

The structure of the 1918 H1N1 neuraminidase was determined to 1.65 Å from crystals with a lattice-translocation defect using uncorrected, as well as corrected, diffraction data.
Keywords: neuraminidases, crystal defects, lattice translocation, pseudo-translation, residual electron density
Abstract
Few examples of macromolecular crystals containing lattice-translocation defects have been published in the literature. Lattice translocation and twinning are believed to be two common but different crystal-growth anomalies. While the successful use of twinned data for structure determination has become relatively routine in recent years, structure determination of crystals with lattice-translocation defects has not often been reported. To date, only four protein crystal structures containing such a crystal defect have been determined, using corrected, but not uncorrected, intensity data. In this report, the crystallization, structure determination and refinement of N1 neuraminidase derived from the 1918 H1N1 influenza virus (18NA) at 1.65 Å resolution are described. The crystal was indexed in space group C2221, with unit-cell parameters a = 117.7, b = 138.5, c = 117.9 Å, and the structure was solved by molecular replacement. The lattice-translocation vector in the 18NA crystal was (0, 1/2, 1/2) or its equivalent vector (1/2, 0, 1/2) owing to the C lattice symmetry. Owing to this special lattice-translocation vector in space group C2221, structure refinement could be achieved in two different ways: using corrected or uncorrected diffraction data. In the refinement with uncorrected data, a composite model was built to represent the molecules in the translated and untranslated layers, respectively. This composite structure model provided a unique example to examine how the molecules were arranged in the two lattice domains resulting from lattice-translocation defects.
1. Introduction
Crystal defects represent imperfections in the regular geometrical arrangement of the atoms in a crystal. In protein crystals, twinning and lattice translocation are two common crystal defects (Yeates, 1997 ▶; Wang, Kamtekar et al., 2005 ▶). While crystal twinning occurs when a single crystal is composed of two or more separate domains or layers which intergrow in different relative orientations (Yeates, 1997 ▶), lattice-translocation defects refer to the random translation of some layers by a fixed constant within the stacked layers (Wang, Kamtekar et al., 2005 ▶). Twinning can be detected by carefully analyzing the processed diffraction data; there is no interference between the diffraction from the individual domains in the twinned crystal as the domain sizes are presumed to be large compared with the coherence length of the X-ray beam (Yeates, 1997 ▶).
In lattice-translocation defects, two identical crystal lattices translated by a specific vector form a single coherent mosaic block and a physical model for this defect has been proposed (Wang, Kamtekar et al., 2005 ▶). It is noteworthy that the two-lattice treatment used here is just a statistical means of describing this defect; it does not mean that there are only two crystal domains. Using this two-lattice treatment, in which one lattice consists of a fraction κ of untranslated crystal layers and the other lattice consists of a fraction (1 − κ) of translated crystal layers, the total observed structure factor F total(h) for diffraction index h is related to the unit structure factor F unit(h) in the two lattice domains and the translocation vector t d (Wang, Kamtekar et al., 2005 ▶),
while the relationship of the corresponding total and unit intensities is
In (2), two parameters need to be determined: the translocation vector t d and the lattice-defect fraction κ. The translocation vector can be determined by the location of an off-origin peak in the native Patterson map and the fraction κ can be determined using layer-averaging intensity calculations and the least-squares minimization method (Wang, Kamtekar et al., 2005 ▶). In some special situations, such as t d = (0, 0, 1/2), the fraction κ can be calculated directly from the peak-height ratio of the Patterson functions at (0, 0, 1/2) and (0, 0, 0) (Wang, Kamtekar et al., 2005 ▶). On the other hand, in some difficult cases the fraction κ can be empirically derived from the subsequent structure-determination processes and native Patterson map examinations by generating a series of ‘corrected’ data sets with different κ-fraction values. Once the translocation vector t d and the lattice-defect fraction κ have been obtained, the unit intensities can be calculated for each reflection and these corrected intensities can then be used as ‘perfect’ diffraction data for structure determination.
The unique feature of lattice translocation is the strong/sharp and weak/diffuse diffraction pattern. According to the lattice-translocation theory (Cochran & Howells, 1954 ▶; Wang, Kamtekar et al., 2005 ▶), if the translation between two layers generates no phase shift or a phase shift of magnitude 2nπ for a particular reflection, then a strong and sharp reflection will be observed. However, a phase shift of magnitude (2n + 1)π will create weak and diffuse reflections, while other phase shifts will give smeary spots.
Another striking feature of lattice translocation is an off-origin native Patterson peak introduced by the strong–weak modulation of X-ray intensities (Wang, Kamtekar et al., 2005 ▶). Although off-origin native Patterson peaks also occur in some other situations, such as pseudo-translation, the combination of this off-origin native Patterson peak with a strong/sharp and weak/diffuse diffraction pattern is a strong indication of possible lattice-translocation defects.
To date, only lattice-translocation diffraction data from ϕ29 DNA polymerase (Kamtekar et al., 2004 ▶), HslV–HslU (Wang, Rho et al., 2005 ▶), S1-RBD-80R (Hwang et al., 2006 ▶) and CcmL (Tanaka et al., 2008 ▶) have been successfully used for structure determination of macromolecules using an algorithm developed by Wang, Kamtekar et al. (2005 ▶) to correct the defect by modifying the observed diffraction intensities. Here, we report the structure determination of native A/Brevig Mission/1/1918 neuraminidase (18NA) at 1.65 Å resolution from a crystal with lattice-translocation defects. The 1918 influenza pandemic has been cited as the most devastating epidemic in recorded world history (Reid & Taubenberger, 1999 ▶). Continuing efforts are being made to target the neuraminidase for structure-based anti-influenza drug design. The crystal structure of 18NA was determined by molecular replacement and refined using the corrected X-ray intensities. Interestingly, in this case the observed uncorrected intensities could also be used in the structural refinement.
2. Materials and methods
2.1. Cloning, expression and purification
A detailed description of 18NA protein production will be published elsewhere (Xu et al., manuscript in preparation). Briefly, cDNA corresponding to residues 83–468 of the 18NA ectodomain was cloned into the baculovirus transfer vector pAcGP67A (BD Biosciences). A construct containing an N-terminal hexahistidine tag and an N-terminal ‘tetramerization domain’ from human vasodilator-stimulated phosphoprotein (VASP; Kuhnel et al., 2004 ▶) was assembled with a thrombin site incorporated between the tetramerization domain and the N-terminus of the 18NA ectodomain. The recombinant 18NA protein was secreted into the cell-culture medium using suspension cultures of insect Sf9 and Hi5 cells. The constructed plasmid was co-transfected into Sf9 cells and the protein was expressed in Hi5 cells with a multiplicity of infection of 5 for 3 d in CellSTACK (Corning) culture chambers at 301 K. Hi5 cells were removed by centrifugation and soluble 18NA was recovered from the cell supernatant by metal-affinity chromatography using Ni–NTA resin (Qiagen). Thrombin digestion followed at 277 K overnight using 3 U thrombin per milligram of protein while the protein was still bound to the Ni–NTA resin and the supernatant mixture was collected and concentrated. The cleaved 18NA was further purified from the thrombin and any other aggregated materials by size-exclusion chromatography with Superdex 200 in 20 mM Tris–HCl, 150 mM NaCl and 2 mM CaCl2 pH 8.0.
2.2. Crystallization and data collection
18NA protein was concentrated to 10 mg ml−1 in 75 mM NaCl, 10 mM phosphate pH 7.3. Initial crystallization hits for the native 18NA protein were obtained with EasyXtal µplate G-RB JCSG+ suite (Qiagen) using the sitting-drop vapor-diffusion method at 295 K. Optimized crystals of native 18NA were grown by mixing 0.5 µl protein solution with 0.5 µl reservoir solution (0.16 M calcium acetate, 14.4% PEG 8000, 20% glycerol, 0.08 M cacodylate pH 6.5). Crystals were usually obtained within 24 h.
X-ray diffraction data were collected on Stanford Synchrotron Radiation Laboratory (SSRL) beamline 9-2 by remote access, using a MAR 325 CCD detector. Crystals of native 18NA were flash-cooled in liquid nitrogen. Prior to data collection, crystal annealing (Harp et al., 1998 ▶) was performed by turning off the cryostream once for 5 s, which significantly reduced the mosaicity of the flash-cooled crystal. Starting with the crystal orientation that gave the image shown in Fig. 1 ▶(a), a total of 214 frames were recorded with an oscillation angle of 0.5°. All data were integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶; Table 1 ▶).
Figure 1.

Diffraction pattern of a native 18NA crystal with lattice-translocation defects. (a) Alternating sharp–diffuse reflections were observed and the streaky axis of the smeary features was determined to be parallel to the b* direction, which is nearly vertical in the plane of the paper. (b) The b* direction was found to be nearly parallel to the X-ray beam and perpendicular to the plane of the paper. The sharp–diffuse sets of reflections were not obvious in this orientation.
Table 1. Data-collection and refinement statistics for native 18NA.
(a).
Data collection. Values in parentheses are for the outer shell.
| Space group | C2221 |
| Unit-cell parameters (Å) | a = 117.7, b = 138.5, c = 117.9 |
| Resolution (Å) | 50.0–1.65 (1.68–1.65) |
| X-ray source | SSRL 9-2 |
| No. of unique reflections | 113144 (4405) |
| Redundancy | 4.2 (2.8) |
| Average I/σ(I) | 22.2 (2.0) |
| Completeness | 97.3 (76.4) |
| Rmerge† | 0.093 (0.528) |
(b).
Refinement.
| Uncorrected data | Corrected data‡ | |
|---|---|---|
| Molecules in ASU | 2 + 2 monomers | 2 monomers |
| Refined residues | 1540 | 770 |
| Refined waters | 1508 | 754 |
| Rcryst§ | 0.193 | 0.180 |
| Rfree¶ | 0.231 | 0.210 |
| Average B values (Å2) | ||
| Protein | 12.3 | 16.6 |
| Waters | 28.8 | 32.0 |
| Ramachandran plot (%) | ||
| Most favored | 84.2 | 84.3 |
| Additionally allowed | 15.1 | 15.1 |
| Generously allowed | 0.8 | 0.6 |
| Disallowed | 0.0 | 0.0 |
| R.m.s.d. bond lengths (Å) | 0.012 | 0.013 |
| R.m.s.d. bond angles (°) | 1.3 | 1.4 |
R
merge = 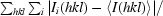
 , where 〈I(hkl)〉 is the average intensity of i symmetry-related observations of reflections with Miller indices hkl.
, where 〈I(hkl)〉 is the average intensity of i symmetry-related observations of reflections with Miller indices hkl.
The refinement statistics for corrected data (PDB code 3beq; Xu et al., manuscript in preparation) are shown here for comparison.
R
cryst = 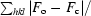
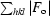 , where F
o and F
c are the observed and calculated structure factors.
, where F
o and F
c are the observed and calculated structure factors.
R free was calculated as for R cryst, but on 5% of data that were excluded prior to refinement.
3. Results and discussion
3.1. Molecular replacement and initial refinement
Crystals of native 18NA diffracted to beyond 1.65 Å resolution using SSRL beamline 9-2 (Table 1 ▶). The crystals were indexed in space group C2221, with unit-cell parameters a = 117.7, b = 138.5, c = 117.9 Å. The solvent content of the crystals, which contain two 18NA subunits per asymmetric unit, was estimated at 52.8% with a Matthews coefficient V M of 2.6 Å3 Da−1.
Molecular replacement was carried out with the program Phaser (McCoy et al., 2005 ▶) using a monomer of the H5N1 avian influenza neuraminidase tetramer (PDB code 2hty) as the search model. Two copies of 18NA were found in the asymmetric unit using Phaser (McCoy et al., 2005 ▶), which was run in a default ‘automated search’ mode, with a Z score of 73.4 and a log-likelihood gain of 8023 using all data between 50.0 and 2.5 Å resolution. The structure was refined with REFMAC v.5.2 (Murshudov et al., 1999 ▶) and the model was rebuilt using the program Coot (Emsley & Cowtan, 2004 ▶). Initial rigid-body refinement gave R cryst = 0.40 with data between 50.0 and 3.0 Å resolution. After several cycles of manual model rebuilding followed by refinement, the two 18NA monomers were essentially complete, but R cryst and R free stalled at 0.38 and 0.40 using data to 1.65 Å resolution. Significantly, substantial regions of the electron density had not been interpreted in the 2F o − F c and F o −F c maps and were subsequently confirmed to arise from two additional 18NA monomers that partially overlapped with the current model.
3.2. Sharp–diffuse diffraction patterns and native Patterson peaks
Without obvious anisotropy, the X-ray diffraction images of the native 18NA crystals displayed the striking feature of a periodic sharp–diffuse pattern of reflections (Fig. 1 ▶). Only half of the Bragg reflections remained well defined as sharp spots and the remaining reflections had comet tail-like streaky features (Fig. 1 ▶ a). Usually, these streaky features render all subsequent data-processing steps problematic, since it is difficult to determine the geometric centroids of the reflections, while the intensities of the reflections are themselves reduced significantly. However, for this data set, with careful optimization of spot size and integration profiles, it was still relatively straightforward to apply the autoindexing routines and the quality of X-ray diffraction data was still sound (Table 1 ▶).
Upon indexing, the streaky axes of the smeary features appeared to be parallel to the b* direction. In Fig. 1 ▶(a), the crystal was oriented such that the comet tail-like features along the b* axis are clearly visible, resulting in elongated lattice points for half of the reflections. In a second orientation (Fig. 1 ▶ b), that is 90° from that in Fig. 1 ▶(a), the streaky defects are no longer visible, so that b* is now nearly perpendicular to the paper plane and nearly parallel to the X-ray beam, resulting in ‘increased mosaicity’ in this orientation for the crystal. The apparent mosaicity, as refined in the process of integration, increased from about 0.34° in the orientation shown in Fig. 1 ▶(a) to about 0.77° in the orientation shown in Fig. 1 ▶(b), while the post-refinement mosaicity of 0.60° for the entire crystal is about the average of both.
Another striking feature in the native 18NA crystal was the appearance of a strong off-origin peak in the native Patterson map at (0, 1/2, 1/2), or its equivalent (1/2, 0, 1/2), owing to C lattice symmetry when the origin was shifted from (0, 0, 0) to (1/2, 1/2, 0). Its peak height was about 54% of the origin peak and was not obviously dependent on the resolution range used for calculation. In contrast, the other additional peak heights in the Patterson map were all below 4% of the origin peak.
The existence of the sharp–diffuse diffraction pattern and a strong off-origin native Patterson peak, as well as the residual electron density in the initial refinement maps, indicated that the native 18NA crystals had a lattice-translocation defect in which two crystal lattices were related by a fixed translation vector and coexist as a single coherent mosaic block (Wang, Kamtekar et al., 2005 ▶). Correlation between these two lattices caused the appearance of an extra strong peak in the native Patterson map: the peak location is a function of the translation vector, while the peak height is a function of the extent of the lattice-translocation defect. Since the streaky axes in the native 18NA crystal were parallel to the b* direction, it was suggested that the layers were stacked along the b* axis and translocation of layers occurred during layer stacking (Wang, Kamtekar et al., 2005 ▶). Examination of the diffraction images showed that reflections with k + l = 2n (or h + l = 2n owing to C lattice symmetry) were sharp and strong and reflections with k + l = 2n + 1 (or h + l = 2n + 1) were diffuse and weak (Figs. 1 ▶ and 2 ▶ a).
Figure 2.

Layer-averaged intensity as a function of index k + l for (a) the observed uncorrected intensities and (b) the corrected intensities using the procedure described in the text.
The lattice-translocation vector is different from the streaky axes of the diffraction pattern. The lattice-translocation vector is the translation vector between translated and untranslated layers, while the streaky axis is related to the layer-stacking direction. No clear relationships could be found between these two directions in protein crystals with known lattice translocations (Table 2 ▶).
Table 2. Comparison of some known protein crystals with lattice-translocation defects.
| Protein† | ϕ29 DNA polymerase | HslV–HslU | S1-RBD-80R | Native 18NA |
|---|---|---|---|---|
| Space group | P21 | H32 | P21 | C2221 |
| Unit-cell parameters (Å, °) | a = 59.9, b = 169.4, c = 68.6, β = 107.6 | a = 181.3, b = 181.3, c = 530.0 | a = 47.5, b = 175.9, c = 67.6, β = 96.6 | a = 117.7, b = 138.5, c = 117.9 |
| Resolution (Å) | 2.8 | 4.2 | 2.3 | 1.65 |
| Average I/σ(I) | 7.6 | 14.7 | 8.8 | 22.2 |
| Completeness (%) | 100 | 100 | 93.8 | 97.3 |
| Rmerge‡ | 0.171 | 0.170 | 0.145 | 0.093 |
| Patterson peak location | (0, 0, 1/2) | (0, 0, 1/3) | (1/3, 0, 0) | (0, 1/2, 1/2) |
| Patterson peak ratio§ (%) | 27 | 26 | 65 | 54 |
| Defect fractions (%) | 83 and 17 | 79 and 21 | 73 and 27 | 77 and 23 |
| Observed ‘streaky’ axis | a* | c* | a* | b* |
Data sources: ϕ29 DNA polymerase, Wang, Kamtekar et al. (2005 ▶); HslV–HslU, Wang, Rho et al. (2005 ▶); S1-RBD-80R, Hwang et al. (2006 ▶).
R
merge =
, where 〈I(hkl)〉 is the average intensity of i symmetry-related observations of reflections with Miller indices hkl.
The Patterson peak ratio is the ratio between the off-origin peak height and the origin peak height.
3.3. Structure determination with corrected intensities using lattice-translocation theory
Lattice-translocation defects yield two associated lattice domains. In the crystal of native 18NA, for the translocation vector t d = (0, 1/2, 1/2), the corresponding observed and unit intensities I obs and I unit are related by
or
where 〈I obs〉k+l and 〈I unit〉k+l are unscaled layer-average measurements for a given index k + l and f is the intensity-modulation factor and is simply a function of the reciprocal-space index k + l as shown in layer-averaged observed intensity (Fig. 2 ▶), while A and B are the parameters that need to be determined by least-squares fitting of the layer-averaged measured intensities by minimizing Q using the equation (Wang, Kamtekar et al., 2005 ▶)
Noticeably, in this least-squares minimization, it was assumed that, in the absence of lattice translocation, the diffraction intensity in three-dimensional reciprocal space will be distributed in a pattern such that the unit intensities of all layers 〈I unit〉k+l will be comparable with one another.
After least-squares fitting using (5), these two parameters were determined to be A = 339.3 and B = 182.3 and the ratio r = B/A was 0.537. The lattice-defect fraction κ could be estimated from A and B as 22.6% and 77.4% using the equation (Wang, Kamtekar et al., 2005 ▶),
Furthermore, in this special situation in which t d = (0, 1/2, 1/2), the fraction κ could be calculated directly from the peak ratio p of the native Patterson functions between (0, 1/2, 1/2) and (0, 0, 0) (Wang, Kamtekar et al., 2005 ▶), each of which could be Fourier-summed through the two intensity groups with either index k + l = odd or k + l = even (including k + l = 0), and this calculation was also independent of resolution,
Actually, using the native 18NA X-ray data, the peak ratio p between (0, 1/2, 1/2) and (0, 0, 0) was calculated to be 0.540, which was almost identical to the value r = 0.537 from the least-squares fitting.
Since the parameters A and B in (3) had been determined, the correction factor 1/f could be calculated for each diffraction spot and the new intensities (I unit) that correspond to the contribution from one lattice could be obtained. Upon intensity correction, the layer-averaged intensity variations in every other index of k + l of the observed intensities were removed (Fig. 2 ▶) and, in the new native Patterson map calculated with the corrected intensities, the peak at (0, 1/2, 1/2) was now completely eliminated (data not shown).
The corrected intensities were then used to carry out the structure determination with the molecular-replacement program Phaser (McCoy et al., 2005 ▶) using a monomer of the H5N1 avian influenza neuraminidase tetramer (PDB code 2hty) as the search model, which yielded two copies of the 18NA monomer per asymmetric unit (Fig. 3 ▶ a) with a Z score of 85.9 and a log-likelihood gain of 10 214 using data between 50.0 and 2.5 Å resolution. The initial model was refined in REFMAC (Murshudov et al., 1999 ▶) and model rebuilding was carried out in Coot (Emsley & Cowtan, 2004 ▶). The additional electron density which was found in the 2F o − F c and F o − F c maps from the uncorrected data was dissipated and final refinement in REFMAC using isotropic B-factor refinement to 1.65 Å resolution gave R cryst = 0.180 and R free = 0.210 (Table 1 ▶; PDB code 3beq).
Figure 3.

18NA molecules in ribbon representation in the crystal lattice domains: (a) in one lattice domain as a dimer (green), (b) in two lattice domains as high-occupancy and low-occupancy dimers (green and grey, respectively).
3.4. Structure determination with uncorrected intensities
In lattice-translocation cases where the lattice-translocation defects do not destroy the symmetry of each lattice domain or the entire crystal, it might be possible to obtain the correct structural solutions without correction of the intensities. Lattice translocation may complicate phasing and refinement, but does not render it impossible. A composite atomic model can be proposed to include two lattice domains with the respective occupancy factors corresponding to the defect-fraction factors. The total calculated structure factor from this composite model is related to the structure factor F calc(h) from the same structure in the untranslated lattice,
By comparison of (1) and (8), there should be perfect agreement between the observed and calculated structure factors in both amplitude and phase, although the resulting electron-density maps would usually be difficult to interpret owing to the overlay of the two-lattice models.
Actually, in this native 18NA case, the molecular-replacement calculation was first carried out by using the uncorrected data to find a dimer in one asymmetric unit, which was positioned in the unit cell almost identically to that of the dimer obtained from the corrected intensity data. The initial refinement to 1.65 Å resolution with R cryst and R free values of 0.38 and 0.40, respectively, showed that the dimer model fitted the electron density reasonably well, except for part of the model where the extra densities were overlapped. Careful analysis of the electron-density maps suggested that this was because of an additional 18NA dimer that overlapped with the current one. This extra dimer actually came from the model of the adjacent lattice domain and was easily built into the electron density (Fig. 4 ▶) using Coot (Emsley & Cowtan, 2004 ▶). The occupancy factors of the overlapped dimers were refined using SHELXH (Sheldrick, 2008 ▶), which gave values of 0.75 and 0.25 for the original dimer and the extra dimer, respectively. As expected, the occupancy factors of these two dimers were very close to the defect-fraction factors of 0.77 and 0.23.
Figure 4.

OMIT electron-density map around Val424–Arg428 from the subsequent low-occupancy 18NA dimer. The electron-density map was calculated using molecular-replacement solutions of the original high-occupancy dimer and contoured at 1.0σ (green mesh). The model fits the electron density well, even before it was included in structural refinement.
Further refinement was carried out using REFMAC (Murshudov et al., 1999 ▶) to an R cryst of 0.27 and R free of 0.30 before well defined water molecules were added, which further decreased R cryst and R free to 0.22 and 0.26, respectively. In this composite model (Fig. 3 ▶ b), it was found that the same portions (about one-third) of each dimer were overlapped with each other. Fortunately, since one dimer had an occupancy factor of 0.75, this dimer model fitted the electron density very well even in the overlapped region and the model for the other dimer could accordingly be rebuilt even without perfect electron density.
The next major obstacle for further model rebuilding was the assignment of water molecules in the overlapped region or around the low-occupancy dimer. Thus, the final refinement was carried out with the program REFMAC (Murshudov et al., 1999 ▶) using the dimer model including water molecules obtained from the corrected intensity data. The occupancy factors of two lattice domains were assigned as 0.77 and 0.23, respectively, and the final model had an R cryst of 0.193 and an R free of 0.231 (Table 1 ▶). In the final density maps, the model of the high-occupancy dimer fitted the electron density very well, while some residues of the low-occupancy dimer did not. Nevertheless, the structures of these two dimers (Fig. 3 ▶ b) were very similar, with an r.m.s.d. of 0.15 Å for main-chain atoms after the ‘best’ computational superimposition and an r.m.s.d. of 0.17 Å for main-chain atoms when the translation (0, 1/2, 1/2) was used to superimpose the two dimers. No significant rotation existed between the high-occupancy and low-occupancy dimers in untranslated and translated layers of the crystal.
In summary, the native 18NA diffraction data were used for refinement by two strategies: corrected or uncorrected. The refinement results verified each other and proved that the intensity correction is appropriate without overcorrection. The structure models obtained from both corrected and uncorrected data are very similar, with r.m.s.d. values of 0.04 or 0.15 Å, respectively, for main-chain atoms after superimposing the dimers from the corrected data (Fig. 3 ▶ a) with the high-occupancy or low-occupancy dimers from the uncorrected data (Fig. 3 ▶ b), respectively. The r.m.s.d. value was calculated to be 0.06 Å for main-chain atoms without any further superimposition between the dimers from the corrected data (Fig. 3 ▶ a) and the high-occupancy dimer from the uncorrected data, while the r.m.s.d. value was 0.18 Å for main-chain atoms between the dimer from the corrected data (Fig. 3 ▶ a) and the low-occupancy dimer from the uncorrected data when the translation (0, 1/2, 1/2) was used to superimpose the dimers. The detailed structural characterization of 18NA will be published elsewhere (Xu et al., in preparation).
4. Concluding remarks
Despite being one of the anticipated common crystal defects, occurrences of lattice translocation seem to be underreported. The key point is perhaps how to detect this defect initially and how to correct the intensities for structural refinement. Here, we report a special case in which the lattice-translocation defects do not destroy the symmetry of each lattice domain or the entire crystal. Structural refinement was successfully carried out with both corrected and uncorrected intensity data and provided a unique example of the ability to use protein crystals with lattice-translocation defects productively. The lattice-translocation defects generate a sharp/diffuse diffraction pattern and reduce the quality of the reflection data. However, as shown here and in some other published cases (Table 2 ▶), it is still possible to use the diffraction data for a successful structure determination. The structure model of native 18NA obtained from the uncorrected data provided a first glimpse of how the protein molecules were arranged in two crystal lattice domains. Although in some ways it is difficult to use only uncorrected data for structure determination, especially in dealing with the water molecules in the overlapped region, the method is still suitable for the verification of the intensity-correction method.
The central feature of lattice translocation is the sharp/strong and diffuse/weak patterns in some reflections. The extent of the lattice translocation and the relationship between the translated lattices can be obtained from the analysis of native Patterson map. A strong Patterson peak should exist in the native Patterson map, which provides evidence for such lattice-translocation defects. Notwithstanding, in this special native 18NA case strong residual electron density could be found to partially overlap with the initial model, providing strong evidence for the presence of this crystal defect.
Furthermore, it is interesting to compare lattice translocation (Wang, Kamtekar et al., 2005 ▶) and pseudo-translation (Chook et al., 1998 ▶). Pseudo-translation refers to translational symmetry which relates multiple molecules in an asymmetric unit. Both lattice translocation and pseudo-translation yield a distribution of systematically strong and weak reflections, which in turn result in extra peaks in the native Patterson maps. However, lattice translocation can be differentiated from pseudo-translation by examination of the unique feature of a sharp/diffuse diffraction pattern in lattice-translocation defects. On the other hand, the crystal packing can be another strong piece of evidence to distinguish between lattice translocation and pseudo-translation. In some special cases, the position of the extra native Patterson peak can provide an indicator that it must be lattice translocation rather than pseudo-translation that is present. In particular, if there is a major Patterson peak near the origin then this is not consistent with pseudo-translation since it is impossible for two molecules to coexist if the shift between them is small. Noticeably, if a crystal has a twofold pseudo-translation (Chook et al., 1998 ▶), the ‘defect-fraction factor’ calculated using the lattice-translocation intensity-correction method (Wang, Kamtekar et al., 2005 ▶) would be around 0.5.
Supplementary Material
PDB reference: 1918 H1N1 neuraminidase, 3cye, r3cyesf
Acknowledgments
We acknowledge the staff of Stanford Synchrotron Radiation Laboratory BL9-2 for assistance during data collection. We thank Drs Robyn Stanfield and Xiaoping Dai for helpful discussions, Dr Todd O. Yeates, UCLA for critical comments and excellent suggestions on the manuscript, and Professor Raymond Dwek for continual support and encouragement. This work was supported by National Institutes of Health grant AI-058113 (IAW) and by the Oxford University (Glycobiology Institute) and TSRI graduate programs. This is manuscript No. 19420-MB from The Scripps Research Institute.
References
- Chook, Y. M., Lipscomb, W. N. & Ke, H. (1998). Acta Cryst. D54, 822–827. [DOI] [PubMed]
- Cochran, W. & Howells, E. R. (1954). Acta Cryst.7, 412–415.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Harp, J. M., Timm, D. E. & Bunick, G. J. (1998). Acta Cryst. D54, 622–628. [DOI] [PubMed]
- Hwang, W. C., Lin, Y., Santelli, E., Sui, J., Jaroszewski, L., Stec, B., Farzan, M., Marasco, W. A. & Liddington, R. C. (2006). J. Biol. Chem.281, 34610–34616. [DOI] [PMC free article] [PubMed]
- Kamtekar, S., Berman, A. J., Wang, J., Lazaro, J. M., de Vega, M., Blanco, L., Salas, M. & Steitz, T. A. (2004). Mol. Cell, 16, 609–618. [DOI] [PubMed]
- Kuhnel, K., Jarchau, T., Wolf, E., Schlichting, I., Walter, U., Wittinghofer, A. & Strelkov, S. V. (2004). Proc. Natl Acad. Sci. USA, 101, 17027–17032. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Reid, A. H. & Taubenberger, J. K. (1999). Lab. Invest.79, 95–101. [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Tanaka, S., Kerfeld, C. A., Sawaya, M. R., Cai, F., Heinhorst, S., Cannon, G. C. & Yeates, T. O. (2008). Science, 319, 1083–1086. [DOI] [PubMed]
- Wang, J., Kamtekar, S., Berman, A. J. & Steitz, T. A. (2005). Acta Cryst. D61, 67–74. [DOI] [PubMed]
- Wang, J., Rho, S.-H., Park, H. H. & Eom, S. H. (2005). Acta Cryst. D61, 932–941. [DOI] [PubMed]
- Yeates, T. O. (1997). Methods Enzymol.276, 344–358. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: 1918 H1N1 neuraminidase, 3cye, r3cyesf