

Separation of two individual lattices within an epitaxially twinned data set allowed the crystal structure of the V3-specific neutralizing antibody 447-52D in complex with a V3 peptide (UG1033) to be determined. The structure confirms that the neutralization breadth of Fab 447-52D is likely to be attributable to the extensive focus on main-chain hydrogen-bond interactions with the peptide that permit the recognition of a range of V3 sequences.
Keywords: antibodies, HIV-1, twinning, V3 loop
Abstract
Although antibodies against the third variable loop (V3) of the HIV-1 viral envelope glycoprotein are among the first neutralizing antibodies to be detected in infected individuals, they are normally restricted in their specificity. X-ray crystallographic studies of V3-specific antibodies have contributed to a more thorough understanding of recognition of this epitope and of conserved features in the V3 loop that could potentially aid in the design of a multi-component vaccine. The human antibody 447-52D exhibits relatively broad neutralization of primary viral isolates compared with other V3-loop antibodies. A crystal structure of Fab 447-52D in complex with a V3 peptide (UG1033) was determined at 2.1 Å resolution. The structure was determined using an epitaxially twinned data set and in-house programs to detect and remove overlapping reflections. Although the processed data have lower than desired completeness and slightly higher than normal R values for the resolution, good-quality electron-density maps were obtained that enabled structure determination. The structure revealed an extended CDR H3 loop that forms a β-sheet with the peptide, with the predominant contacts being main-chain hydrogen bonds. The V3 peptide and Fab show high structural homology with the previously reported structures of other Fab 447-52D complexes, reinforcing the idea that the V3 loop may adopt a small set of conserved structures, particularly around the crown of the β-hairpin.
1. Introduction
The human immunodeficiency virus type 1 (HIV-1) envelope is a highly glycosylated trimer of heterodimers, composed of gp120, which mediates binding to the cell-surface CD4 receptor and chemokine co-receptors, and gp41, a transmembrane protein that is involved in membrane fusion. Despite its exposure on the viral surface, the HIV-1 envelope spike, which is the target of neutralizing antibodies, has evolved many diverse mechanisms to escape the humoral immune response. Extensive glycosylation of the exposed envelope protein, which gives rise to an immunologically silent ‘glycan shield’, and exposure of regions of gp120 that are highly variable in sequence, termed the V1–V5 hypervariable loops, are among the major mechanisms that contribute to the ability of HIV-1 to evade the antibody response during infection (Burton et al., 2005 ▶; Pantophlet & Burton, 2006 ▶; Wei et al., 2003 ▶; Wyatt et al., 1998 ▶). The glycan shield results in a general dampening of the immune response, whereas the immunodominant variable loops tend to elicit neutralizing antibodies that exhibit strain-specific neutralization.
The third hypervariable (V3) loop of gp120 was initially known as the principal neutralizing determinant of HIV-1 owing to its ability to readily elicit anti-HIV-1 antibodies (Gao et al., 2005 ▶; Goudsmit et al., 1988 ▶; Javaherian et al., 1989 ▶, 1990 ▶; Rusche et al., 1988 ▶). The anti-V3 antibodies induced early in infection and after limited exposure to immunogens are relatively type-specific; however, anti-V3 antibodies that display broader reactivity have been reported (Binley et al., 2004 ▶; Gorny et al., 2002 ▶, 2006 ▶; Krachmarov et al., 2005 ▶; Pantophlet et al., 2007 ▶; Richman et al., 2003 ▶). The neutralization activity of anti-V3 antibodies may be affected by the propensity of the V3 region to undergo sequence changes that alter neutralizing-antibody epitopes and give rise to escape mutants during the course of infection (Hartley et al., 2005 ▶; Richman et al., 2003 ▶), epitope masking by the V1/V2 domain (Hartley et al., 2005 ▶; Pantophlet & Burton, 2006 ▶; Pinter et al., 2004 ▶) and/or the presence of glycans (Back et al., 1994 ▶; Koch et al., 2003 ▶; McCaffrey et al., 2004 ▶).
However, human V3-specific monoclonal antibodies (mAbs) have been described that exhibit neutralization of multiple primary HIV-1 isolates, suggesting that this region may indeed be a worthwhile target in a multi-component HIV-1 vaccine. The antibody 447-52D, which was originally isolated from an HIV-1-infected individual (Gorny et al., 1992 ▶, 1993 ▶), is one of several anti-V3 monoclonal antibodies (Gorny et al., 2004 ▶, 2006 ▶) that exhibit significant neutralizing breadth (Binley et al., 2004 ▶). Immunochemical studies mapped the core epitope to a GPxR motif in the crown of the V3 loop (Keller et al., 1993 ▶). Although this motif is prevalent within subtype B isolates, the dominant crown motif of non-clade-B isolates is GPGQ (http://www.hiv.lanl.gov/content/hiv-db). In one analysis, Fab 447-52D neutralized 50% of 32 clade-B viruses (Binley et al., 2004 ▶); in a second study, 92% of 38 primary isolates containing the GPGR crown motif could be neutralized (Zolla-Pazner et al., 2004 ▶). The V3-specific mAb 2219, which was also derived from an HIV-1-infected individual, similarly exhibits neutralization of a relatively large proportion of HIV-1 isolates (Gorny et al., 2002 ▶, 2004 ▶). More recently, human mAb F425-B4e8 has been reported to exhibit cross-neutralizing activity comparable to those of Fab 447-52D and Fab 2219 (Pantophlet et al., 2007 ▶).
In addition to being a potential target for a neutralizing epitope in an HIV-1 vaccine, the V3 loop plays an important role in virus infectivity. The sequence of the V3 loop can influence cell tropism (Hoffman & Doms, 1999 ▶) and this loop is thought to interact with the chemokine co-receptors CCR5 or CXCR4 during infection (Basmaciogullari et al., 2002 ▶; Rizzuto & Sodroski, 2000 ▶; Rizzuto et al., 1998 ▶). The V3 region is composed of approximately 35 residues and contains a conserved disulfide bridge at the base of the loop. A recent crystal structure of an HIV-1 gp120 core containing an intact V3 loop reveals that this region contains three structurally distinct domains: a conserved base, a crown or tip region (apex) and a flexible stem that links the base and crown (Huang et al., 2005 ▶). Antibody-binding studies performed in the presence of soluble CD4 suggest that the V3 loop may become more accessible upon CD4 binding (Mbah et al., 2001 ▶). Such an extended structure could help to explain the immunodominance of the V3 loop. Cryo-electron tomography studies performed with SIV virions suggest two possible models for the location of the V3 loop within the context of the trimeric envelope. In the first, the V3 loop is solvent-exposed and positioned away from the trimer interface along the outer side of gp120 (Zhu et al., 2006 ▶). Alternatively, the V3 region may be packed into the trimer interface and become exposed upon CD4-envelope ligation owing to structural rearrangement of the trimer (Zanetti et al., 2006 ▶).
To gain a better understanding of the conformation of the V3 loop and of the basis for neutralization by V3 mAbs, structural studies of anti-V3 mAbs in complex with V3 peptides have been undertaken. To date, X-ray crystallographic structures of four murine anti-V3 Fabs (50.1, 58.2, 59.1, 83.1) have been determined for the Fabs in complex with V3 peptides corresponding to the HIV-1 isolate MN (Ghiara et al., 1994 ▶, 1997 ▶; Rini et al., 1993 ▶; Stanfield et al., 1993 ▶, 1999 ▶, 2003 ▶). The crystal structure of human Fab 447-52D has also been determined in complex with an MN peptide (Stanfield et al., 2004 ▶) and, more recently, crystal structures of Fab 2219 in complex with three different V3 peptides, corresponding to isolates UG1033, MN and UR29, have been reported (Stanfield, Gorny et al., 2006 ▶).
The MN V3 peptide adopts a similar conformation in complex with the murine Fabs 50.1, 59.1 and 83.1, whereas a different V3-crown structure is observed in the Fab 58.2 complex. In the former, residues upstream of the crown form an extended β-sheet, with a type I or II β-turn around the GPGR crown motif. The V3 peptide in the Fab 58.2 complex adopts a different conformation at the crown that contains two consecutive turns. The V3 peptides in the Fab 447-52D and Fab 2219 crystal structures show structural homology to these murine Fab complexes, adopting β-hairpin structures, although the relative orientation of the N- and C-terminal strands in the Fab 2219 peptides differs owing to a differing torsion angle of a single residue within the N-terminal strand. These V3 structures are also comparable to the conformation observed in the V3-containing core gp120 structures (Huang et al., 2005 ▶, 2007 ▶; Stanfield & Wilson, 2005 ▶). Finally, NMR studies of the Fv fragment of 447-52D in complex with V3 peptides corresponding to isolates MN and IIIb show that the peptides in both of these complexes form β-hairpin structures and exhibit the same N-terminal conformation (Rosen et al., 2005 ▶; Sharon et al., 2003 ▶).
Here, we report the processing strategy for an epitaxially twinned data set (Table 1 ▶) and the details of the resulting 2.1 Å structure of the UG1033 V3 peptide bound to Fab 447-52D. We also discuss insights gained from a comparative analysis of this new structure with those previously reported for other V3-peptide complexes of Fab 447-52D.
Table 1. Comparison of the six different data-processing and initial test refinements of the epitaxially twinned data.
| Data set no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Lattice 1 | Lattice 2 | Lattice 1 + 2 | Lattice 1† | Lattice 2† | Lattice 1 + 2† | |
| No. of spots‡ | 2346478 | 2350364 | 4696842 | 1810397 | 1814960 | 3625357 |
| No. of overlapping spots deleted by in-house program | 0 | 0 | 0 | 536081 | 535405 | 1071485 |
| No. of observations§ | 206766 | 215665 | 427782 | 153463 | 159567 | 310899 |
| No. of observations rejected by SCALEPACK | 26884 | 29981 | 89699 | 15236 | 15350 | 49923 |
| No. of unique reflections¶ | 68033 | 68897 | 69984 | 56663 | 57482 | 60586 |
| Completeness (%) | 96.9 | 97.4 | 98.9 | 80.6 | 81.3 | 85.7 |
| Redundancy | 3.0 | 3.1 | 6.1 | 2.7 | 2.8 | 5.1 |
| I/(I) | 23.6 | 24.1 | 34.4 | 19.5 | 19.7 | 27.1 |
| R merge | 6.7 (32.9) | 6.7 (34.5) | 8.3 (39.4) | 6.8 (38.7) | 6.9 (38.8) | 8.4 (44.5) |
| R rigid †† | 38.1 | 37.6 | 37.7 | 36.7 | 36.6 | 36.4 |
| R cryst/R free | 30.9/34.4 | 30.4/33.7 | 30.3/34.2 | 29.7/33.2 | 29.4/33.9 | 29.1/32.9 |
The last three columns are with overlaps manually deleted, whereas the first three columns are with no manual deletion of overlapping spots.
Spots are defined as the two-dimensional diffraction peak from a single image frame. In most cases, these are partial reflections.
Observations are the integrated intensity formed by the profile-fitted summation of the partial spots.
Reflections are the average of multiply measured and symmetry-related observations.
R rigid is the R cryst value from the initial rigid-body refinement with resolution limits of 154.
2. Experimental
2.1. Fab production and purification
IgG 447-52D (IgG3, λ) was produced and purified as previously reported (Gorny et al., 1989 ▶, 1991 ▶). Briefly, pooled peripheral blood cells from two HIV-1-infected patients were transformed and fused to a human/mouse hetero-myeloma cell line (SHM-D33). The Mab 447-52D heterohybridoma was selected based on its ability to bind a peptide corresponding to a portion of the V3 sequence of the HIV-1 isolate MN (YNKRKRIGIGPGRAFYTTKNIIG) and then repeatedly cloned until monoclonality was achieved. Fab fragments were prepared by IgG 447-52D cleavage with 4% papain in 10 mM l-cysteine for 4 h. Fab 447-52D was purified on Protein A, Superdex 200 (10/30 column; 0.2 M sodium acetate pH 5.5) and Mono S columns (10/10 column; buffer A = 50 mM sodium acetate pH 4.5; buffer B = 50 mM sodium acetate, 1 M sodium chloride pH 4.5).
2.2. Crystallization
A 23-mer V3 peptide, UG1033 (NNTRKSIHLGPGRAFYATGDIIG), was used for cocrystallization. Fab 447-52D (20 mg ml−1) and UG1033 peptide were mixed in a 1:5 molar ratio and crystals were grown using the standard sitting-drop vapor-diffusion method. Thin intergrown crystal plates which could not be readily separated were grown from 1.4 M ammonium sulfate, 0.1 M cacodylate pH 6.5 and did not diffract beyond 2.5 Å. However, screening of these conditions with Hampton Additive Screen 1 revealed larger single-crystal plates with CuCl2 as an additive. Crystals that visually appeared to be single were grown using a final reservoir solution of 1.58 M ammonium sulfate, 0.1 M Tris pH 7.88, 1 mM CuCl2. Crystals were cryopreserved by brief immersion into a solution consisting of the reservoir solution augmented with 25% glycerol. Data were collected to 2.1 Å resolution on SSRL beamline 11-1 with a 0.25° step size and a crystal-to-detector distance of 250 mm on an ADSC Q315 detector and reduced in space group P21 with unit-cell parameters a = 70.5, b = 75.9, c = 114.1 Å, β = 101.5°. The Matthews coefficient was 3.0 Å3 Da−1 (Matthews, 1985 ▶), corresponding to 59% solvent content. Data were indexed, integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶) to 2.1 Å resolution (Table 2 ▶). Multiple lattices were detected in all crystals that were screened. Twinned data sets were resolved using DENZO/SCALEPACK (Otwinowski & Minor, 1997 ▶) and in-house programs, as described below in §3.
Table 2. Processing and final refinement statistics for data set 6.
Values in parentheses are for the last shell.
| Data collection | |
| Space group | P21 |
| Unit-cell parameters (, ) | a = 70.5, b = 75.9, c = 114.1, = 101.5 |
| Resolution () | 502.1 (2.152.10) |
| No. of observations | 310899 (13751) |
| No. of unique reflections | 60586 (3035) |
| Completeness (%) | 85.7 (86.0) |
| Redundancy | 5.1 (4.5) |
| I/(I) | 27.1 (4.0) |
| R merge | 8.4 (44.5) |
| Refinement statistics | |
| R cryst | 24.1 (29.9) |
| R free | 29.8 (37.5) |
| No. of waters | 188 |
| R.m.s.d. from ideal geometry | |
| Bond lengths () | 0.017 |
| Angles () | 1.6 |
| B values† (2) | |
| Fab 1 | |
| VL | 29.4 |
| CL | 31.1 |
| VH | 34.1 |
| CH1 | 31.3 |
| Peptide | 57.8 |
| Fab 2 | |
| VL | 53.7 |
| CL | 52.2 |
| VH | 49.8 |
| CH1 | 39.9 |
| Peptide | 82.8 |
| Waters | 34.6 |
| Overall | 40.8 |
| Ramachandran plot | |
| Most favored (%) | 88.5 |
| Additionally allowed (%) | 11.0 |
| Generously allowed (%) | 0.1 |
| Disallowed (%) | 0.4 |
B values are the sum of TLS and residual B factors.
2.3. Structural determination and refinement
The structure was determined by molecular replacement using the refined structure of Fab 447-52D (PDB code 1q1j) as a search model. Two Fab–peptide complexes were present in the asymmetric unit and were related by an NCS twofold around x (or z). Alternating cycles of model building and positional refinement were carried out using TOM/FRODO (Jones, 1982 ▶) and CNS (Brünger et al., 1998 ▶). Tight NCS restraints were used early in refinement and then gradually released. Final rounds of refinement were carried out using REFMAC (Collaborative Computational Project, Number 4, 1994 ▶) with eight TLS groups corresponding to CL, VL, CH1 and VH + peptide for each Fab complex. B values for all protein domains and solvent molecules are reported in Table 2 ▶. An R free test set of 5% of the reflections was maintained throughout refinement. Electron-density maps used for rebuilding included σA-weighted 2F o − F c and F o − F c maps. Hydrogen bonds and van der Waals contacts were evaluated with HBPLUS (McDonald & Thornton, 1994 ▶) and CONTACSYM (Sheriff et al., 1987 ▶). Buried molecular-surface areas were calculated using the program MS (Connolly, 1993 ▶) with a 1.4 Å probe radius.
3. Results and discussion
3.1. Lattice separation in the epitaxially twinned crystals
Although the crystals grown in the presence of CuCl2 diffracted strongly, they contained two separate lattices of approximately equivalent intensity with the same unit-cell parameters, as if two crystals in different orientations had intergrown (Fig. 1 ▶). Multiple crystals were tested, but none were obtained with a single lattice. Attempts to obtain strongly diffracting crystals under alternative growth conditions also failed. As a result, data from the epitaxially twinned crystals were collected and processed with DENZO/SCALEPACK (Otwinowski & Minor, 1997 ▶) and in-house programs, as detailed below, to include data from both lattices. While published descriptions of computer programs for treating epitaxially twinned data processed with XDS (Lietzke et al., 1996 ▶) and XENGEN (Schulze-Gahmen et al., 1993 ▶) are available, we were unable to find any similar programs that worked with DENZO/SCALEPACK. Since this work was carried out, a web-based server has been developed in another laboratory that works with DENZO/SCALEPACK using a similar scheme to that which we describe here (http://ultr23.vub.ac.be/untangle).
Figure 1.

Raw data from the epitaxially twinned data set. The frames are displayed with the XDISPLAYF (Otwinowski & Minor, 1997 ▶) software, and the images have been cropped to show only the strongly diffracting spots. (a) The first diffraction image (crystal to detector distance = 250 cm, oscillation angle = 0.25°). (b) Spots (diffraction maxima) corresponding to the first lattice are shown in green, after separation of the spot file into two files for the two different lattices, as described in §3. (c) Spots (green) corresponding to the second lattice. (d) The same image with all spots (green) from both lattices selected for auto-indexing by XDISPLAYF.
Firstly, a standard DENZO/XDISP peak search was used to find spots on the first diffraction image. The term ‘spots’ will be used throughout to define two-dimensional diffraction peaks from a single image that result, in most cases, from partial reflections. The spots representing both lattices were used in the initial DENZO autoindexing. The autoindexing routine then calculates a unit cell and orientation matrix for one of the two lattices and using this information ‘spots’ from ‘lattice 1’ only were integrated. An in-house program was then used to compare the x, y centroid position of each integrated spot from lattice 1 with the spot positions found by XDISP. The XDISP spots were then separated into two files, one containing the spots within two pixels (1 pixel = 0.1024 mm) of integrated spots from lattice 1 and a second file containing the remaining spots, which predominantly belong to lattice 2 (Fig. 1 ▶). Each of these XDISP spot files was used as input to the autoindexing routine in DENZO, resulting in a unit cell and orientation matrix for each lattice. All frames were then integrated twice, once with each orientation matrix, resulting in files containing integrated spots from each of the two lattices. The two lattices are related by an approximate twofold rotation around the c axis of the crystal.
To deal with the problem of overlapping spots that arise from both lattices, another program was written to read in the files containing the integrated spots for each lattice on a frame-by-frame basis and then delete spots whose predicted centroid positions (in pixel units) are within two pixels (1 pixel = 0.1024 mm) of each other. The partial spots were then summed and profile-fitted to create the integrated intensities (observations) and scaled using SCALEPACK (to obtain unique reflections) for each of the individual lattices and also for the merged lattices (lattice 1 + 2) with and without overlapping-spot deletion. We generated six data sets using these criteria (see Table 1 ▶) in order to evaluate the best method for treating these data. Two to three rounds of initial model refinement with CNS were then carried out with each data set to determine which would yield the best quality model, as judged by R values and stereochemistry. Structures could be readily refined with all six data sets (see §2) with electron-density maps of similar quality. The R cryst and R free values are also very similar (Table 1 ▶). The final structure reported here was determined at 2.1 Å from the sixth data set in Table 1 ▶, which was chosen because it had the lowest R cryst/R free values of the six data sets. The final statistics listed in Table 2 ▶ are for the sixth data set, but with model rebuilding and complete refinement in addition to the preliminary refinements reported for the initial tests of the six data sets in Table 1 ▶.
3.2. Structure of Fab 447-52D
Fab 447-52D was crystallized with a 23-mer V3 peptide designated UG1033 (NNTRKSIHLGPGRAFYATGDIIG). Processing and refinement statistics for the 2.1 Å structure are reported in Table 2 ▶. The crystallographic asymmetric unit contains two Fab complexes related by a noncrystallographic (NCS) twofold axis around x (or z). Slightly higher B values and weaker electron density were observed in the second Fab complex, indicating a higher degree of flexibility for this complex. The Cα r.m.s. deviations for the equivalent variable light-chain (VL) and heavy-chain (VH) regions are 0.31 and 0.22 Å, respectively, and 0.43 Å for the V3 peptide. Because the two structures are so similar, only the first of the two molecules is discussed here unless noted otherwise. The Fab chains are numbered according to the Kabat and Wu convention (Kabat et al., 1987 ▶) and the peptide is numbered according to the HIV-1 HxBc2 numbering convention (Ratner et al., 1987 ▶). The heavy and light chains are designated chains H and L for Fab 1 and chains I and M for Fab 2, respectively, and the peptides are designated chains P and Q for Fabs 1 and 2, respectively. The elbow angles of the two Fabs are 212° and 207°, which are consistent with rather large elbow angles frequently observed for other Fabs with λ light chains (Stanfield, Zemla et al., 2006 ▶). Only three residues in the two complexes fall in the disallowed region of the Ramachandran plot. AsnL51 and AsnM51 are in a highly conserved γ-turn that is found in almost all Fabs (Stanfield et al., 1999 ▶) and AspM151 is in a loop which also has well ordered density. One residue, SerH134, lies within the generously allowed region of the Ramachandran plot and is within in a poorly ordered region of CH1 that tends to be disordered in most Fabs. Although CuCl2 was part of the crystallization solution, no density was found that could be assigned to copper or chloride ions.
An overview of the Fab 447-52D structure is illustrated in Fig. 2 ▶. As expected based on length and sequence, the light-chain CDR1 and CDR2 belong to canonical classes λL1-1 and L2-2 and the heavy-chain CDR1 and CD2 to classes 1 and 4, respectively. As reported previously, the light-chain CDR3 of Fab 447-52D contains a three-residue insertion (L95a–L95c) that makes it longer than other CDR L3 loops classified to date (Stanfield et al., 2004 ▶). The 20-residue heavy-chain CDR3 (H3) forms an extended structure, approximately 32 Å from base to tip, which projects outwards from the molecule. The extended structure is stabilized by a hydrogen-bonding network between the N- and C-terminal regions of the CDR H3 loop.
Figure 2.

Stereoview of the Fab 447-52D–UG1033 peptide complex. The Fab is colored gray, with the light chain oriented to the left and the heavy chain oriented to the right. Complementarity-determining regions (CDRs) L1, L2 and L3 are colored purple, orange and cyan, respectively. CDRs H1, H2 and H3 are colored blue, green and red, respectively. The UG1033 V3 peptide is colored yellow. This and subsequent figures were generated with PyMOL (DeLano, 2002 ▶).
3.3. Structure of the UG1033 V3 peptide
The majority of clade A and C isolates contain the V3-crown sequence GPGQ. However, the clade A UG1033 sequence corresponds to the dominant crown motif, GPGR, typically found in clade B. Although the UG1033 peptide is 23 residues in length, strong interpretable electron density was only present for ten residues (Fig. 3 ▶). Weaker electron density was present for an additional three residues (PheP317, TyrP318 and AlaP319), which are also included in the final model. The remaining C-terminal residues could not be built into the final model owing to very weak electron density, which is likely to be a consequence of the conformational flexibility of this region of the peptide, as frequently observed in other V3-loop complexes with Fabs (Ghiara et al., 1994 ▶, 1997 ▶; Rini et al., 1993 ▶; Stanfield et al., 1993 ▶, 1999 ▶, 2003 ▶, 2004 ▶; Stanfield, Gorny et al., 2006 ▶).
Figure 3.

Stereoview of electron density of the UG1033 peptide bound to Fab 447-52D. The 2F o − F c electron density (gray) for the UG1033 V3 peptide (yellow) after refinement is contoured at 1.0σ.
The V3 peptide binds alongside the extended CDR H3 (Fig. 2 ▶), forming a three-stranded mixed β-sheet with CDR H3 (Fig. 4 ▶ a). The majority of contacts between the Fab and peptide are main-chain hydrogen bonds between the N-terminal half of the peptide and the C-terminal portion of the CDR H3 (Fig. 4 ▶ b), although with fewer regular hydrogen bonds to the fourth strand. Five main-chain hydrogen bonds are formed between the following Fab and V3-peptide residues in the two independent complexes: AspH100f N—LysP305 O (3.2/3.1 Å), AspH100f O—IleP307 N (2.8/2.6 Å), TyrH100h N—IleP307 O (2.8/2.8 Å), TyrH100h O—LeuP309 N (2.8/2.8 Å), TyrH100j N—LeuP309 O (3.0/3.0 Å). A sixth hydrogen bond is made between the side chains of HisP308 Nδ1 and TyrH100j OH (2.5/2.6 Å). The peptide is also anchored into the Fab-binding site by a salt bridge between the side chains of ArgP315 NH1 and AspH95 Oδ2 (2.5/2.5 Å; Fig. 4 ▶ c) and ArgP315 also forms π–cation interactions with TrpH33 and TyrH100j. The guanidinium moiety of ArgP315 is sandwiched between the indole ring of TrpH33 and the phenyl ring of TyrH100j, with distances of approximately 3.7 and 3.5 Å from the N∊ atom of ArgP315 to the center of the tyrosine and tryptophan rings, respectively. ProP313 is cradled between the indoles of TrpL91 and TrpL96 in the antibody-combining site (Fig. 4 ▶ c). The conserved V3-crown motif, GPGR, forms a type II β-turn (ϕi+1 = −71°, ψi+1 = 138° for ProP313, ϕi+2 = 78°, ψi+2 = 2° for GlyP314), as previously observed in the Fab 447-52D in complex with an MN V3 peptide (Stanfield et al., 2004 ▶).
Figure 4.

Binding-site interactions between the UG1033 V3 peptide and Fab 447-52D. The Fab CDR H3 is shown in violet and the UG1033 peptide in yellow. (a) The mixed three-strand β-sheet formed by CDR H3 and the V3 peptide is shown. (b) The main-chain hydrogen-bond interactions in the three-strand β-sheet are represented by black dotted lines. (c) Side-chain interactions between the UG1033 peptide and the Fab light chain (cyan) and heavy chain (violet).
110 and 141 van der Waals interactions are made in the two Fab–peptide complexes, respectively. The buried molecular-surface areas of the Fab and peptide are 553/587 and 522/487 Å2 for the two Fab–peptide complexes. The buried surface area of the Fab is largely formed by aromatic tyrosine and tryptophan residues (69%/65%), followed by acidic (19%/20%), basic (6.4%/6.2%), polar (2.4%/4.1%) and small and/or hydrophobic residues (3.3%/4.9%).
3.4. Comparison with the Fab 447-52D–MN V3-peptide complex
Only minor differences are observed between the structure of the Fab 447-52D complex reported above and the Fab 447-52D–MN V3 peptide complex previously reported by our group (Stanfield et al., 2004 ▶) based on comparisons calculated using the first molecule of the asymmetric unit of each crystal structure. When the most highly ordered residues in the peptides are superimposed (LysP305–AlaP316), the r.m.s. deviation (r.m.s.d.) of the Cα backbone for the different V3 peptides is 0.45 Å, with the largest deviation (1.0 Å) being at AlaP316 (Fig. 5 ▶). The UG1033 V3-peptide structure extends an additional three residues beyond AlaP316, although the density for the extra three residues is weak. In the UG1033 V3-peptide structure, TyrP318 contacts a symmetry-related LysL156 in the second molecule in the asymmetric unit, but not in the first molecule. This contact is not made in the MN V3-peptide complex owing to a slight difference in the packing of the molecules. It is not clear whether this contact has helped to stabilize these residues or whether the stronger peptide density is a function of the better diffraction of the UG1033 V3-peptide complex crystals. The GPGR crown motif also adopts a type II β-turn in each of the two structures. The two different Fab 447-52D structures also do not differ significantly, with Cα backbone r.m.s.d. values of 0.24 Å for the VL domain and 0.34 Å for the VH domain. Notably, inclusion of an additional three residues in the C-terminal portion of the UG1033 peptide also does not increase the peptide-binding interactions with Fab 447-52D.
Figure 5.
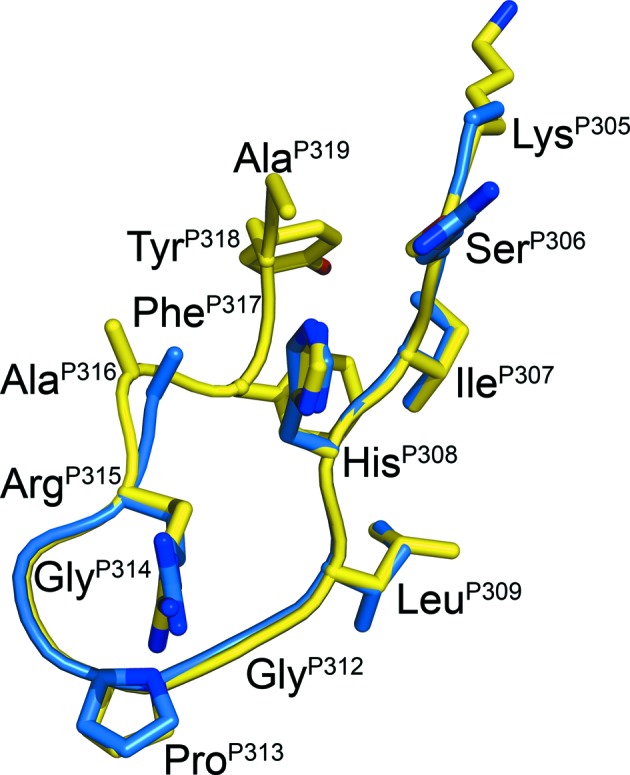
Comparison of the UG1033 and MN peptides bound to Fab 447-52D. A superposition of the Cα backbones of the UG1033 V3 peptide bound to Fab 447-52D and the MN V3-peptide structure previously solved in complex with Fab 447-52D is shown. The UG1033 peptide is shown in yellow and the MN peptide in blue. The UG1033-peptide residues are labeled.
The UG1033 V3-peptide sequence differs from the MN peptide at two positions. An arginine is present at position P306 and an isoleucine at position P309 in the MN peptide, whereas serine and leucine are found in these positions, respectively, in the UG1033 peptide. However, as expected, these differences in sequence do not affect the main-chain hydrogen bonds that the peptide forms with Fab 447-52D CDR H3. Although P317–P319 at the C-terminus of the UG1033 peptide do not interact with the Fab, they contact the N-terminal strand of the V3 peptide through main-chain hydrogen bonds to form a more regular β-hairpin-type structure. An evaluation of hydrogen-bond contacts detects additional hydrogen bonds in each peptide between PheP317 O and HisP308 N (2.8/2.6 Å) and a second hydrogen bond in the second peptide between AlaQ319 N and SerQ306 O (2.7 Å). The distance between AlaP319 N and SerP306 O increases slightly in the first peptide to 3.1 Å, although the density at the C-terminus is weaker.
3.5. Comparison with Fab 2219–UG1033 and gp120 V3 core structures
The UG1033 peptide structure has also been determined in complex with the V3-specific Fab 2219 (Stanfield, Gorny et al., 2006 ▶). As observed here, the GPGR motif also forms a type II β-turn in the Fab 2219 structure, with a Cα r.m.s.d. of 0.21 Å to the Fab 447-52D structure (GlyP312–ArgP315). However, a difference in one of the dihedral angles (ψ) of LeuP309 results in a change in the orientation of the peptide backbone (Table 3 ▶). Hence, the carbonyl O atoms of the N-terminal residues preceding LeuP309 are rotated by about 180° in the Fab 2219 complex with respect to the conformation seen in the Fab 447-52D complex (Fig. 6 ▶). Despite this difference, the N- and C-terminal strands of the hairpin independently show close structural homology with the UG1033 peptide in the Fab 447-52D structure, with Cα backbone r.m.s.d. values of 0.60 Å (LysP305—GlyP312) and 0.86 Å (AlaP316—AlaP319).
Table 3. Backbone torsion angles for the UG1033 peptide structures complexed with Fab 447-52D and Fab 2219 and the equivalent V3 loop from a gp120 structure.
| (), UG1033 V3 in | (), UG1033 V3 in | |||||
|---|---|---|---|---|---|---|
| Residue | Fab 447-52D | Fab 2219† | gp120 V3‡ | Fab 447-52D | Fab 2219† | gp120 V3‡ |
| 305 | n/a | 78 | 124 | 135 | 30 | 12 |
| 306 | 140 | 70 | 147 | 125 | 138 | 146 |
| 307 | 103 | 109 | 118 | 138 | 120 | 160 |
| 308 | 125 | 85 | 129 | 114 | 106 | 121 |
| 309 | 113 | 73 | 84 | 94 | 39 | 140 |
| 312 | 170 | 165 | 158 | 171 | 175 | 131 |
| 313 | 71 | 60 | 59 | 138 | 126 | 99 |
| 314 | 78 | 71 | 100 | 2 | 12 | 8 |
| 315 | 91 | 139 | 172 | 134 | 144 | 149 |
| 316 | 69 | 158 | 13 | 190 | 154 | 167 |
| 317 | 21 | 109 | 133 | 61 | 157 | 153 |
| 318 | 87 | 54 | 60 | 158 | 38 | 65 |
| 319 | 156 | 88 | 150 | n/a | 170 | 2 |
Figure 6.

Comparison of V3 crystal structures. The Cα-backbone structures are shown in a similar orientations for (a) the UG1033 V3 peptide bound to Fab 447-52D (b) the UG1033 V3 peptide bound to Fab 2219 (PDB code 2b1a) and the V3 loops from two gp120 core complexes: (c) PDB code 2b4c and (d) PDB code 2qad. Two conformations were observed for the V3 crown in the core gp120 V3 structure with PDB code 2b4c and both of these conformations are shown in (c).
When bound to Fab 2219, the majority of contacts are made through side chains of residues in the N-terminal strand of the UG1033 peptide, although two side-chain contacts from C-terminal residues are also observed. Despite making contact with the Fab 2219, N-terminal residues ThrP303–SerP306 are still solvent-exposed. It is likely that Fab 2219 is able to tolerate diversity in side chains at these positions because of the solvent-accessible nature of these residues. In contrast, the use of main-chain hydrogen-bond contacts allows Fab 447-52D to tolerate diversity within the N-terminal region of the V3. In both structures, ArgP315 makes side-chain contacts with the Fab and forms salt bridges to AsnL31 in Fab 2219 and to AspH95 in Fab 447-52D. However, the UG1033 V3 crown is also largely accessible in the Fab 2219 complex, whereas it is less accessible and oriented into the antibody-binding pocket in the Fab 447-52D complex reported here.
The structure of the UG1033 V3 peptide in the Fab 447-52D complex also reveals structural homology to the V3 region of gp120-containing V3 crystal structures (Fig. 6 ▶). The Cα backbone r.m.s.d. values of the UG1033 peptide with the corresponding V3 loop in the reported gp120 V3 structure (Huang et al., 2005 ▶) are 0.56 and 0.85 Å for the N-terminal (LysP305–GlyP312) and C-terminal (AlaP316–AlaP319) strands respectively, and 0.59 Å for the GPGR crown motif. A gp120-containing V3 structure in complex with a tyrosine-sulfated antibody has recently been reported (Huang et al., 2007 ▶). The GPGR crown of the V3 loop in this structure exhibits even greater structural homology with the crown of the UG1033 V3 peptide, with a Cα backbone r.m.s.d. value of only 0.16 Å.
3.6. Comparison with NMR structures
The structures of MN and IIIB V3 peptides in complex with 447-52D Fv have also been determined by NMR methods. Solution (Sharon et al., 2003 ▶) and solid-state (Sharpe et al., 2004 ▶) NMR studies of MN V3 peptides (residues 303–325 and 305–322 of the MN V3 loop, respectively) in complex with 447-52D Fv show an overall similarity to the structure reported here (Fig. 7 ▶). In the NMR studies, the MN peptide forms a β-hairpin stabilized by intrapeptide hydrogen-bond interactions (Gly312 O—Gly314 N, Arg315 O—Thr319 N, Thr319 O—Arg315 N, His308 O—Phe317 N and Phe317 O—His308 N) with an inverse γ-turn at the crown. The extended C-terminal portion of the β-hairpin is not observed here, which is likely to be the consequence of flexibility of the C-terminal residues in the UG1033 peptide. However, the overall shape of the peptides is similar (Fig. 7 ▶), with a Cα r.m.s.d. of 1.7 Å for LysP305–AlaP319. In the MN–447-52D Fv solution NMR structure, strong interactions between the Fv and peptide are reported for N-terminal peptide residues LysP305–ArgP315 and weaker interactions are reported for the C-terminal AlaP316–ThrP320. Although the same C-terminal interactions are not observed here, the N-terminal residues of the UG1033 peptide still dominate binding to Fab 447-52D. Moreover, in the solid-state NMR structure of the MN V3–447-52D Fv complex, the C-terminal interactions could not be observed. The side chains of GlyP312, ProP313 and ArgP315 are also reported to interact with aromatic rings of the Fv in the NMR structures, which is in agreement with our observation that ProP313 is flanked by the indoles of TrpL91 and TrpL96 and that ArgP315 makes π–cation interactions with TyrH100j and TrpH33.
Figure 7.

Comparison of the crystal structure of the UG1033 peptide bound to Fab 447-52D with the NMR structures of the MN and IIIB peptides bound to 447-52D Fv. The Cα-backbone structures are shown in a similar orientation for (a) the UG1033 V3 peptide from the crystal structure and (b) MN (PDB code 1n1z) and (c) IIIB (PDB code 1u6u) from NMR structures. The N-terminus of each peptide is oriented to the left.
The NMR structure of a IIIB V3 peptide (residues 303–322) in complex with 447-52D Fv has also been reported (Rosen et al., 2005 ▶). The V3 loop of HIV-1IIIB contains a two-residue insertion (QR) before the GPGR crown sequence. In the IIIB V3–Fv structure the peptide forms a β-hairpin, although the QR insertion creates a kink before the β-turn at the crown. Despite the two-residue insertion, the resulting structure is very similar to the UG1033 crystal structure and the MN NMR structure (Fig. 7 ▶) discussed above, with the N-terminal residues dominating binding to 447-52D. The similarity of the overall peptide conformation is evident when the UG1033 N-terminal strand (LysP305–LeuP309), β-turn (GlyP312–ArgP315) and C-terminal strand (AlaP316–AlaP319) are superimposed with the corresponding IIIB peptide residues, giving Cα backbone r.m.s.d. values of 0.64, 0.30 and 1.82 Å, respectively. Based on a 2D-homonuclear TOSCY spectrum, Rosen and coworkers also conclude that IIIB residues IleP309 and GlyP312 are likely to interact with aromatic tyrosine side chains of the Fv and ProP313 and GlyP314 are likely to interact with tryptophan residues. In the UG1033 structure, LeuP309 is positioned near TyrH100i and TyrL32, and GlyP312, ProP313 and GlyP314 are close to TrpL91, TrpL96 and TrpH33. These aromatic contacts are not identical to those proposed in the IIIB–447-52D Fv NMR structure. However, as previously suggested (Rosen et al., 2005 ▶), the QR insertion in the IIIB peptide may alter the positioning of the peptide in the 447-52D binding site.
4. Conclusions
The structure of the UG1033 peptide in complex with Fab 447-52D has been determined from an epitaxially twinned data set where the individual lattices were separated and overlapping reflections were removed. The final R cryst and R free values of the model are higher than expected at this resolution, which is likely to be the consequence of some residual overlapping spots (diffraction maxima) that were not removed by the procedure. However, given the quality and resolution of the electron-density maps, the Fab–peptide complex can still be determined with confidence. Moreover, this structure shows very high structural homology to the crystal structure of the MN V3 peptide in complex with Fab 447-52D previously reported by our group (Ghiara et al., 1994 ▶, 1997 ▶; Rini et al., 1993 ▶; Stanfield et al., 1993 ▶, 1999 ▶, 2003 ▶, 2004 ▶; Stanfield & Wilson, 2005 ▶) and to the NMR structure (Sharon et al., 2003 ▶), further validating the final model built from the epitaxially twinned data set.
The structure of the Fab 447-52D complex reported here confirms our previous conclusion that the breadth of this neutralizing antibody is attributable to the use of main-chain hydrogen bonds to contact the N-terminal region of the V3-loop backbone (Stanfield et al., 2004 ▶). This mode of antigen recognition enables 447-52D to bind a diverse range of V3 sequences. We observe here that a two-residue sequence difference between the UG1033 peptide and the MN peptide previously determined in complex with Fab 447-52D (ArgP306–SerP306 and IleP309–LeuP309) does not alter the binding interactions between the V3 peptides and Fab. The reported dependence of antigen recognition on the presence of the conserved crown motif GPxR (Keller et al., 1993 ▶) can be explained in part by the observation that the crown and in particular ArgP315 form specific interactions with the antibody-binding site. Finally, the V3-peptide structure shows a high level of structural homology with previously reported V3 crystal and NMR structures, supporting the hypothesis that the V3 loop of gp120 may adopt a conserved structure or limited set of structures, particularly at the crown (Zolla-Pazner, 2004 ▶; Ghiara et al., 1994 ▶, 1997 ▶; Rini et al., 1993 ▶; Stanfield et al., 1993 ▶, 1999 ▶, 2003 ▶, 2004 ▶; Stanfield, Gorny et al., 2006 ▶; Stanfield & Wilson, 2005 ▶; Cardozo et al., 2007 ▶; Rosen et al., 2006 ▶). Future structural studies of such V3-specific neutralizing antibodies in complex with a diverse range of V3 peptides will help to further clarify conserved structural features of the V3 loop, as well as the precise mechanism by which V3 antibodies recognize their epitopes. This information will in turn provide valuable insight into how the V3 region of gp120 may be utilized as an anti-HIV-1 immunogen that may provide some efficacy in combination with immunogens corresponding to other epitopes.
Supplementary Material
PDB reference: anti-HIV-1 Fab 447-52D–peptide complex, 3c2a, r3c2asf
Acknowledgments
We gratefully acknowledge support by NIH grants GM-46192 (to IAW and RLS), AI36085 (SZP) and HL59725 (SZP), by the NYU Center for AIDS Research (AI27742 to SZP), by the Bill and Melinda Gates Foundation (38631 to SZP), by research funds from the Department of Veterans Affairs (SZP), by the Natural Sciences and Engineering Research Council of Canada (AKD) and by the International AIDS Vaccine Initiative Neutralizing Antibody Consortium (IAW). This is manuscript No. 19279 from The Scripps Research Institute.
References
- Back, N. K., Smit, L., De Jong, J. J., Keulen, W., Schutten, M., Goudsmit, J. & Tersmette, M. (1994). Virology, 199, 431–438. [DOI] [PubMed]
- Basmaciogullari, S., Babcock, G. J., Van Ryk, D., Wojtowicz, W. & Sodroski, J. (2002). J. Virol. 76, 10791–10800. [DOI] [PMC free article] [PubMed]
- Binley, J. M., Wrin, T., Korber, B., Zwick, M. B., Wang, M., Chappey, C., Stiegler, G., Kunert, R., Zolla-Pazner, S., Katinger, H., Petropoulos, C. J. & Burton, D. R. (2004). J. Virol. 78, 13232–13252. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Burton, D. R., Stanfield, R. L. & Wilson, I. A. (2005). Proc. Natl Acad. Sci. USA, 102, 14943–14948. [DOI] [PMC free article] [PubMed]
- Cardozo, T., Kimura, T., Philpott, S., Weiser, B., Burger, H. & Zolla-Pazner, S. (2007). AIDS Res. Hum. Retroviruses, 23, 415–426. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Connolly, M. L. (1993). J. Mol. Graph. 11, 139–141. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Gao, F. et al. (2005). J. Virol. 79, 1154–1163. [DOI] [PMC free article] [PubMed]
- Ghiara, J. B., Ferguson, D. C., Satterthwait, A. C., Dyson, H. J. & Wilson, I. A. (1997). J. Mol. Biol. 266, 31–39. [DOI] [PubMed]
- Ghiara, J. B., Stura, E. A., Stanfield, R. L., Profy, A. T. & Wilson, I. A. (1994). Science, 264, 82–85. [DOI] [PubMed]
- Gorny, M. K., Conley, A. J., Karwowska, S., Buchbinder, A., Xu, J. Y., Emini, E. A., Koenig, S. & Zolla-Pazner, S. (1992). J. Virol. 66, 7538–7542. [DOI] [PMC free article] [PubMed]
- Gorny, M. K., Gianakakos, V., Sharpe, S. & Zolla-Pazner, S. (1989). Proc. Natl Acad. Sci. USA, 86, 1624–1628. [DOI] [PMC free article] [PubMed]
- Gorny, M. K., Revesz, K., Williams, C., Volsky, B., Louder, M. K., Anyangwe, C. A., Krachmarov, C., Kayman, S. C., Pinter, A., Nadas, A., Nyambi, P. N., Mascola, J. R. & Zolla-Pazner, S. (2004). J. Virol. 78, 2394–2404. [DOI] [PMC free article] [PubMed]
- Gorny, M. K., Williams, C., Volsky, B., Revesz, K., Cohen, S., Polonis, V. R., Honnen, W. J., Kayman, S. C., Krachmarov, C., Pinter, A. & Zolla-Pazner, S. (2002). J. Virol. 76, 9035–9045. [DOI] [PMC free article] [PubMed]
- Gorny, M. K., Williams, C., Volsky, B., Revesz, K., Wang, X. H., Burda, S., Kimura, T., Konings, F. A., Nadas, A., Anyangwe, C. A., Nyambi, P., Krachmarov, C., Pinter, A. & Zolla-Pazner, S. (2006). J. Virol. 80, 6865–6872. [DOI] [PMC free article] [PubMed]
- Gorny, M. K., Xu, J. Y., Gianakakos, V., Karwowska, S., Williams, C., Sheppard, H. W., Hanson, C. V. & Zolla-Pazner, S. (1991). Proc. Natl Acad. Sci. USA, 88, 3238–3242. [DOI] [PMC free article] [PubMed]
- Gorny, M. K., Xu, J. Y., Karwowska, S., Buchbinder, A. & Zolla-Pazner, S. (1993). J. Immunol. 150, 635–643. [PubMed]
- Goudsmit, J., Debouck, C., Meloen, R. H., Smit, L., Bakker, M., Asher, D. M., Wolff, A. V., Gibbs, C. J. Jr. & Gajdusek, D. C. (1988). Proc. Natl Acad. Sci. USA, 85, 4478–4482. [DOI] [PMC free article] [PubMed]
- Hartley, O., Klasse, P. J., Sattentau, Q. J. & Moore, J. P. (2005). AIDS Res. Hum. Retroviruses, 21, 171–189. [DOI] [PubMed]
- Hoffman, T. L. & Doms, R. W. (1999). Mol. Membr. Biol. 16, 57–65. [DOI] [PubMed]
- Huang, C. C., Lam, S. N., Acharya, P., Tang, M., Xiang, S. H., Hussan, S. S., Stanfield, R. L., Robinson, J., Sodroski, J., Wilson, I. A., Wyatt, R., Bewley, C. A. & Kwong, P. D. (2007). Science, 317, 1930–1934. [DOI] [PMC free article] [PubMed]
- Huang, C. C., Tang, M., Zhang, M. Y., Majeed, S., Montabana, E., Stanfield, R. L., Dimitrov, D. S., Korber, B., Sodroski, J., Wilson, I. A., Wyatt, R. & Kwong, P. D. (2005). Science, 310, 1025–1028. [DOI] [PMC free article] [PubMed]
- Javaherian, K., Langlois, A. J., LaRosa, G. J., Profy, A. T., Bolognesi, D. P., Herlihy, W. C., Putney, S. D. & Matthews, T. J. (1990). Science, 250, 1590–1593. [DOI] [PubMed]
- Javaherian, K., Langlois, A. J., McDanal, C., Ross, K. L., Eckler, L. I., Jellis, C. L., Profy, A. T., Rusche, J. R., Bolognesi, D. P., Putney, S. D. & Matthews, T. J. (1989). Proc. Natl Acad. Sci. USA, 86, 6768–6772. [DOI] [PMC free article] [PubMed]
- Jones, T. A. (1982). FRODO: A Graphics Fitting Program for Macromolecules. Oxford: Clarendon Press.
- Kabat, E. A., Wu, T. T., Reid-Miller, M., Perry, H. M. & Gottesman, K. S. (1987). Sequences of Proteins of Immunological Interest, 4th ed. Bethesda: National Institutes of Health.
- Keller, P. M., Arnold, B. A., Shaw, A. R., Tolman, R. L., Van Middlesworth, F., Bondy, S., Rusiecki, V. K., Koenig, S., Zolla-Pazner, S., Conard, P., Emini, E. A. & Conley, A. J. (1993). Virology, 193, 709–716. [DOI] [PubMed]
- Koch, M., Pancera, M., Kwong, P. D., Kolchinsky, P., Grundner, C., Wang, L., Hendrickson, W. A., Sodroski, J. & Wyatt, R. (2003). Virology, 313, 387–400. [DOI] [PubMed]
- Krachmarov, C., Pinter, A., Honnen, W. J., Gorny, M. K., Nyambi, P. N., Zolla-Pazner, S. & Kayman, S. C. (2005). J. Virol. 79, 780–790. [DOI] [PMC free article] [PubMed]
- Lietzke, S. E., Carperos, V. E. & Kundrot, C. E. (1996). Acta Cryst. D52, 687–692. [DOI] [PubMed]
- McCaffrey, R. A., Saunders, C., Hensel, M. & Stamatatos, L. (2004). J. Virol. 78, 3279–3295. [DOI] [PMC free article] [PubMed]
- McDonald, I. K. & Thornton, J. M. (1994). J. Mol. Biol. 238, 777–793. [DOI] [PubMed]
- Matthews, B. W. (1985). Methods Enzymol. 114, 176–187. [DOI] [PubMed]
- Mbah, H. A., Burda, S., Gorny, M. K., Williams, C., Revesz, K., Zolla-Pazner, S. & Nyambi, P. N. (2001). J. Virol. 75, 7785–7788. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pantophlet, R., Aguilar-Sino, R. O., Wrin, T., Cavacini, L. A. & Burton, D. R. (2007). Virology, 364, 441–453. [DOI] [PMC free article] [PubMed]
- Pantophlet, R. & Burton, D. R. (2006). Annu. Rev. Immunol. 24, 739–769. [DOI] [PubMed]
- Pinter, A., Honnen, W. J., He, Y., Gorny, M. K., Zolla-Pazner, S. & Kayman, S. C. (2004). J. Virol. 78, 5205–5215. [DOI] [PMC free article] [PubMed]
- Ratner, L., Fisher, A., Jagodzinski, L. L., Mitsuya, H., Liou, R. S., Gallo, R. C. & Wong-Staal, F. (1987). AIDS Res. Hum. Retroviruses, 3, 57–69. [DOI] [PubMed]
- Richman, D. D., Wrin, T., Little, S. J. & Petropoulos, C. J. (2003). Proc. Natl Acad Sci. USA, 100, 4144–4149. [DOI] [PMC free article] [PubMed]
- Rini, J. M., Stanfield, R. L., Stura, E. A., Salinas, P. A., Profy, A. T. & Wilson, I. A. (1993). Proc. Natl Acad. Sci. USA, 90, 6325–6329. [DOI] [PMC free article] [PubMed]
- Rizzuto, C. & Sodroski, J. (2000). AIDS Res. Hum. Retroviruses, 16, 741–749. [DOI] [PubMed]
- Rizzuto, C. D., Wyatt, R., Hernandez-Ramos, N., Sun, Y., Kwong, P. D., Hendrickson, W. A. & Sodroski, J. (1998). Science, 280, 1949–1953. [DOI] [PubMed]
- Rosen, O., Chill, J., Sharon, M., Kessler, N., Mester, B., Zolla-Pazner, S. & Anglister, J. (2005). Biochemistry, 44, 7250–7258. [DOI] [PubMed]
- Rosen, O., Samson, A. O., Sharon, M., Zolla-Pazner, S. & Anglister, J. (2006). Structure, 14, 649–651.
- Rusche, J. R., Javaherian, K., McDanal, C., Petro, J., Lynn, D. L., Grimaila, R., Langlois, A., Gallo, R. C., Arthur, L. O., Fischinger, P. J., Bolognesi, D. P., Putney, S. D. & Matthews, T. J. (1988). Proc. Natl Acad. Sci. USA, 85, 3198–3202. [DOI] [PMC free article] [PubMed]
- Schulze-Gahmen, U., Rini, J. M. & Wilson, I. A. (1993). J. Mol. Biol. 234, 1098–1118. [DOI] [PubMed]
- Sharon, M., Kessler, N., Levy, R., Zolla-Pazner, S., Gorlach, M. & Anglister, J. (2003). Structure, 11, 225–236. [DOI] [PubMed]
- Sharpe, S., Kessler, N., Anglister, J. A., Yau, W. M. & Tycko, R. (2004). J. Am. Chem. Soc. 126, 4979–4990. [DOI] [PubMed]
- Sheriff, S., Hendrickson, W. A. & Smith, J. L. (1987). J. Mol. Biol. 197, 273–296. [DOI] [PubMed]
- Stanfield, R., Cabezas, E., Satterthwait, A., Stura, E., Profy, A. & Wilson, I. (1999). Structure, 7, 131–142. [DOI] [PubMed]
- Stanfield, R. L., Ghiara, J. B., Ollmann Saphire, E., Profy, A. T. & Wilson, I. A. (2003). Virology, 315, 159–173. [DOI] [PubMed]
- Stanfield, R. L., Gorny, M. K., Williams, C., Zolla-Pazner, S. & Wilson, I. A. (2004). Structure, 12, 193–204. [DOI] [PubMed]
- Stanfield, R. L., Gorny, M. K., Zolla-Pazner, S. & Wilson, I. A. (2006). J. Virol. 80, 6093–6105. [DOI] [PMC free article] [PubMed]
- Stanfield, R. L., Takimoto-Kamimura, M., Rini, J. M., Profy, A. T. & Wilson, I. A. (1993). Structure, 1, 83–93. [DOI] [PubMed]
- Stanfield, R. L. & Wilson, I. A. (2005). Hum. Antibodies, 14, 73–80. [PubMed]
- Stanfield, R. L., Zemla, A., Wilson, I. A. & Rupp, B. (2006). J. Mol. Biol. 357, 1566–1574. [DOI] [PubMed]
- Wei, X., Decker, J. M., Wang, S., Hui, H., Kappes, J. C., Wu, X., Salazar-Gonzalez, J. F., Salazar, M. G., Kilby, J. M., Saag, M. S., Komarova, N. L., Nowak, M. A., Hahn, B. H., Kwong, P. D. & Shaw, G. M. (2003). Nature (London), 422, 307–312. [DOI] [PubMed]
- Wyatt, R., Kwong, P. D., Desjardins, E., Sweet, R. W., Robinson, J., Hendrickson, W. A. & Sodroski, J. G. (1998). Nature (London), 393, 705–711. [DOI] [PubMed]
- Zanetti, G., Briggs, J. A., Grunewald, K., Sattentau, Q. J. & Fuller, S. D. (2006). PLoS Pathog. 2, e83. [DOI] [PMC free article] [PubMed]
- Zhu, P., Liu, J., Bess, J. Jr, Chertova, E., Lifson, J. D., Grize, H., Ofek, G. A., Taylor, K. A. & Roux, K. H. (2006). Nature (London), 441, 847–852. [DOI] [PubMed]
- Zolla-Pazner, S. (2004). Nature Rev. Immunol. 4, 199–210. [DOI] [PMC free article] [PubMed]
- Zolla-Pazner, S., Zhong, P., Revesz, K., Volsky, B., Williams, C., Nyambi, P. & Gorny, M. K. (2004). AIDS Res. Hum. Retroviruses, 20, 1254–1258. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: anti-HIV-1 Fab 447-52D–peptide complex, 3c2a, r3c2asf