

Abstract
Previously, we demonstrated that protein kinase D (PKD) plays a protective role during H2O2-induced intestinal cell death. Here, we sought to determine whether this effect is mediated by nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs). Treatment with H2O2 activated NF-κB in RIE-1 cells; H2O2 also induced the translocation of NF-κB p65 as well as phosphorylation of IκB-α. PKD1 siRNA inhibited H2O2-induced activation, translocation of NF-κB and phosphorylation of IκB-α. We also found that overexpression of wild type PKD1 attenuated H2O2-induced phosphorylation of p38 MAPK and its upstream activator, MAPK kinase (MKK) 6 and 3, whereas the phosphorylation was increased by PKD1 siRNA or kinase-dead PKD1. Phosphorylation of neither extracellular signal-regulated kinases (ERK) 1/2 nor c-Jun N-terminal kinases (JNK) was altered by PKD1 plasmids or siRNA. Our findings suggest that PKD protects intestinal cells through up-regulation of NF-κB and down-regulation of p38 MAPK.
Keywords: Protein kinase D, Oxidative stress, NF-κB, p38 MAPK
INTRODUCTION
The ability of the cells to resist injury from reactive oxygen species (ROS) is a critical survival mechanism in response to a variety of environmental stresses [1]. ROS participates in the activation of intracellular signaling pathways, including NF-κB and MAPKs. The contribution of oxidative stress to intracellular signaling pathways has become a common theme of investigation in the area of infection/inflammation [2]. NF-κB is a transcription factor consisting of a group of five proteins. In the resting state, NF-κB is sequestered in the cytoplasm with specific inhibitory proteins, IκB [3]. In response to various stimuli, the IκBs are rapidly phosphorylated, leading to rapid translocation of NF-κB to the nucleus to activate transcription of specific target genes [4]. Because oxidative stress and NF-κB activation both have important roles in inflammation, the effects of ROS on NF-κB or their pathways have received considerable attention [3].
MAPKs encompass a large number of kinases involved in regulating a wide array of cellular processes. Based on structural differences, they are divided into three multimember subfamilies: ERK1/2, JNK, and p38 MAPK. They have all been shown to activate in response to oxidant injury [2]. We have demonstrated that H2O2 treatment results in increased JNK, ERK1/2 and p38 MAPK phosphorylation in intestinal epithelial cells; the inhibition of this process protected these cells from apoptosis [5; 6]. The ERK1/2, JNK, and p38 MAPK subfamilies are activated via independent (at times overlapping) signaling cascades involving a MKK that is responsible for phosphorylation of the MAPK, and a MAPK kinase kinase (MKKK) that phosphorylates and activates MKK [2]. MAPK activity is activated by specific MKK: MEK1/2 for ERK1/2, MKK3/6 for the p38 MAPK, MKK4/7 for the JNK [7].
PKD1, also known as protein kinase Cµ [8], is a serine/threonine protein kinase with unique structural, enzymological, and regulatory properties that are different from those of the PKC family members. PKD has been implicated in many important intracellular signal transduction pathways via PKC-dependent mechanisms [9; 10]. The survival effect of PKD was demonstrated to occur through activation of NF-κB [11]. Inhibition of PKD function blocked NF-κB activation and sensitized cells to death from H2O2 [11]. Therefore, a model has been proposed in which PKD acts as a central integrator of mitochondrial oxidative stress responses, such that NF-κB modulates the induction of MnSOD and promotes cell survival [12]. We have previously shown that PKD plays an important protective role for cell survival during oxidative stress-induced injury [13]. However, the downstream mechanisms of PKD activation have not been identified upon oxidative stress in these cells.
In this study, we sought to determine the role of NF-κB and MAPKs signaling in intestinal epithelial cell line, RIE-1, and to identify which MAPKs subfamily is involved in the PKD-mediated survival pathway. Lastly, we attempted to assess whether the anti-apoptosis effects of PKD is mediated through modulation of NF-κB or MAPKs.
MATERIALS AND METHODS
Reagents and antibodies
The GST-tagged PKD1 plasmids were provided by Dr. Vivek Malhotra (University of California, San Diego). PKD1 siRNA and the non-specific control siRNA were obtained from Dharmacon, Inc. (Lafayette, CO). The luciferase reporter gene construct containing the NF-κB promoter element was designed (SBE-Luc). Lipofectamine 2000 reagent was purchased from Invitrogen (Carlsbad, CA, USA). PKD, IκBα, NF-κB p65 polyclonal antibodies, goat anti-mouse and rabbit antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-p38, p38, phospho-p44/42 MAPK, phospho-SAPK/JNK, phospho-IκBα, phospho-MKK3/6, MKK3 antibodies were purchased from Cell Signaling (Beverly, MA). Alexa Fluor 488 antibody was from Molecular Probes (Eugene, OR). Dual-Luciferase Reporter Assay System was from Promega (Madison, WI). All other reagents were purchased from Sigma (St. Louis, MO).
Cell culture and transfection
The RIE-1 cell lines (originally provided by K. Brown, Babraham Institute, Cambridge, UK) are a diploid, non transformed, crypt-like cell line derived from rat small intestine [14]. For all experiments, cells were used between passages 23-39 and were maintained in DMEM supplemented with 5% fetal bovine serum (FBS) in 5% CO2 at 37°C. Cells were plated in 60-mm dishes and grown to 80-90% confluence and treated with the indicated concentrations of H2O2 at 37°C. siRNA was transfected by electroporation (400V, 500 μF) using GenePulser XCell (Bio-Rad, Hercules, CA). For plasmid experiments, cells were transiently transfected using Lipofectamine 2000 according to the manufacturer's instructions.
Luciferase Reporter Assays
Cells were plated at 2× 104/cm2 cells in 24-well plates the day before transfection using Lipofectamine plus. The pRL-TK vector (Promega) containing the herpes simplex virus thymidine kinase (HSV-TK) promoter driving the expression of a renilla luciferase reporter was used as an internal control for transfection efficiency. Cells were harvested, lysed at 48 h post-transfection. The firefly and renilla luciferase activities were measured using the Promega Dual luciferase assay system with 20 μl of cell extract according to the manufacturer's instructions.
Protein preparation and Western blotting
For total protein, cells were washed with cold phosphate buffered saline (PBS), scraped into lysis buffer containing proteinase inhibitors, and incubated on ice for 30 min. The protein concentration of the supernatants was determined by a Bio-Rad assay (Hercules, CA). Equal amounts of protein were resolved on 4-12% Bis Tris gels (Invitrogen) and electrophoretically transferred to polyvinylidene difluoride membranes. After the nonspecific binding sites were blocked with 5% dried skimmed milk in TBST (120 mM Tris · HCl, pH 7.4, 150 mM NaCl, and 0.05% Tween 20) for 1 h, the membranes were incubated with primary antibodies overnight at 4°C followed by secondary antibodies. Membranes were developed using the ECL detection system.
Immunofluorescent staining and fluorescent microscopy
Cells were grown in chamber slides for two days, and then treated either with or without 500 μM of H2O2 over a time course. After treatment, cells were fixed with 100% cold methanol for 20 min at 4°C. Three washes with PBS, the cells were blocked with 1% bovine serum albumin (BSA)-PBS for 10 min and were incubated with rabbit polyclonal anti-NF-κB p65 antibody diluted 1:100 with 1% BSA-PBS for 45min. Cells were washed three times with PBS and incubated with Alexa 488-conjugated anti-rabbit secondary antibody diluted 1:500 in 1% BSA-PBS. The fluorescence of NF-κB p65 immunoreactivity was observed under a fluorescent microscope.
Statistical analysis
All experiments were repeated at least three times and data are reported as mean ± SEM. Data was analyzed using the Kruskal-Wallis test due to heterogeneous variability in each group. All tests were assessed at the 0.05 level of significance. All statistical computations were conducted using the SAS® system, Release 8.2 (SAS Institute Inc. SAS/STAT® User's Guide, Version 8. Cary, NC: SAS Institute Inc., 1999).
RESULTS
H2O2 induces the activation of NF-κB and IκBα phosphorylation in RIE-1 cells
We assessed NF-κB promoter activity stimulated with H2O2 by transfection of the NF-κB-luciferase plasmid, which contains four tandem copies of the NF-κB consensus sequence. Figure 1A shows that H2O2 treatment significantly increased NF-κB luciferase activity compared with no treatment. Immunofluorescence imaging showed nuclear localization of NF-κB using anti-NF-κB p65 antibody. In untreated cells, the NF-κB p65 was mainly present in the cytoplasm (Fig. 1B; upper left panel). Immediately upon treatment with H2O2 at 1 min, NF-κB p65 localized to the perinuclear area of the cells (Fig. 1B; upper right panel). The nuclear localization of activated NF-κB p65 occurred as early as 5 min after H2O2 exposure and this continued to increase over a 15 min period (Fig. 1B; lower panels). Upon stimulation, IκBα phosphorylation occurred and this resulted in release and translocation of NF-κB to the nucleus [15]. To determine the phosphorylation of IκBα, RIE-1 cells were treated with 500 μM of H2O2 over a time course and Western blotting was performed. IκBα phosphorylation occurred rapidly at 5 min and peaked at 20 min after H2O2 treatment; this rapid increase in IκBα phosphorylation continued to the 60 min timepoint (Fig.1C; upper row).
Fig. 1. H2O2 induces the activation of NF-κB, translocation of NF-κB p65 and IκBα phosphorylation in RIE-1 cells.
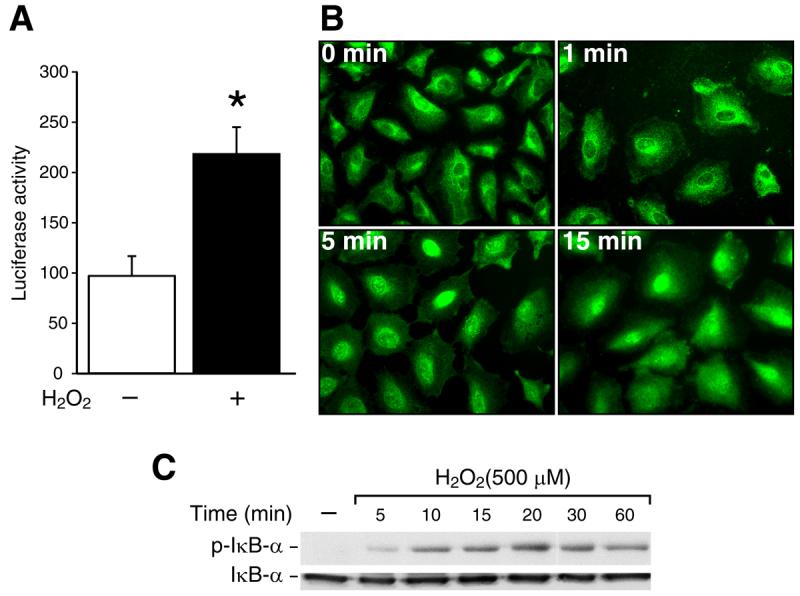
(A) Cells were transfected with 1 μg of a plasmid containing the NF-κB promoter fragment. After 42 h incubation, the transfected cells were treated with H2O2 (100 μM) for 6 h and luciferase activity measured in the cell lysates. The results were normalized for transfection efficiency using the pRL-Tk-luc plasmid [*=p< 0.05 vs. control (−)]. (B) Cells were grown on chamber slides for 2 days then treated with H2O2 (500 μM) or vehicle for 1, 5, 15 min. A representative photomicrograph is shown demonstrating NF-κB p65 localization in using immunofluorescent imaging. (C) After cells were treated with H2O2 (500 μM) over a time course, IκBα phosphorylation was detected by Western blotting using anti-phospho-IκBα (Ser32) antibody. IκBα was used as a loading control.
PKD1 siRNA inhibits H2O2-induced NF-κB activation and IαBαphosphorylation
To determine whether the cellular protective effect of PKD is mediated through the NF-κB pathway, RIE-1 cells were co-transfected with PKD1 siRNA and NF-κB promoter luciferase reporter construct and treated with H2O2. At 72 h after transfection, NF-κB promoter activity was measured by luciferase assay (Fig. 2A; upper panel). H2O2 significantly enhanced NF-κB activity in the cells transfected with control siRNA. Importantly, H2O2-induced NF-κB activity was reduced in the cells transfected with PKD1 siRNA compared with the cells transfected with control siRNA, thus strongly suggesting that the suppression of endogenous PKD1 decreases H2O2-induced NF-κB activity. Western blotting confirms the specificity of PKD1 siRNA transfection (Fig. 2A; lower panel) Next, immunofluorescent staining was performed using anti-p65 antibody in cells transfected with PKD1 siRNA. In untreated cells, NF-κB was noted in the cytoplasm as well as the nucleus in the cells transfected with either control or PKD1 siRNA. After treatment with H2O2, NF-κB p65 translocated from cytoplasm to the nucleus in cells transfected with control siRNA (Fig. 2B; upper right panel). In contrast, NF-κB p65 remained largely in the cytoplasm in cells transfected with PKD1 siRNA (Fig. 2B; bottom right panel). Furthermore, H2O2-induced IκBα phosphorylation was suppressed in PKD1 siRNA-transfected cells, as determined by Western blotting (Fig. 2C; middle row). Altered total PKD protein expression indicates specificity of PKD1 siRNA (Fig. 2C; top row).
Fig. 2. PKD1 is required for H2O2-stimulated NF-κB activation, translocation and IκBα phosphorylation.

(A) RIE-1 cells were co-transfected with a NF-κB plasmid and control siRNA or PKD1 siRNA. After 42 h incubation, the cells were treated with H2O2 for 6 h, and harvested and luciferase activity measured in the cell lysates [*=p< 0.05 vs. control siRNA (−); †=p< 0.05 vs. control siRNA transfected-cells with H2O2 treatment (upper panel)]. PKD expression was examined using anti-PKD antibody (lower panel). (B) RIE-1 cells were transfected with control or PKD1 siRNA and grown on chamber slides for 2 days prior to treatment with H2O2 (500 μM) for 15 min. A representative photomicrograph is shown demonstrating NF-κB localization. (C). Cells were transfected with control or PKD1 siRNA. After 3 days, cells were treated with H2O2 (500 μM) for 15 min and Western blotting was performed. PKD expression was examined using anti-PKD antibody (top row). Phosphorylation of IκBα was detected (middle row) and IκBα was used as a loading control (bottom row).
PKD1 mediates p38 MAPK phosphorylation in oxidative stress signaling
Previously, we demonstrated that the MAPK pathway plays a pro-apoptotic role during oxidative stress-induced intestinal epithelial cell injury [5; 6]. To further examine the potential role of PKD in the cross-talk regulatory process with the MAPK pathway, RIE-1 cells were co-transfected with the wild type (PKD1WT) or the kinase-dead PKD1 (PKD1KD) plasmids as well as the control vector. Figure 3A shows that PKD1KD modestly increased the basal and H2O2-stimulated phosphorylation of p38 MAPK compared with cells transfected with the vector. PKD1WT prevented the increase in p38 MAPK phosphorylation induced by H2O2. However, neither ERK1/2 nor JNK phosphorylation was altered by PKD1. Next, we transfected RIE-1 cells with either control or PKD1 siRNA. Similar to PKD1KD, inhibition of PKD1 activity with PKD1 siRNA increased the basal and H2O2-stimulated phosphorylation levels of p38 MAPK compared to cells transfected with control siRNA; whereas, phosphorylation of neither ERK1/2 nor JNK was altered by PKD1 siRNA (Fig. 3B). These data demonstrate that p38 MAPK is a downstream effector of PKD signaling pathway upon oxidative stress.
Fig. 3. PKD1 affects H2O2-induced p38 MAPK phosphorylation but not ERK1/2 and JNK>.

(A) RIE-1 cells were transfected with PKD1WT, PKD1KD. At 48 h after transfection, cells were treated with H2O2 (500 μM) for 30 min, and protein was extracted for Western blot analysis. PKD overexpression was demonstrated using anti-GST antibody. Phosphorylation of ERK1/2, JNK and p38 MAPK was determined with anti-phospho-ERK1/2, JNK and p38 antibodies. (B) RIE-1 cells were transfected with control or PKD1 siRNA. After 3 days, cells were treated with H2O2 (500 μM) for 30 min, and protein was extracted for Western blotting. Inhibition of PKD expression by PKD1 siRNA was shown with anti-PKD antibody (top row). Phosphorylation of ERK1/2, JNK and p38 MAPK was determined with anti-phospho-ERK1/2, JNK and p38 antibodies. β-actin was used as a loading control.
PKD1-mediated p38 MAPK phosphorylation in oxidative stress requires MKK3/6
To investigate whether MKK6 and MKK3 are involved in H2O2-induced p38 MAPK activation, RIE-1 cells were treated with H2O2 (500 μM) over a time course and then phosphorylation of MKK6 and MKK3 was detected by Western blotting. H2O2-induced MKK3 activation occurred at 5 min, with a significant increase at 10 min; MKK6 activity was increased at 30 min and markedly enhanced at 60 min (Fig. 4A). Next, RIE-1 cells were transfected with PKD1WT, PKD1KD and the empty vector (Fig. 4B). PKD1WT inhibited MKK3/6 phosphorylation induced by H2O2. Furthermore, MKK3/6 phosphorylation was increased by PKD1 siRNA in the presence of H2O2 compared with the control siRNA (Fig. 4C; second panel).
Fig. 4. Phosphorylation of p38 MAPK induced by H2O2 mediates through MKKs.

(A) RIE-1 cells were treated with H2O2 (500 μM) in normal growth medium over a time course; phosphorylation of MKK3/6 was detected by Western blotting (top panel). β-actin was used as a loading control. (B) RIE-1 cells were transfected with PKD1WT, PKD1KD. At 48 h after transfection, cells were treated with H2O2 (500 μM) for 30 min, and protein was extracted for Western blotting. PKD overexpression was demonstrated using anti-PKD antibody (top panel). Phosphorylation of MKK3/6 was determined with anti-phospho-MKK3/6 antibody (second panel). (C) RIE-1 cells were transfected with PKD1 or control siRNA. After 3 days, cells were treated with H2O2 (500 μM) for 30 min, and protein was extracted for Western blotting. Inhibition of PKD expression by PKD1 siRNA was determined using anti-PKD antibody (top panel). Phosphorylation of MKK3/6 was determined with anti-phospho-MKK3/6 antibody (second panel). β-actin was used as a loading control.
DISCUSSION
NF-κB activation has been linked to increased cellular survival in cells exposed to a variety of insults, including oxidative stress [4]. The inducers and products of NF-κB activation are highly relevant to some intestinal inflammation diseases, including necrotizing enterocolitis (NEC) [16]. However, there are conflicting reports on the role of NF-κB as a regulator of apoptosis in different model systems and different cell types [2]. In our present study, we showed that H2O2 increased NF-κB activity in rat intestinal cells. We have also demonstrated that H2O2 can induce NF-κB translocation from cytoplasm to the nucleus as well as IκBα phosphorylation. Furthermore, H2O2-induced NF-κB activation was blocked by PKD1 siRNA; inhibition of PKD1 activation abolished NF-κB nuclear translocation by H2O2. Moreover, inhibition of PKD1 with PKD1 siRNA attenuated IκBα phosphorylation induced by H2O2. Consistent with findings in another model of oxidative stress in HeLa cells [11], our data strongly indicate a role for a PKD-dependent pathway in the regulation of NF-κB expression in rat intestinal cells.
Upon ROS stimulation, IκBα in the cytoplasm is phosphorylated and then rapidly degraded. This process then exposes p65 to undergoes phosphorylation, leading to nuclear translocation and binding to a specific sequence in DNA to regulate gene transcription [17]. In keeping with previous studies in other cell lines, treatment with H2O2 resulted in the phosphorylation of IκBα in RIE-1 cells. Our results showed that as rapidly as 5 min after treatment, H2O2 induces IκBα phosphorylation. Thus, stimulation of intestinal cells with H2O2 can lead to activation of NF-κB via the classical IκBα pathway in intestinal cells.
Activation of NF-κB by oxidative stress is not observed in all cell types, suggesting that NF-κB activation by H2O2 may not occur through a common mechanism, but rather requiring multiple signaling pathways converging upstream of NF-κB [4]. Some studies have provided evidence for a protective function of NF-κB in response to oxidative stress [18; 19]. Storz and colleagues [11] described a signaling pathway that results in the activation of NF-κB in cells specifically exposed to H2O2. In this pathway, a key player is the serine/threonine kinase PKD, which relays a signal from ROS to the activation of the IKK/NF-κB pathway. The consequence is an increase in the survival of cells exposed to increasing concentrations of oxidative stress [11]. In the present study, through the use of PKD1siRNA, we have addressed the relative contributions of PKD1 in influencing the activity of NF-κB in H2O2-treated RIE-1cells. Inhibition of PKD1 significantly altered the activity and cellular location of NF-κB in H2O2-treated RIE-1cells, which demonstrates an important role of NF-κB in the PKD protection of oxidative stress-induced intestinal epithelial cell injury.
In contrast to the anti-apoptotic signals of the PKD1 pathway, H2O2 also stimulates the activation of the stress-activated p38 MAPK, which appears to be an important modulator of the pro-apoptotic program in various cell types [2]. In this paper, we provide evidence regarding the existence of cross-talk between the p38 MAPK and PKD1 pathways under oxidative stress. We show that blockade of PKD1 signaling with PKD1 siRNA or PKD1KD enhances MKK3/6 and p38 MAPK phosphorylation. On the other hand, PKD1WT attenuates MKK3/6 and p38 MAPK phosphorylation. We have previously reported that PKD activation protects intestinal epithelial cells from oxidative stress-induced apoptosis [13] and that p38 MAPK has a pro-apoptotic role in H2O2-induced RIE-1 cell death [5; 13]. Therefore, our results from this study confirmed that the regulation of H2O2-induced apoptosis by PKD1 likely acts through down-regulation of p38 MAPK. Our data strongly suggest that cross-talk between PKD1 and p38 signaling pathways contributes to the balance of pro- and anti-apoptotic signaling that occurs in the intestinal epithelial cells.
JNK and p38 MAPK are often grouped together and referred to as stress-activated protein kinases (SAPK). However, our findings showed that phosphorylation of JNK was not altered by transfection of either PKD1 siRNA or PKD1 plasmids. The pathways leading to JNK and p38 MAPK activation are extremely complex and involve multiple MKKs, more than 10 MKKKs, and a variety of other interacting regulatory proteins. Therefore, we speculate that the reason for the lack of JNK phosphorylation is that H2O2 may act at multiple levels in the signaling pathway to regulate their activities [20]. Although some investigators reported that PKD selectively mediates JNK signaling induced by oxidative stress in certain cell lines [21], our results confirmed that the resistance of rat intestinal cells to oxidative stress is dependent on PKD-mediated down-regulation of p38 MAPK. It has been shown that dual phosphorylation of the upstream MKK3/6 is required for p38 activation [22]. We also found that PKD1WT attenuated H2O2-induced phosphorylation of MKK3/6, whereas PKD1siRNA or PKD1KD increased the phosphorylation. Thus, PKD can mediate the phosphorylation of p38 MAPK at the MKKs level, downstream to MKKKs.
In summary, based on our previous studies [5; 13] and our present observations, we propose the signaling cascade in H2O2-induced intestinal cell death is as follows: H2O2 activates and ultimately leads to PKD activation. PKD can mediate IκBα phosphorylation and therefore, NF-κB activation. On the other hand, PKD, in turn, inactivates p38 MAPK. In this manner, PKD participates in the regulation of cell survival pathway in oxidative stress-induced intestinal epithelial cell injury. These findings provide novel insights to the development of unique therapeutic strategy targeted to intracellular processes for the treatment of a variety of oxidant-induced inflammatory disorders of the gastrointestinal tract, including NEC in premature infants.
ACKNOWLEDGEMENTS
The authors thank Karen Martin for manuscript preparation and Tatsuo Uchida for assistance with statistical analyses. This work was supported by the grants RO1 DK61470, RO1 DK48498, RO1 CA104748, PO1 DK 35608 from the National Institutes of Health and a grant #8580 from the Shriners Hospital for Children.
REFERENCES
- 1.Gabbita SP, Robinson KA, Stewart CA, Floyd RA, Hensley K. Redox regulatory mechanisms of cellular signal transduction. Arch Biochem Biophys. 2000;376:1–13. doi: 10.1006/abbi.1999.1685. [DOI] [PubMed] [Google Scholar]
- 2.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 4.Li N, Karin M. Is NF-kappaB the sensor of oxidative stress? Faseb J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- 5.Zhou Y, Wang Q, Mark Evers B, Chung DH. Oxidative stress-induced intestinal epithelial cell apoptosis is mediated by p38 MAPK. Biochem Biophys Res Commun. 2006;350:860–865. doi: 10.1016/j.bbrc.2006.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Wang Q, Evers BM, Chung DH. Signal transduction pathways involved in oxidative stress-induced intestinal epithelial cell apoptosis. Pediatr Res. 2005;58:1192–1197. doi: 10.1203/01.pdr.0000185133.65966.4e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 8.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waldron RT, Iglesias T, Rozengurt E. Phosphorylation-dependent protein kinase D activation. Electrophoresis. 1999;20:382–390. doi: 10.1002/(SICI)1522-2683(19990201)20:2<382::AID-ELPS382>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 10.Storz P, Doppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Storz P, Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. Embo J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song J, Li J, Lulla A, Evers BM, Chung DH. Protein kinase D protects against oxidative stress-induced intestinal epithelial cell injury via Rho/ROK/PKC-{delta} pathway activation. Am J Physiol Cell Physiol. 2006;290:C1469–1476. doi: 10.1152/ajpcell.00486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blay J, Brown KD. Characterization of an epithelioid cell line derived from rat small intestine: demonstration of cytokeratin filaments. Cell Biol Int Rep. 1984;8:551–560. doi: 10.1016/0309-1651(84)90054-7. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 16.Chung DH, Ethridge RT, Kim S, Owens-Stovall S, Hernandez A, Kelly DR, Evers BM. Molecular mechanisms contributing to necrotizing enterocolitis. Ann Surg. 2001;233:835–842. doi: 10.1097/00000658-200106000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 18.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 20.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Zheng S, Storz P, Min W. Protein kinase D specifically mediates apoptosis signal-regulating kinase 1-JNK signaling induced by H2O2 but not tumor necrosis factor. J Biol Chem. 2005;280:19036–19044. doi: 10.1074/jbc.M414674200. [DOI] [PubMed] [Google Scholar]
- 22.Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]