8.6 (63.0) | 9.5 (65.7) |
| I/σ(I) | 14.3 (2.8) | 14.1 (2.9) |
| Completeness (%) | 100 (100) | 100 (100) |
| Multiplicity | 6.8 (6.8) | 7.1 (7.3) |
| Unit-cell parameters (Å) | a = b = c = 112.4 | a = b = c = 112.4 |
R
merge = 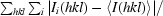
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the mean intensity.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the mean intensity.
The structure of the AMPPCP complex of MtFPGS was solved by molecular replacement with Phaser (McCoy et al., 2005 ▶) using the MtFPGS–ADP structure as the search model. The structure was refined and built with Coot and REFMAC, using TLS refinement in the final steps. Refinement statistics for both structures are presented in Table 2 ▶. The quality of each model was inspected using the program PROCHECK (Laskowski et al., 1993 ▶). All figures were generated using PyMOL (DeLano, 2002 ▶).
Table 2. Structure-refinement statistics.
| MtFPGS–ADP | MtFPGS–AMPPCP | |
|---|---|---|
| Resolution range (Å) | 39.0–2.0 | 50.3–2.3 |
| Rwork/Rfree† (%) | 15.7/19.0 | 17.6/22.3 |
| Total atoms, protein | 3241 | 3215 |
| Total atoms, solvent | 168 | 122 |
| B factors, protein | 42.9 | 41.0 |
| B factors, solvent | 42.2 | 40.7 |
| R.m.s. deviation from ideality | ||
| Bonds (Å) | 0.020 | 0.023 |
| Angles (°) | 1.84 | 2.06 |
| Ramachandran plot (nonglycine residues) | ||
| Residues in most favored regions (%) | 97 | 97 |
| Residues in disallowed regions (%) | — | — |
R = 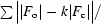
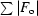 × 100. R
free was calculated with 5% of the reflections.
× 100. R
free was calculated with 5% of the reflections.
3. Results
3.1. Three-dimensional structure of MtFPGS
MtFPGS is folded into two distinct domains, an N-terminal ATPase domain and a C-terminal Rossmann-fold domain, which are joined by a flexible linker (Fig. 2 ▶). The N-terminal domain (residues 1–334) is folded around a central seven-stranded twisted β-sheet with six parallel strands and one antiparallel strand. This β-sheet is flanked by six α-helices on one side and three α-helices on the other. A small three-stranded antiparallel β-sheet packs against the back of this domain and forms an expanded open barrel with strands from the central β-sheet. The C-terminal domain (residues 342–489) comprises a central six-stranded twisted β-sheet, with five parallel strands and a single antiparallel strand, bounded by five α-helices.
Figure 2.

Ribbon diagram of MtFPGS in stereoview. The N-domain is shown in grey and the C-domain is shown in light cyan. The P-loop is shown in red, the β5–α6 loop in dark blue and the interdomain-connecting peptide in orange. Dashed lines represent sections of the polypeptide chain lacking electron density, including the disordered DHP-binding loop (α1–α2) and several other mobile loops. The small three-stranded β-sheet is shown in teal and the bound ADP is shown in magenta in stick mode. Co atoms are shown as dark blue spheres, with Co1 and Co2 showing the location of the cobalt ions involved in crystal contacts.
The structures of the ADP and AMPPCP complexes of MtFPGS superimpose very closely on each other, with a root-mean-square difference (r.m.s.d.) of 0.35 Å for 381 Cα positions. Both structures contain similar disordered regions which are not included in the model. These include the first 23 residues and residues from four mobile loops: the α1–α2 loop (residues 40–46 missing), the α4–α5 loop (residues 144–148 missing), the α6–β6 loop (residue 227 missing) and the α12–β16 loop (residues 458–464 missing) (Fig. 2 ▶). Of these loops, only the α1–α2 loop appears to be functionally important, having been shown to be involved in pteroate binding in E. coli FPGS (Mathieu et al., 2005 ▶).
The relative orientations of the two domains of MtFPGS correspond to the ‘activated’ conformation observed for the folate complexes of E. coli FPGS (EcFPGS; PDB code 1w78; Mathieu et al., 2005 ▶) and L. casei FPGS (LcFPGS; PDB code 1jbw; Sun et al., 2001 ▶). MtFPGS shares 27.5% and 31.5% sequence identity, respectively, with these enzymes and superposition with SSM (Krissinel & Henrick, 2004 ▶) shows that the complete MtFPGS molecule can be superimposed onto their respective folate complexes with r.m.s.d.s of 1.55 Å for 381 Cα positions (EcFPGS) and 1.67 Å for 378 Cα positions (LcFPGS). Corresponding values when the individual domains of MtFPGS were superimposed on the E. coli and L. casei proteins were 1.60 Å (255 Cα) and 1.56 Å (276 Cα), respectively, for the N-terminal domains and 1.27 Å (120 Cα) and 1.88 Å (105 Cα), respectively, for the C-terminal domains.
Cobalt chloride was essential for the crystallization of MtFPGS, and four bound Co2+ ions have been identified on the basis of their density and associated anomalous signal. Two of these cobalt ions are involved in crystal contacts. One sits on the crystallographic threefold axis, where it is coordinated by two ligands (a water molecule and Asp326 Oδ1) from each of the three symmetry-related molecules (Fig. 3 ▶ a). The other cobalt ion forms a bridge between the side chains of Asp455 and Asp457 on helix α12 of the C-terminal domain and is also coordinated by His370 from helix α10 of the C-terminal domain of an adjacent molecule and by three water molecules (Fig. 3 ▶ b). This appears to anchor the end of helix α12, which is significantly longer in MtFPGS (17 residues) than in either EcFPGS or LcFPGS, in which it is only ten residues in length. The loop following this anchor point is larger than in either E. coli or L. casei FPGS and is disordered in MtFPGS. The other two cobalt ions are associated with residues in the active site and will be discussed in more detail later.
Figure 3.

Coordination of cobalt ions involved in protein crystal contacts. (a) Co1, which is situated on the crystal threefold axis and mediates contacts with Asp326 in the N-terminal domains of three adjacent FPGS molecules. (b) Co2, which is coordinated between the C-terminal domains of two adjacent protein molecules. Cobalt ions (Co) are represented as dark blue spheres and water molecules are represented as red spheres.
3.2. Nucleotide binding
Crystals of MtFPGS only grew in the presence of the nucleotides ADP, AMPPNP or AMPPCP. Extensive efforts were made to crystallize either apoprotein or folate complexes, but to date these have been unsuccessful. In both the ADP and AMPPCP structures the contacts between protein and nucleotide are essentially the same (Table 3 ▶). The nucleotide occupies a narrow channel between the N- and C-terminal domains (Fig. 4 ▶), where it adopts the same ‘activated’ conformation as observed in EcFPGS (Mathieu et al., 2005 ▶). The adenine ring sits in a deep pocket formed by the glycine-rich P-loop (Gly73–Gly76), strand β5 and helix α8, where it stacks between the P-loop and His299 and is anchored by hydrogen bonding to Asn303, which is conserved in all FPGS enzymes. The ribose moiety hydrogen bonds to a conserved aspartate residue (Asp353) from strand β12. In the ADP structure, Asn75 from the P-loop is closer to the ribose moiety than in the AMPPCP complex and the E. coli or L. casei structures and forms an additional hydrogen bond to one of the ribose O atoms.
Table 3. Hydrogen-bond contacts between MtFPGS and bound nucleotides.
Water-mediated contacts are excluded.
| MtFPGS–ADP | MtFPGS–AMPPCP | |||
|---|---|---|---|---|
| Atom | Distance (Å) | Atom | Distance (Å) | |
| Adenine-ring moiety | ||||
| N6 | Asn303 Oδ1 | 2.95 | Asn303 Oδ1 | 3.10 |
| N7 | Asn303 Nδ2 | 2.97 | Asn303 Nδ2 | 3.01 |
| Ribose moiety | ||||
| O2 | Asp353 Oδ1 | 2.85 | Asp353 Oδ2 | 2.88 |
| O3 | Asp353 Oδ1 | 2.90 | Asp353 Oδ1 | 2.78 |
| Asp353 Oδ2 | 2.94 | Asp353 Oδ2 | 3.20 | |
| O4 | Asn75 Nδ2 | 3.16 | ||
| α-Phosphate | ||||
| O1A | Arg340 N∊ | 2.84 | Arg340 N∊ | 2.69 |
| Arg340 Nη2 | 2.80 | Arg340 Nη2 | 3.07 | |
| O2A | Ser79 N | 2.99 | Ser79 N | 3.27 |
| Ser79 Oγ | 2.78 | Ser79 Oγ | 2.78 | |
| O3A | Gly76 N | 2.94 | Gly76 N | 2.90 |
| β-Phosphate | ||||
| O1B | Thr78 N | 2.71 | Thr78 N | 2.82 |
| Thr78 Oγ1 | 3.03 | Thr78 Oγ1 | 3.30 | |
| Co2 | 2.02 | Co2 | 2.13 | |
| O2B | Lys77 N | 2.80 | Lys77 N | 3.02 |
| Lys77 Nζ | 2.79 | Lys77 Nζ | 2.64 | |
| γ-Phosphate | ||||
| O1G | His356 N∊2 | 3.07 | ||
| O2G | Arg340 Nη2 | 3.17 | ||
| Thr78 Oγ1 | 3.14 | |||
| Co2 | 1.97 | |||
Figure 4.

Stereoview showing the contacts made between AMPPCP and MtFPGS. AMPPCP is shown in stick mode, coloured by atom type. The active-site cobalt ion is represented as a dark blue sphere and water as a red sphere. Broken lines represent contacts that have the appropriate geometry (distances and angles) and partners to represent hydrogen bonds.
The α-phosphate of the nucleotide (both ADP and AMPPCP) sits at the N-terminus of helix α3, in contact with the main-chain N atoms of Gly76 and Ser79, the side-chain O atom of Ser79 and the side chain of Arg340 from the linker region between the N- and C-terminal domains. The β-phosphate is adjacent to the P-loop and is anchored by hydrogen bonds to the peptide NH groups of Lys77 and Thr78, Lys77 Nζ and Thr78 Oγ2 (Fig. 4 ▶). The γ-phosphate group of the AMPPCP points into the inter-domain cleft towards the external solvent and is hydrogen bonded to the imidazole N atom of His356 from the loop between β12 and α10, Arg340 Nη1, Thr78 Oγ2 and a water molecule.
3.3. Divalent-cation interactions
In both the EcFPGS and LcFPGS structures two magnesium ions are found in the active site, where they are believed to play important functional roles. The first is the ‘classical’ magnesium ion, which is associated with the β- and γ-phosphates of ATP and which is common to the FPGS enzymes and many related ATPases. In MtFPGS this is displaced by a cobalt ion which has a similar, although not identical, coordination. In the AMPPCP structure this cobalt ion is octahedrally coordinated to two O atoms from the β- and γ-phosphate groups, the carbonyl O atom of Ser100 from the Ω-loop and the O∊1 atom of Glu176 from strand β4 (these four atoms forming a plane), with a water molecule and Thr78 Oγ2 in the axial positions (Fig. 4 ▶). In the ADP complex two waters substitute for γ-phosphate O atoms, with one of them replacing the γ-phosphate O atom in the cobalt coordination sphere.
In most FPGS sequences, the residue equivalent to Thr78 is a glycine (Smith et al., 2006 ▶) and, as shown in the E. coli and L. casei structures, this metal-coordination site is occupied by a water molecule. MtFPGS appears to be representative of a subgroup comprising members of the order Actinomycetales, which includes the Corynebacteriaeae (Corynebacteria and Mycobacteria) and Streptomycineae suborders and whose FPGS enzymes have a threonine at this position and a consensus P-loop sequence NGKTS. In this regard, the FPGS enzymes from these bacterial species have a P-loop that is more similar to purine nucleotide triphosphatases (NTPases) such as ras P21, the G proteins and other ATP-dependent amide-bond ligases such as the mur family of bacterial cell-wall ligases. All of these enzymes have a hydroxyl residue (serine or threonine) at this position, the side chain of which forms part of the classical magnesium ion-binding site (Smith & Rayment, 1996 ▶; Smith, 2006 ▶).
The second Mg atom (Mg2) found in other FPGS structures is bound to a conserved histidine in the active site, His173 in EcFPGS or His170 in LcFPGS (His203 in MtFPGS), and is linked via two water ligands to a conserved carbamylated lysine (Lys218 in MtFPGS), as discussed later. Mg2 is rather variably positioned in different FPGS complexes, but an equivalent magnesium ion is also found in some of the mur ligases. It is believed that this magnesium may be important for counterbalancing the negative charge of the γ-phosphate and the incoming carboxylate group and further stabilizing the acyl-phosphate intermediate (Sun et al., 2001 ▶).
In both the ADP and AMPPCP complexes of MtFPGS a cobalt ion is bound to the equivalent histidine, His203, as well as Asp205 and two waters (Fig. 5 ▶ a). There is a major difference in comparison with the other FPGS structures, however, in that in MtFPGS the β5–α6 loop (residues 197–210), to which His203 belongs, undergoes a conformational change that flips His203 out of the active site. This movement, together with the changed carbamylation status of Lys218, appears to be part of a coordinated series of changes that regulate activity.
Figure 5.

Ribbon diagrams showing bonding-stabilizing interactions involving loop β5–α6 (residues 197–210) in its ‘open’ conformation. (a) A Co2+ ion (Co4, shown as a dark blue sphere) is bound by His203 and Asp205; residues 201–204 also participate in a hydrogen-bond network with residues 357 and 386–387 from the C-terminal domain. (b) Hydrogen-bond interactions between glycerol and residues lining the cavity created by the movement of loop β5–α6 in MtFPGS. Water molecules are shown as red spheres.
3.4. Carbamylation of Lys218
A common feature of both the EcFPGS and LcFPGS structures and of the majority of the mur ligase superfamily is the presence of a carbamylated lysine (Lys188 in EcFPGS, Lys185 in LcFPGS). In most structures it is a second-shell ligand of the second magnesium ion, Mg2, hydrogen-bonded to two of the waters in the coordination sphere of Mg2. In the MtFPGS structures, however, the equivalent residue, Lys218, is not carbamylated. This is also true of both T. maritima FPGS (PDB code 1o5z) and the apo structures of LcFPGS (1fgs, 2gca). Analysis of the crystallization conditions of all FPGS and mur ligase enzymes shows a correlation between the pH of crystallization and the presence of a carbamylated lysine. In all structures crystallized at pH 7 and above, with the singular exception of MurF (1gg4), the equivalent lysine is carbamylated, whereas in those structures crystallized below pH 7 it remains unmodified.
Similar pH-dependent carbamylation of lysine has been previously documented with the class D OXA-10 β-lactamases, which depend on a carbamylated lysine for the hydrolysis of β-lactam moieties. Crystal structures of this enzyme show a clear correlation between the pH of the crystallization buffer and the degree of carbamylation, with an increase in carbamylation of specific lysine residues from pH 6.0 through to full carbamylation of these residues at pH 8.5 (Golemi et al., 2001 ▶). The carbamylation of lysine in FPGS enzymes may explain the nonspecific activation of FPGS by sodium bicarbonate (Bolanowska et al., 1990 ▶) and also the effect of pH on the activity of FPGS; little activity is observed below pH 7.5 (Shane, 1980 ▶).
3.5. Loop movement in MtFPGS
A significant difference between the two MtFPGS structures and the structures of other FPGS enzymes is found in the loop connecting strand β5 to helix α6 (residues 197–210). This loop is a key part of the active site, as it includes the histidine that binds Mg2 (His203 in MtFPGS) and an aromatic residue (Trp176 in EcFPGS) which helps form the pocket that binds the pterin ring of folate in EcFPGS. It also mediates important contacts between the C-terminal domain and the pterin-binding pocket in the N-terminal domain.
In the majority of FPGS structures, the β5–α6 loop forms a single-turn helix (α5b in EcFPGS) and packs tightly against the N-terminal domain. However, in MtFPGS this loop has moved away from the N-terminal domain and adopts an ‘open’ conformation stabilized by hydrogen-bonding interactions with the loop between strand β13 and helix α11, involving residues Val204–Asp386, His203–Asp386, Asp202–Lys387 and Ile201–Asn357 (Fig. 5 ▶ a). In both T. maritima FPGS (1o5z) and the L. casei MgATP complex (1fgs) the β5–α6 loop is disordered, which suggests that it may be moving between an ‘open’ state, represented by the MtFPGS structures, and a ‘closed’ state characteristic of the EcFPGS structures and the folate-bound LcFPGS structure (Fig. 6 ▶). In MtFPGS a glycerol molecule occupies the cavity left open by the movement of the β5–α6 loop and is hydrogen-bonded to Lys218 Nζ, Thr74 Oγ2, the carbonyl O atom of His203, the amide N atoms of Asp202 and His203 and a water molecule (Fig. 5 ▶ b).
Figure 6.

The DHP-binding loop and its environment. (a) Electron density (from an F o − F c OMIT map contoured at 1.5σ) for the DHP-binding loop of MtFPGS (residues 36–50). No interpretable density is found between Arg39 and Asp46. The positions of these latter residues suggest that the loop has a different conformation from that of EcFPGS, which is shown as a green ribbon. (b) Stereoview showing changes in the DHP-binding loop and the β5–α6 loop in the MtFPGS (teal) and EcFPGS (green) structures. Residues Arg39 and Asp46, which flank the disordered part of the DHP-binding loop in MtFPGS, are circled. The E. coli DHP-binding loop, in contrast, is ordered and is closed over the substrate. DHP binding in EcFPGS leads to the ‘closing’ of the DHP-binding loop from an ‘open’ position similar to that seen in MtFPGS. The ‘closed’ position is stabilized by hydrophobic interactions involving Ile28 and Leu30 from the DHP-binding loop and Trp176 from the β5–α6 loop. In MtFPGS, the open β5–α6 loop causes Tyr206, corresponding to Trp176 in EcFPGS, to move 11 Å away. Other accompanying changes in MtFPGS compared with EcFPGS are the rotation of His203 away from the Mg2-binding site, the loss of this magnesium ion and the noncarbamylation of Lys218. In the EcFPGS structure, phosphorylated DHP (DHPP), Mg2 (green sphere), its water ligands (red spheres) and the carbamylated Lys188, which forms part of the second-shell coordination of Mg2, can all be observed. ADP is situated below the DHPP and is identically positioned in MtFPGS and EcFPGS.
4. Discussion
Polyglutamylation of folate substrates by FPGS is believed to follow a ter-ter sequential mechanism in which MgATP binds first, folate second and glutamate last (Shane, 1980 ▶). The structure of MtFPGS is entirely consistent with this mechanism and, together with the previously determined structures of EcFPGS (Mathieu et al., 2005 ▶) and LcFPGS (Sun et al., 1998 ▶, 2001 ▶) and modelling studies (Tan & Carlson, 2005 ▶), points to some of the conformational changes along the reaction pathway.
The ADP and AMPPCP complexes of MtFPGS show that these structures represent the ‘activated’ conformation in which the enzyme is prepared for folate binding; the adenine and ribose rings are fully buried, with only the triphosphate moiety exposed to the incoming folate. At least two different folate-binding sites have been observed in FPGS: that identified in EcFPGS, which is believed to be associated with DHFS activity (Mathieu et al., 2005 ▶), and a second observed in LcFPGS, which may be used as the polyglutamate tail lengthens (Sun et al., 2001 ▶; Tan & Carlson, 2005 ▶). These clearly map to a long surface groove in MtFPGS (Fig. 7 ▶). While both sites appear to be fully accessible, we were not able to prepare folate complexes of MtFPGS either by cocrystallization or by soaking. This suggests that binding of substrate to MtFPGS requires a conformational change which cannot be accommodated in the current crystal form or that the crystal pH (5.5) is not favourable for binding or both. FPGS is inactive at the crystal pH, having little activity below pH 7.5 (Shane, 1980 ▶), but all attempts to grow crystals above pH 5.5 or soak crystals at higher pH were unsuccessful. While the β5–α6 loop does make crystal contacts in the MtFPGS structure, which may restrain its movement, we further suggest that the noncarbamylation of Lys218 at the crystal pH may also contribute to the failure to prepare stable folate complexes, as discussed below.
Figure 7.

Surface representation of MtFPGS (a) and EcFPGS (b), showing the DHP-binding site, with DHPP from the EcFPGS structure modelled into the MtFPGS structure. The N- and C-terminal domains of MtFPGS are coloured teal and light teal, respectively, and those of EcFPGS green and light green, respectively. The β5–α6 loop is coloured red and moves from an ‘open’ state as in MtFPGS to a closed state as in EcFPGS, where it stabilizes the closed conformation of the DHP-binding loop (blue), forming a deep narrow pocket around DHPP. Without the stabilizing interaction of a closed β5–α6 loop, the DHP-binding loop in MtFPGS is mobile and a disordered region joins the two ends (shown in blue). An open cavity is exposed, allowing ready binding or dissociation of DHP or DHF. The nucleotide is almost fully buried in each protein, with only its β-phosphate seen, pointing into the inter-domain cleft towards the phosphate group on DHPP. To the right of the DHPP a long groove (indicated by a curved solid black line) extends around to the back of the protein. This is thought to be the cleft associated with successive additions of glutamate residues and contains 5,10-methylene-tetrahydrofolate in the LcFPGS structure.
Folate binding in EcFPGS is associated with the closure of a loop (residues 25–32) over the pteridine ring to generate a narrow pocket (Mathieu et al., 2005 ▶). This loop is referred to as the dihydropteroate (DHP) binding loop. Deletion of this loop in an N-terminal deletion mutant of EcFPGS severely reduces but does not completely abolish activity (Kimlova et al., 1991 ▶), indicating its functional importance. In the MtFPGS structures, the equivalent loop (residues 42–49) is in an open conformation and is partially disordered (Fig. 6 ▶ a). The disordered region includes residues Ile45 and Pro47, which correspond to E. coli residues Ile28 and Leu30, which stack against the pterin ring. In E. coli these hydrophobic residues are stabilized by their interactions with Trp176 on helix α5b (Mathieu et al., 2005 ▶). In MtFPGS the corresponding stabilizing residue (Tyr206) has moved 11 Å away as the single-turn helix α5b unwinds in the ‘open’ form of the β5–α6 loop (Fig. 6 ▶ b).
The accepted mechanism for the formation of a new amide bond in the DHFS and FPGS enzymes begins with the ATP-dependent activation of the free carboxylate group on DHP or the folate substrate. Since this requires the close approach of the negatively charged γ-phosphate and carboxylate groups, a divalent cation is necessary to counter some of the negative charge and thus facilitate acyl-phosphate formation (Sun et al., 2001 ▶). This cation, typically magnesium (Mg2), is found in both the FPGS enzymes and the related mur ligases (Smith, 2006 ▶), albeit with some variation in its binding site. The mobility of this metal ion may be essential to its role in stabilizing the charges in the active site.
The MtFPGS structures presented here suggest that the highly mobile β5–α6 loop may provide a mechanistic switch linking the coordination of Mg2 with DHP binding through stabilization of the DHP-binding loop. In EcFPGS the β5–α6 loop (which contains the α5b helix) is involved in Mg2 coordination through His173 and with stabilization of the DHP-binding loop through Trp176. This is also likely to be the case in MtFPGS, as EcFPGS and MtFPGS have both been shown to have FPGS and DHFS activity (A. Bognar, personal communication). However, in MtFPGS the ‘open’ conformation of the β5–α6 loop results in both residues (His203 and Tyr206) being flipped away.
The carbamylation of the conserved lysine (Lys218 in MtFPGS) also appears to be essential for the stabilization of Mg2 in FPGS enzymes and mur ligases. When there is no carbamylation of this lysine, no Mg2 is associated with the histidine (His203 in MtFPGS) and the β5–α6 loop is either disordered or has substantially shifted. We propose that there is a synergistic relationship between lysine carbamylation, Mg2 binding and the movement of the β5–α6 loop and thence the DHP-binding loop which helps regulate the DHFS activity of these enzymes.
A possible mechanistic sequence for DHFS activity in MtFPGS would see ATP binding first, with the concomitant binding of Mg1 and Mg2 and the closure of the β5–α6 loop from an ‘open’ state as seen in MtFPGS to a ‘closed activated’ state as seen in EcFPGS. This in turn stabilizes the DHP loop through hydrophobic stacking interactions mediated by Tyr206 and facilitates stable binding of the DHP substrate. There is evidence from mutagenesis and binding studies that this site, formed by closure of the DHP loop, accommodates both DHP and monoglutamylated folate derivatives (Sheng et al., 2008 ▶) and thus supports the DHFS reaction and the first FPGS glutamylation. Diglutamylated and polyglutamylated derivatives, in contrast, are likely to use the extended cleft shown in Fig. 7 ▶, as in the LcFPGS–folate structure, thus explaining how the enzyme accommodates the growing substrate during successive additions of glutamate residues. The number of residues added by the FPGS of a particular organism may then depend on the length of this cleft.
The structural results presented here on the MtFPGS enzyme have allowed some insight into the mode by which this enzyme could be activated and the means by which substrate binding and the presence of the product are communicated throughout the active site. It also adds to the growing understanding of how the growing polyglutamate tail is accommodated by the enzyme. Finally, the structural and molecular features presented by FPGS suggest considerable potential for drug design. Many potent inhibitors are available that target ATP-binding sites, for example in kinases, and have appropriate pharmacokinetic properties for drug development. These could provide leads directed at the highly conserved nucleotide-binding site in FPGS, with further selectivity and affinity being added by the elaboration of such compounds to exploit the nearby DHP pocket and surface groove on FPGS.
Supplementary Material
PDB reference: MtFPGS–ADP, 2vos, r2vossf
PDB reference: MtFPGS–AMPPCP, 2vor, r2vorsf
Acknowledgments
This work was supported by a program grant from the New Zealand Health Research Council (ENB, PM and CAS). It is based in part upon research conducted at the Stanford Synchrotron Radiation Laboratory (SSRL), which is funded by the Department of Energy (BES, BER) and the National Institutes of Health (NCRR, NIGMS).
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed]
- Bailey, L. B. (1995). Folate in Health and Disease. New York: Marcel Dekker.
- Bolanowska, W. E., Russell, C. A. & McGuire, J. J. (1990). Arch. Biochem. Biophys.281, 198–203. [DOI] [PubMed]
- Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M. & Paciorek, W. (2003). Acta Cryst. D59, 2023–2030. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Durand, P., Prost, M. & Blache, D. (1998). Clin. Chem. Lab. Med.36, 419–429. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Golemi, D., Maveyraud, L., Vakulenko, S., Samama, J. P. & Mobashery, S. (2001). Proc. Natl Acad. Sci. USA, 98, 14280–14285. [DOI] [PMC free article] [PubMed]
- Kim, Y.-I. (1999). J. Nutr. Biochem.10, 66–88. [DOI] [PubMed]
- Kimlova, L. J., Pyne, C., Keshavjee, K., Huy, J., Beebakhee, G. & Bognar, A. L. (1991). Arch. Biochem. Biophys.284, 9–16. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Lowe, K. E., Osborne, C. B., Lin, B.-F., Kim, J.-S., Hsu, J.-C. & Shane, B. (1993). J. Biol. Chem.268, 21665–21673. [PubMed]
- Lu, Y.-Z., Aiello, P. D. & Matthews, R. G. (1984). Biochemistry, 23, 6870–6878. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Mathieu, M., Debousker, G., Vincent, S., Viviani, F., Bamas-Jacques, N. & Mikol, V. (2005). J. Biol. Chem.280, 18916–18922. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Osborne, C. B., Lowe, K. E. & Shane, B. (1993). J. Biol. Chem.268, 21657–21664. [PubMed]
- Perrakis, A., Harkiolaki, M., Wilson, K. S. & Lamzin, V. S. (2001). Acta Cryst. D57, 1445–1450. [DOI] [PubMed]
- Pitkin, R. M. (2007). Am. J. Clin. Nutr.85, 285S–288S. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Ravanel, S., Cherest, H., Jabrin, S., Grunwald, D., Surdin-Kerjan, Y., Douce, R. & Rébeillé, F. (2001). Proc. Natl Acad. Sci. USA, 98, 15360–15365. [DOI] [PMC free article] [PubMed]
- Ray, J. G. & Laskin, C. A. (1999). Placenta, 20, 519–529. [DOI] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Mol. Microbiol.48, 77–84. [DOI] [PubMed]
- Schirch, V. & Strong, W. B. (1989). Arch. Biochem. Biophys.269, 371–380. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shane, B. (1980). J. Biol. Chem.255, 5663–5667. [PubMed]
- Shane, B. (1989). Vitam. Horm.45, 263–335. [DOI] [PubMed]
- Sheng, Y., Cross, J. A., Shen, Y., Smith, C. A. & Bognar, A. L. (2002). Arch. Biochem. Biophys.402, 94–103. [DOI] [PubMed]
- Sheng, Y., Khanam, N., Tsaksis, Y., Shi, X., Lu, Q. & Bognar, A. L. (2008). Biochemistry, 47, 2388–2396. [DOI] [PubMed]
- Smith, C. A. (2006). J. Mol. Biol.362, 640–655. [DOI] [PubMed]
- Smith, C. A., Cross, J. A., Bognar, A. L. & Sun, X. (2006). Acta Cryst. D62, 548–558. [DOI] [PubMed]
- Smith, C. A. & Rayment, I. (1996). Biophys. J.70, 1590–1602. [DOI] [PMC free article] [PubMed]
- Sun, X., Bognar, A. L., Baker, E. N. & Smith, C. A. (1998). Proc. Natl Acad. Sci. USA, 95, 6647–6652. [DOI] [PMC free article] [PubMed]
- Sun, X., Cross, J. A., Bognar, A. L., Baker, E. N. & Smith, C. A. (2001). J. Mol. Biol.310, 1067–1078. [DOI] [PubMed]
- Synold, T. W., Willits, E. M. & Barredo, J. C. (1996). Leuk. Lymphoma, 21, 9–15. [DOI] [PubMed]
- Tan, X. J. & Carlson, H. A. (2005). J. Med. Chem.48, 7764–7772. [DOI] [PubMed]
- Toy, J. & Bognar, A. L. (1994). Arch. Biochem. Biophys.314, 344–350. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2006). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]
- Young, P. G., Smith, C. A., Sun, X., Baker, E. N. & Metcalf, P. (2006). Acta Cryst. F62, 579–582. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: MtFPGS–ADP, 2vos, r2vossf
PDB reference: MtFPGS–AMPPCP, 2vor, r2vorsf