

Abstract
Previously, we demonstrated an important role for insulin in protection of endothelial cells against hyperglycemic stress through maintaining cellular glutathione (GSH) redox balance. The current study focuses on the contribution of insulin to transcriptional control of endothelial cell GSH recovery during acute oxidative challenge and the influence of low glucose. The results show that insulin induced an approximate 2-fold increase in expression of γ-glutamyl-cysteine ligase catalytic subunit (GCLc) mRNA and protein; interestingly, cellular GSH levels were not elevated accordingly. However, upon tert-butylhydroperoxide challenge, insulin-treated cells demonstrated a robust GSH recovery that was attributed to a greater capacity for de novo synthesis via elevated GCLc levels. Notably, the effects of insulin were observed under low, but not normal, glucose conditions. Our results implicate a role for Nrf2 involvement in both constitutive and inducible endothelial GCLc expression and GSH synthesis, while PI3K/Akt/mTOR signaling appears to participate only in insulin-inducible GSH synthesis. Collectively, these results support the functional importance of insulin in Nrf2-dependent transcriptional upregulation of GCLc in GSH recovery during oxidative challenge and suggest a possible role for hypoglycemia in promoting insulin-mediated GCLc upregulation.
Introduction
γ-glutamylcysteinyl glycine, or glutathione (GSH), is a thiol tripeptide that plays important roles in a number of cellular functions including antioxidant defense and thiol redox signaling. The redox potential of the glutathione/ glutathione disulfide (GSH/GSSG) redox couple is typically indicative of the overall cellular redox environment and is often used as a quantitative assessment of oxidative stress [1]. Cellular GSH has a profound impact on vascular endothelial function and phenotype, such as modulation of endothelial permeability [2], chemotaxis, tube formation, and angiogenesis [3, 4], constitutive and inducible ICAM-1 expression and leukocyte adhesion [5], as well as endothelial dependent vasodilation [6]. These effects of GSH are likely through scavenging of reactive oxygen species (ROS), which play second messenger roles in endothelial function, and through post translational modification of signaling proteins via glutathiolation of redox sensitive cysteine residues within these proteins. Notably, animal models of a number of cardiovascular diseases including hypertension, atherosclerosis, and diabetes, which are characterized by vascular oxidative stress and endothelial dysfunction, display prominent decreases in vascular tissue GSH levels [7-9]. In this regard, intravenous GSH infusion has been shown to improve endothelial dependent vascular reactivity in patients at risk for or diagnosed with coronary artery disease [6, 10] and those with coronary spastic angina [11]. These observations underscore an important role for GSH in protection against vascular oxidative stress and cardiovascular disease.
Intracellular GSH levels are maintained by de novo synthesis, redox cycling, and transmembrane transport. The importance of de novo synthesis is underscored by observations that inhibition of γ-glutamylcysteine ligase (GCL), the rate limiting enzyme in the first of two steps in GSH formation, can significantly deplete GSH in cultured cells and tissues within hours [12-15]. The GCL holoenzyme is a heterodimer composed of a catalytic (GCLc) and a modulatory (GCLm) subunit, and while GCLc alone possesses all of the catalytic activity of the enzyme, dimerization with GCLm increases enzymatic efficiency [16]. GCLc transcription is a primary means for increasing enzyme activity in response to a number of growth factors, cytokines, and electrophillic compounds. A particular single nucleotide polymorphism (-129 C/T) within the GCLc promoter decreases GCL activity and has been linked to endothelial dysfunction, coronary artery disease, and myocardial infarction in several ethnic groups [17-19]. Collectively, these findings support a role for transcriptional regulation of GCLc in maintaining normal vascular function.
Evidence suggests that GCLc transcription is regulated primarily by the transcription factor nuclear factor erythroid 2 related factor 2 (Nrf2), whether induction is via mechanical stimuli such as laminar flow [20], or electrophilic agents such as β-naphthoflavone (β-NF) [21], peroxynitrite (ONOO-) [22] or tert-butylhydroquinone (tBHQ) [23]. Nrf2 is a member of the cap-n-collar (CNC) family of basic leucine zipper (bZIP) transcription factors and is expressed ubiquitously in most cell types. Normally sequestered in the cytosol by Kelch associated protein 1 (Keap1), Nrf2 translocates to the nucleus under conditions of oxidative stress and activates transcription of a number of Phase II cytoprotective genes involved in antioxidant defense and maintenance of redox homeostasis, including GCLc. Different investigators have demonstrated a role for growth factor-induced PI3K signaling in Nrf2 mediated upregulation of Phase II detoxication enzymes [24].
Insulin plays an important cytoprotective role in endothelial cells via Akt- dependent inhibition of apoptosis via inactivation of proapoptotic molecules such as FKHRL1, Bad, and caspase 9, coincident with activation of eNOS and nitric oxide (NO•) production [25]. An underlying feature common to diabetes, hypertension, and atherosclerosis is a decreased vascular response to insulin. The fact that endothelial dysfunction and oxidative stress accompany cardiovascular pathologies associated with decreased insulin sensitivity suggests that insulin plays an important role in the regulation of the endothelial redox environment. Insulin treatment has been shown to increase GSH in rat hepatocytes via transcriptional upregulation of GCLc expression [16, 26], suggesting that insulin signaling contributes to cellular GSH redox balance. In the present study, we sought to determine the effect of insulin on endothelial GCLc expression and cellular GSH and the response to hydroperoxide challenge. Here we show that while insulin per se has no effect on endothelial GSH, it increases GCLc expression and promotes rapid recovery of cellular GSH from an acute oxidative stress. We further found that low extracellular glucose stimulates the responses to insulin.
Methods
Reagents
Porcine insulin, tert-butylhydroperoxide (tBH), L-buthionine-(S, R)-sulfoximine (BSO), rapamycin B, insulin-transferrin-sodium selenite solution, hygromycin B, leupeptin, aprotonin, and Medium 199 were purchased from Sigma (St. Louis, MO). LY294002, okadaic acid, and Akt Inhibitor (SH-5) were purchased from Calbiochem (San Diego, CA). Anti-GCLc rabbit polyclonal antibody was obtained from NeoMarkers (Fremont, CA), anti-actin mouse monoclonal antibody from BD Biosciences (San Jose, CA), and anti-Nrf2 rabbit polyclonal antibody from Santa Cruz (# SC722, Santa Cruz, CA). Total and phospho-specific Akt, mTOR, and p70S6K antibodies were purchased from Cell Signaling Technologies (Danvers, MA). HRP-linked goat-anti-rabbit and goat-anti-mouse antibodies and ECL plus chemiluminescent detection reagents were purchased from Amersham Biopharmacia (Piscataway, NJ). The empty (pEF), WT (pEF-Nrf2) and dominant negative Nrf2 (pEF-dnNrf2) expression vectors were kind gifts from Dr. Jeffrey Johnson at the University of Wisconsin - Madison, and the hygromycin resistance vector (pTK-Hyg) was a gift from Dr. Lynn Harrison at LSUHSC-S. All other chemicals were of reagent grade and purchased from Sigma.
Cell Culture Conditions
An immortalized human brain endothelial cell line (IHEC) was a kind gift from Dr. Danica Stanimirovic of the National Research Council Canada’s Institute for Biological Sciences and was propagated by Dr. Steven J. Alexander at LSUHSC. IHECs were cultured in M-199 with 10% FBS, 1% insulin-transferrin-sodium selenite solution, and 1x antibiotic/antimycotic at 37°C in 5% CO2. For experiments, confluent endothelial monolayers were serum starved overnight in M-199 base media supplemented with antibiotic/antimycotic followed by pharmacological treatment in fresh base media for the specified time periods. For experiments utilizing stable stably transfected cell lines, cells were maintained and passaged in M-199 with 10% FBS, 1% insulin-transferrin-sodium selenite solution, 1x antibiotic/antimycotic, and 50 μg/mL hygromycin B. Cells were serum starved in the presence of 50 μg/mL hygromycin B, and experiments were performed in M-199 base media with antibiotic/antimycotic and 50 μg/mL hygromycin B. Cell viability was routinely checked and was unaffected under conditions of serum starvation.
Cell incubations for GSH determination
Confluent IHEC monolayers in 6 well plates were treated with 100 nM insulin for up to 72 hrs. For experiments with tert-butylhydroperoxide (tBH), IHEC were treated with insulin for 48 hrs prior to 100 μM tBH treatment for 3 hrs. For BSO treatments, cells were treated with insulin for 48 hrs, then pretreated with 300 μM BSO for 1 hr prior to tBH exposure. In studies of PI3K/Akt/mTOR involvement, cells were pretreated with the respective kinase inhibitors for 1 hr prior to insulin co-administration for 48 hrs and then treated with tBH. Cells were lysed in 5% TCA at designated time points and scraped into 1.5 mL microcentrifuge tubes. GSH concentrations were measured as previously described [27, 28, see below].
Quantification of GSH
Cellular GSH content was determined by high-performance liquid chromatography (HPLC, 24). In all determinations, the acid supernatants were derivatized with 6mM iodoacetic acid and 1% 2,4-dinitrophenyl fluorobenzene to yield the S-carboxymethyl and 2,4-dinitrophenyl derivative of GSH. Separation of GSH derivatives was performed on a 250mm × 4.6 mm Alltech Lichrosorb NH2 10micron anion exchange column. Cellular GSH contents were quantified by comparison to standards derivatized in the same manner. Protein pellets were resuspended in 1 mL 0.1 M NaOH for protein quantitation.
Western Analysis of GCLc, Akt, mTOR, and p70S6K
Cells were treated with 100 nM insulin in serum free media for up to 72 hrs and lysed in RIPA with aprotinin, PMSF, okadaic acid, and leupeptin at the designated time points. Total protein (40 μg) per sample were loaded onto 10% acrylamide gels and separated at 100 V followed by transfer to PVDF overnight at 30V at 4°C. Membranes were blocked in 5% milk TBS-T at room temperature for 1-2 hrs and then incubated with rabbit anti-GCLc (1:1000) in 5% milk TBS-T for 1 hr at room temperature, or anti-phospho-AktSer473 (1:1000), anti-phospho-mTORSer2448 (1:1000), or anti-phospho-p70S6KinaseSer241/Thr244 (1:1000) in 5% BSA TBS-T for overnight at 4°C. After washing, membranes were incubated in HRP conjugated goat-anti-rabbit secondary for 2-3 hrs at room temperature followed by washing and 5 min incubation with ECL reagents. For phospho-specific western blots, membranes were then stripped and blotted for total expression of the respective kinases (total Akt, mTOR, and p70S6K were used at 1:1000 in 5% BSA TBS-T and membranes were incubated overnight at 4°C). Expression was normalized to actin using a mouse monoclonal antibody (1:5000).
Isolation of Nuclear Protein and Western Analysis of Nuclear Nrf2 Expression
IHEC nuclear protein was isolated as described previously [29]. Briefly, confluent IHEC monolayers in T-75 flasks were serum starved overnight and then treated with 100 nM insulin for up to 24 hrs. At each time point, the monolayers were scraped in PBS, pelleted, and then resuspended in a hypotonic buffer (10 mM HEPES pH 7.9, 1.5 mM MgCL2, 10 mM KCl, 0.5 mM DTT, 0.2 mM PMSF). Cell pellets were resuspended in the same hypotonic buffer with 10% IGEPAL, and kept on ice for 5 min prior to disruption of the plasma membranes in a glass homogenizer. The resultant suspension was centrifuged for 1 min at 12000rpm, 4°C and the supernatant (cytosolic protein) was stored at -20°C. The remaining pellet was resuspended in a low salt buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 25% glycerol, 0.5 mM DTT, 0.2 mM PMSF) and a hypertonic solution (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 1 M KCl, 0.2 mM EDTA, 25% glycerol, 0.5 mM DTT, 0.2 mM PMSF) was added drop wise at half the volume of the low salt buffer (for 1 T-75, 100 μL low salt buffer and 50 μL hypertonic solution added slowly). The suspension was centrifuged at 12000 rpm for 5 min at 4°C and the supernatant (nuclear protein) was stored at -20°C. Nuclear proteins (20 μL) were separated on a 10% gel, followed by transfer to PVDF overnight at 30V and 4°C. Western blotting was conducted at described above for GCLc with the exception that incubation with the Nrf2 antibody (rabbit polyclonal, 1:500) was overnight at 4°C. The results were normalized to histone H1 protein (mouse monoclonal, 1:2000).
Semiquantitative PCR Analysis of GCLc mRNA
Confluent IHEC monolayers were serum starved overnight and then treated with 100 nM insulin for various times. At designated time points, mRNA was isolated using a Qiagen RNeasy kit and quantitated on an Agilent 2100 Bioanalyzer. cDNA was synthesized from 1 μg of each sample using Applied Biosystems reverse transcription reagents per the manufacturer’s instructions, and 20 μL of each cDNA reaction was used for PCR. The primers and reaction conditions were as follows: GCLc – sense primer: 5’-CGGGATCCTCCAGTTCCTGC-3’, antisense primer: 5’-ACCTCGGGCAGTGTGAACCC-3’, PCR conditions - 40 cycles of : 94°C for 30 sec, 55°C for 30 sec, 72°C for 60 sec; GAPDH – sense primer: 5’-ATGGGGAAGGTGAAGGTCGG-3’, antisense primer: 5’-ATGACCTTGCCCACAGCCTT GG-3’, PCR conditions - 30 cycles of : 94°C for 30 sec, 55°C for 30 sec, 72°C for 60 sec.
Generation of Stable IHEC Cell lines overexpressing WT or dominant negative Nrf2
IHEC were trypsinized and 1×106 cells were pelleted and resuspended in 100 μL Amaxa nucleofection buffer along with 0.2 μg pTK-Hyg and 2 μg of either an empty mammalian expression vector (pEF) or the same vector containing cDNA for either the wild type (pEF-Nrf2) or a dominant negative (pEF-dnNRF2 [30]) Nrf2 transcription factor. For each of these nucleofections, cells were plated in 100 mm dishes in full M-199 media supplemented with 100 μg/mL hygromycin B (Sigma, St. Louis, MO). After 4-6 weeks, isolated colonies were transferred to 35 mm dishes and cultured as needed. Clone selection was based on Western analyses of protein expression of Nrf2 in the various cell lines.
Other assays
Cell viability was determined by trypan blue exclusion. Confluent IHEC monolayers in 6 well plates were trypsinized and cells resuspended in PBS. An aliquot of the cell suspension was mixed with an equal volume of 0.4% trypan blue (3 min), and counted using a hemacytometer. Viability was expressed as percent of the ratio of cells that excluded the dye to total cell number.
Media glucose was measured in duplicate using an Accu-Chek Advantage glucometer and Accu-Chek Comfort Curve test strips.
Statistical Analysis
All results are expressed as mean +/- SE, and all data was analyzed by one way ANOVA using Bon Ferroni’s post test for comparison. P<0.05 was considered statistically significant.
Results
Insulin Treatment Increases GCLc mRNA and protein expression in IHECs
To investigate the effects of insulin on endothelial GCLc protein and mRNA expression, confluent IHEC were treated with 100 nM insulin for up to 72 hrs. Western blot analysis of GCLc expression showed a significant increase by 36 hrs (Figure 1A) that was preceded by a significant increase in GCLc mRNA at 24 hrs (Figure 1B). The kinetics of GCLc expression over the same time period were unchanged in untreated cells (Figure 1A), consistent with an insulin specific effect. This finding that insulin enhances GCLc expression in endothelial cells is in agreement with studies in hepatocytes [16, 26].
Figure 1. Insulin increases GCLc protein and mRNA expression in IHECs.

Confluent IHEC monolayers were treated with 100 nM insulin for the indicated time periods and GCLc protein and mRNA expression were determined by western blotting and semiquantitative PCR, respectively. A. GCLc protein expression was normalized to β-actin (upper panel) and expressed as fold change in expression from 0 hrs (lower panel). * p<0.05 vs 100 nM Insulin at 0 hrs, # p<0.05 vs No insulin at respective time point. B. GCLc mRNA expression was normalized to GAPDH mRNA (upper panel) and expressed as fold change from 0 hrs (lower panel). * p<0.05 vs 0 hrs.
Insulin-induced GCLc expression enhances the potential for GSH production in IHECs during tert-butylhydroperoxide challenge
To determine whether the increase in GCLc expression resulted in elevated cell GSH, GSH levels were measured over the 72 hr time period. Interestingly, insulin treatment did not elevate GSH levels (Figure 2A). Over 72 hrs, there was a steady, parallel decline in GSH levels in untreated and insulin-treated cells, demonstrating that while insulin increased GCLc expression, the enhancement of protein expression did not translate into an increase in cell GSH levels. These results are consistent with an efficient GSH feedback inhibition at the level of GCL activity when GSH concentrations are sufficiently high. To test whether insulin-induced GCLc expression affected GSH concentrations during oxidative stress, insulin-treated IHECs were challenged with tBH (100 μM). The results in Figure 2B show that tBH exposure significantly decreased cell GSH by 30 min in both control and insulin-treated cells. However, GSH in insulin treated cells began to recover at 1 hr and essentially reached pre-oxidant levels by 3 hrs. In contrast, GSH in untreated cells continued to decrease throughout the 3 hr incubation with tBH (Figure 2B). To validate that insulin-mediated recovery of GSH was due to de novo GSH synthesis, we pretreated cells with 300 μM BSO prior to tBH challenge. The results show that pretreatment with BSO, a known inhibitor of the activity of GCL, but not of the expression of GCLc protein [29], completely prevented insulin-induced GSH recovery (Figure 2C), indicating that the insulin-induced GSH recovery was GCL-dependent. Thus, insulin-induced GCLc expression promoted restoration of the intracellular GSH pool following tBH challenge, suggesting that insulin functions to enhance endothelial cell capacity to maintain cellular GSH during oxidative stress.
Figure 2. Insulin enhances GSH recovery in IHECs after acute tBH stress in a GCL-dependent manner.
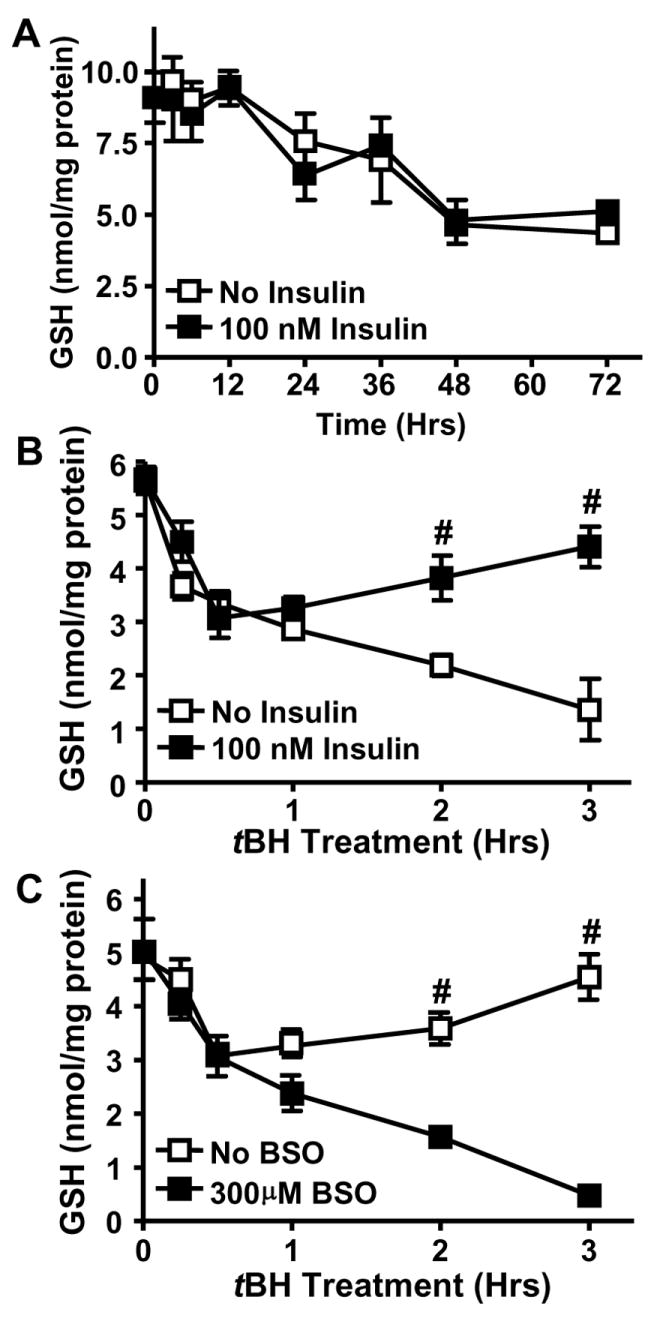
Confluent IHECs were treated with 100 nM insulin for 72 (A) or for 48 hrs followed by exposure to tBH (B and C). Total cellular GSH was measured by HPLC as described in Methods. A. Time course of GSH levels without or with insulin treatment for 72 hrs. B. Cells were treated with insulin for 48 hrs and challenged with 100 μM tBH for 3 hrs. Cellular GSH was determined at designated times. # p<0.05 vs No Insulin at the same time point. C. 48-hr insulin treated cells were incubated with 300 μM BSO for 1 hr prior to 100 μM tBH administration for 3 hrs. Cellular GSH was determined at designated times. # p<0.05 vs 300 μM BSO at respective time point.
Enhanced GSH recovery in insulin treated cells is dependent on PI3K signaling
Our recent studies have shown that insulin-induced endothelial GCLc expression during hyperglycemia was associated with activation of the PI3K/Akt/mTOR/Nrf2 pathway [29]. We tested whether this pathway was involved in insulin-mediated GCLc expression and protection against tBH induced oxidative stress by examining the phosphorylation status of Akt, mTOR, and p70S6K. The results in Figure 3 revealed that insulin treatment resulted in rapid phosphorylation of each kinase as early as 5 minutes post exposure without affecting total protein levels, in agreement with our recent study [29]. To further explore the involvement of the kinase pathway, confluent IHECs were pretreated with inhibitors of PI3K (LY294002), Akt (SH-5), or mTOR (rapamycin) for 1 hr prior to insulin co-treatment for 48 hrs. Pretreatment with the kinase inhibitors decreased kinase phosphoprotein levels (data not shown) and insulin-induced Nrf2 nuclear accumulation and GCLc protein expression [29]. Accordingly, these inhibitors significantly attenuated insulin-mediated GSH recovery after tBH treatment (Figure 4A); notably, the GSH levels in cells pretreated with kinase inhibitors were similar to those caused by BSO. Interestingly, cellular GSH levels were unaffected by 48 hr treatment with the inhibitors alone or inhibitors plus insulin in the absence of tBH challenge (Figure 4B), consistent with the suggestion that PI3K signaling plays a minimal role in regulating constitutive GSH levels, but is important in the insulin-mediated GSH recovery post oxidative challenge. It was noted that 48 hr PI3K inhibition followed by 3 hr tBH exposure resulted in IHEC cytotoxicity which may have contributed to the noticeably lower cell GSH levels in these cells as compared to the other kinase inhibitors and BSO (Figure 4A). However, cytotoxicity was not evident in cells treated with LY294002 alone in the absence of tBH (Figure 4C), or in cells treated with SH-5 or rapamycin with or without tBH (data not shown).
Figure 3. Insulin activates the PI3K/Akt/mTOR/p70S6K pathway in IHECs.
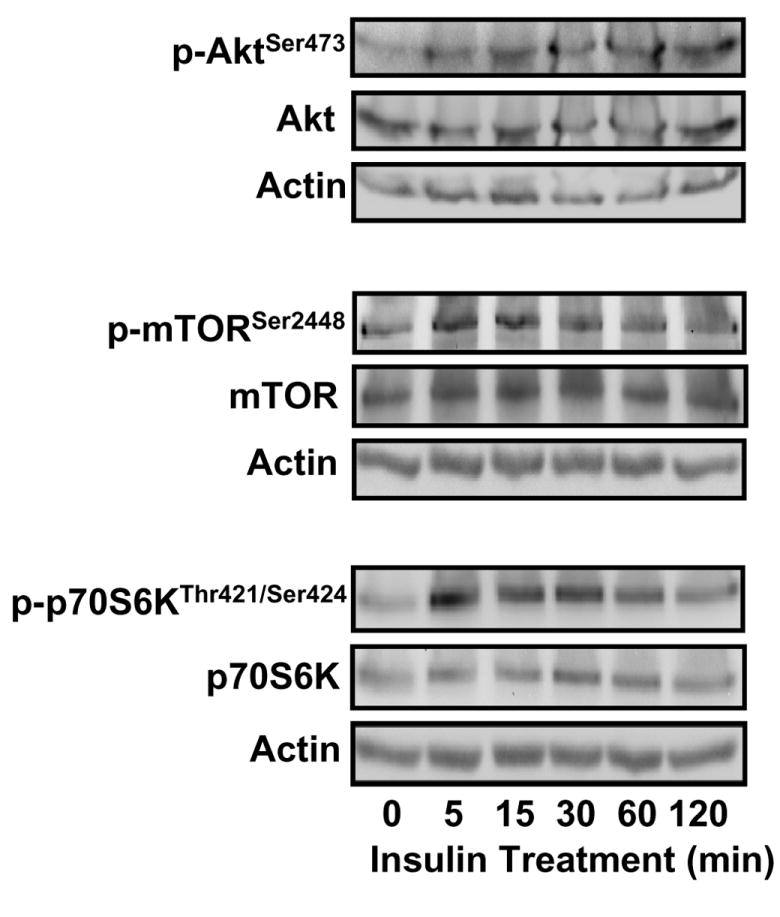
IHECs were treated with 100 nM insulin and at designated times up to 2 hrs, the phosphorylation status of Akt Ser473, mTOR Ser2448, and p70S6K Thr421 and Ser424 was assessed by Western blotting. The membranes were stripped and reprobed for total kinase protein as well as actin to verify equal protein loading in each lane. One representative of three Western blots is shown for each kinase.
Figure 4. PI3K/Akt/mTOR/p70S6K signaling regulates insulin-inducible GSH recovery after tBH challenge.
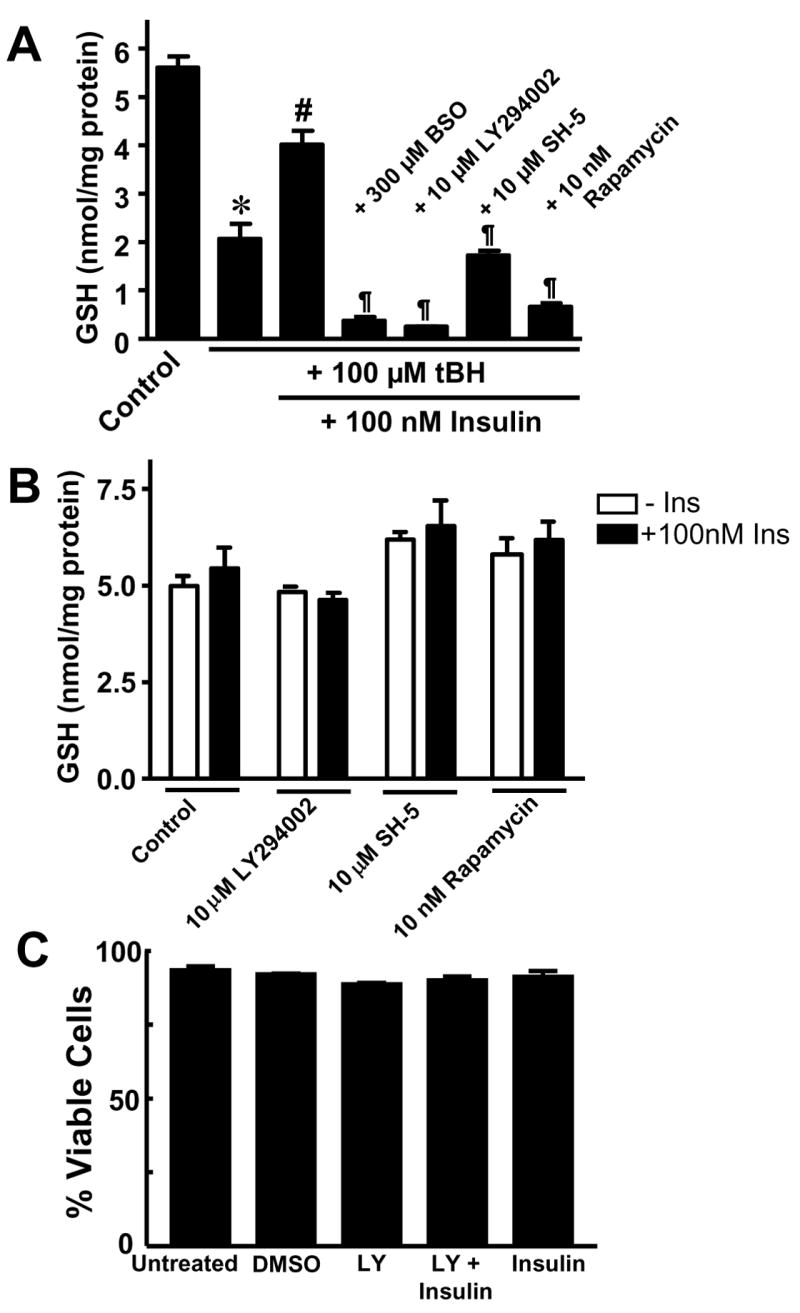
A. Confluent IHEC monolayers were pretreated with either 10 μM LY294002, 10 μM SH-5, or 10 nM rapamycin for 1 hr prior to co-incubation with 100 nM insulin for 48 hrs. Thereafter cells were challenged with 100 μM tBH for 3 hrs and cellular GSH levels were measured at designated times. BSO controls were performed as described in Figure 2C. * p<0.05 vs untreated control; # p<0.05 vs plus 100 μM tBH plus 100 nM Insulin; ¶ p<0.05 vs plus 100 μM tBH plus 100 nM Insulin without kinase inhibitors. B. IHEC monolayers were pretreated with either 10 μM LY294002, 10 μM SH-5, or 10 nM rapamycin for 1 hr prior to co-incubation with 100 nM insulin for 48 hrs, and then cellular GSH levels were measured. C. IHECs were treated with or without 10 μM LY294002, 100 nM Insulin or in combination for 48 hrs and cell viability was assessed by trypan blue exclusion. DMSO served as solvent control. For B & C there was no significant difference among treatment groups by one way ANOVA.
Insulin enhances post-tBH GSH recovery in an Nrf2 dependent manner
Figure 5A illustrates the relationship between insulin and expression of nuclear Nrf2. The results show that insulin induced a time-dependent increase in nuclear Nrf2 expression between 4-24 hrs that achieved statistical significance at 24 hrs, a time that corresponded to significant increases in GCLc message and preceded expression of GCLc protein (see Figure 1A). To explore the functional role of Nrf2, we generated stable IHEC cells lines that overexpress either vector control (pEF1), WT Nrf2 (pEF-Nrf2) or a dominant negative mutant Nrf2 (pEF-dnNrf2). Figure 5B shows that under control, non-insulin stimulated conditions Nrf2 was primarily localized to the nucleus and was barely detectable in the cytosol in pEF1 cells. Increased Nrf2 protein was detected in cells overexpressing wild-type and dominant negative Nrf2, pEF-Nrf2 and pEF-dnNrf2 respectively, which localized to both cytosolic and nuclear compartments (Figure 5B). The presence of nuclear Nrf2 in the absence of insulin indicates the existence of a constitutive Nrf2 pool within the nucleus, in agreement with recent suggestions that Nrf2 resides primarily in the nucleus under non-stimulated conditions [31].
Figure 5. Cytosolic-nuclear distribution of Nrf2 and insulin stimulation of Nrf2 nuclear translocation and GSH recovery post tBH challenge.
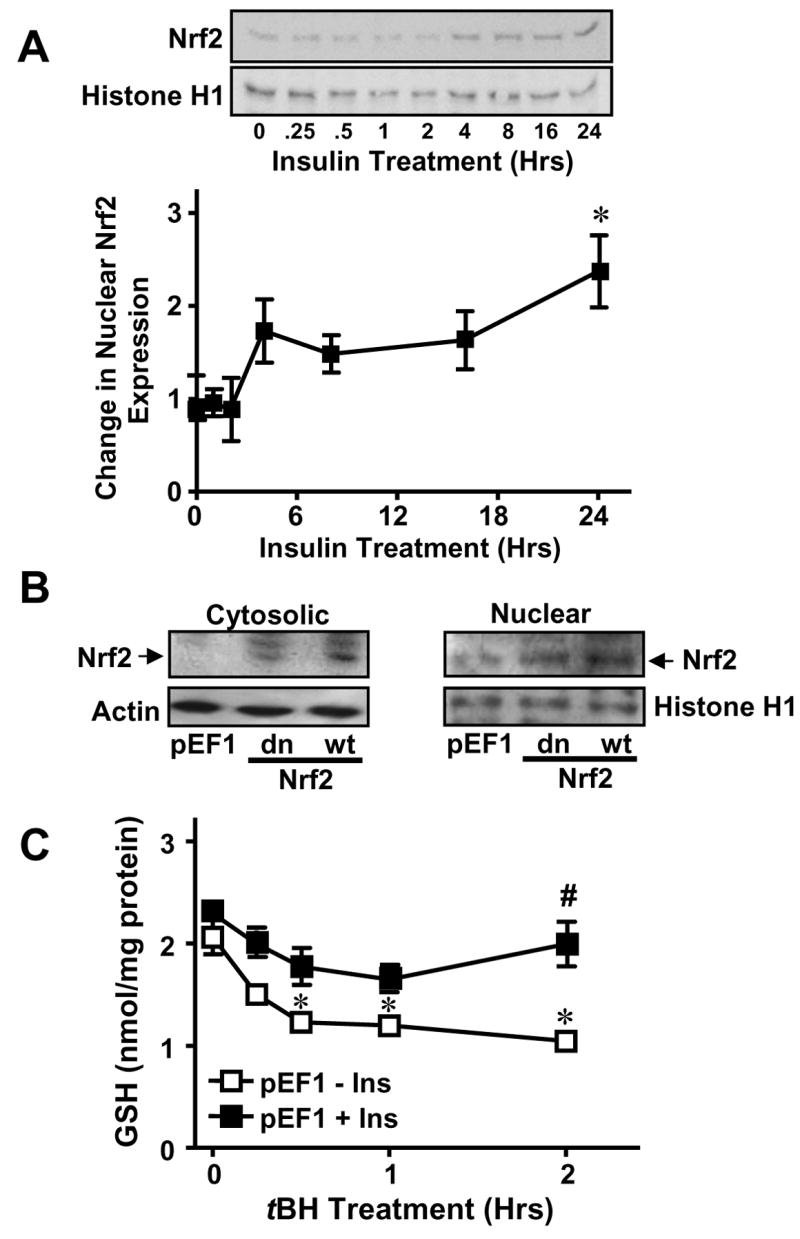
A. Confluent IHECs were treated with 100 nM insulin for 24 hrs and nuclear protein was isolated at the indicated time points for assessment of nuclear Nrf2 expression by Western blotting (Top panel). Nrf2 expression was normalized to Histone H1 and expressed as fold change vs 0 hrs (Lower panel). * p<0.05 vs 0 hrs. B. Cytosolic and nuclear fractions were prepared from IHECs stably transfected with empty mammalian expression vector (vector control, pEF1), or with vector containing either dominant negative (dn) or wild-type (wt) Nrf2. Nrf2 protein contents in the subcellular compartments for each IHEC clones were examined by Western analyses. The membranes were reprobed with actin or histone H1, respectively to verify equal protein loading in the cytosolic and nuclear compartments. One representative of two Western blots is shown. C. IHEC cells stably transfected with an empty expression vector (pEF1) were treated with 100 nM insulin for 48 hrs prior to challenge with 100 μM tBH for 2 hrs. Cellular GSH contents were measured at designated times. * p<0.05 vs pEF1 minus Ins at 0 hrs. # p<0.05 vs pEF 1 minus Ins at respective time point.
Figures 5C and 6 illustrate the results of Nrf2 overexpression and functional knockdown on insulin-mediated GSH recovery after tBH challenge. In pEF1 cells, tBH induced time-dependent decreases in cell GSH over 2 hrs which were restored to pre-tBH levels by insulin pretreatment (Figure 5C), resembling the findings in WT IHEC cells (see Figure 2B). In the absence of insulin, pEF-Nrf2 cells exhibited similar baseline GSH levels (Figure 6A, C) as vector controls (pEF1, Figure 5C), but tBH induced a more gradual GSH loss in Nrf2 overexpressing cells (Figure 6A). Remarkably, 48 hr insulin pretreatment doubled GSH levels in these cells which were maintained over the time course of 2-hr tBH treatment (Figure 6A). In contrast, overexpression of dominant negative Nrf2 (pEF-dnNrf2) reduced basal GSH concentrations by half, and were essentially unchanged by exposure to tBH (Figure 6B, D). Pretreatment with insulin did not significantly raise cellular GSH levels except at 2hr post tBH challenge (Figure 6B, D). It is notable that GSH levels in pEF-dnNrf2 cells were consistently ~50% of that in vector controls with or without the presence of insulin and/or tBH (Figure 6C and D). Taken together, the results indicate a role for Nrf2 in insulin-mediated enhancement of GSH production after acute oxidative stress and suggest that Nrf2 regulates both constitutive and inducible GSH levels in IHECs.
Figure 6. Role of Nrf2 in insulin-induced GSH recovery after tBH treatment.

Stable IHEC clones overexpressing either wild-type (A) or dominant negative (B) Nrf2 were treated with 100 nM insulin for 48 hrs prior to treatment with 100 μM tBH for 2 hrs. Cellular GSH contents were determined at designated times. A. # p<0.05 vs pEF-Nrf2 12 minus Ins at respective time point. B. # p<0.05 vs pEF-dnNrf2 16 minus Ins at respective time point. The numerical designates of 12 and 16, respectively refer to specific stable IHEC clones overexpressing wt or dominant negative Nrf2. Panels C and D compare GSH levels between the 3 cell lines at 0 hr (C) and 2 hr (D) after tBH treatment. Data are taken from panels A and B and Figure 5C. C. * p<0.05 vs pEF 1 minus Ins, # p<0.05 vs pEF 1 plus Ins. D. # p<0.05 vs pEF 1 plus Ins.
Low glucose enhances the insulin-mediated increase in GCLc expression
The findings that insulin induction of GCLc occurred at 36 hrs and that IHEC GSH decreased over 72 hrs in the absence of fresh media change (Figure 2A) suggest the development of a hypoglycemic state and oxidative stress that contributed to GCLc regulation. To test this, we measured media glucose and its status on GCLc expression. Figure 7A shows that media glucose concentrations steadily declined over 48 hrs in cell incubation without or with insulin treatment with a slightly greater decrease in the presence of insulin at all time points. Media glucose levels were decreased by 75% and 87.5%, respectively at 48 hrs (Figure 7A); media glucose levels were essentially undetectable at 72 hrs, but cell viability was maintained at 95% (data not shown). The coincidence of 24-hr nuclear Nrf2 accumulation and GCLc mRNA expression (Figures 1B, 5A) with significant media glucose depletion at this time (Figure 7A) suggests that low glucose could be permissive for insulin action on GCLc expression. To address the influence of low glucose, cells were incubated first for 14 hrs to decrease media glucose before the addition of insulin. Under these reduced glucose conditions, insulin enhanced GCLc expression within 6 hrs, which peaked at 8 hrs and remained elevated for 12 hrs (Figure 7B). These results suggest that the lengthened kinetics of insulin-induced GCLc expression at 36 hrs under normal glucose conditions (Figure 1A) was due to a time-dependent development of a low glucose state which appears to be requisite for GCLc upregulation.
Figure 7. Effect of low glucose on insulin-mediated GCLc induction.

A. Wild type IHECs were incubated for 0-48 hrs without or with 100 nM insulin and media glucose concentrations were determined at designated times using a glucometer. # p<0.05 vs IHEC minus insulin at respective time point. B-D. Wild type IHECs (B) or those overexpressing wt Nrf2, pEF-Nrf2 (C) or dominant negative Nrf2, pEF-dnNrf2 (D) were incubated for 12 hrs in serum free media prior to addition of 100 nM insulin. GCLc protein was measured at the indicated time points by Western blotting. One representative of 3 Western blots is shown. GCLc expression was normalized to β actin and expressed as fold change from 0 hrs. * p<0.05 vs 0 hrs.
To examine the role of Nrf2 in insulin induction of GCLc under low glucose conditions, media glucose was first decreased for 14 hrs in pEF-Nrf2 and pEF-dnNrf2 cell incubations prior to insulin treatment. Figure 7C shows that under these conditions, Nrf2 overexpression significantly stimulated GCLc expression at 4 hrs after insulin administration, and the elevated GCLc levels were maintained for 12 hrs. In contrast, GCLc levels were undetectable at all time points in pEF-dnNrf2 cells (Figure 7D). Also notable was that baseline Nrf2 expression was suppressed in pEF-dnNrf2 cells (Figure 7D), suggesting that Nrf2 is responsible for constitutive and insulin-induced GCLc expression in IHECs under conditions of decreased glucose.
Discussion
The current study demonstrates that insulin induced Nrf2-dependent GCLc expression, which enhanced endothelial cell GSH recovery during oxidative stress. Our results further show that the insulin effect is promoted by low glucose. These conclusions are supported by several lines of evidence. While insulin alone was insufficient to increase GSH, it elicited significant increases in GCLc message and protein levels that enhanced the cellular capacity for GSH recovery following acute tBH challenge. The finding that insulin-induced GSH recovery was blocked by inhibition of de novo GSH synthesis is consistent with a GCL-dependent event. A functional role for Nrf2 in insulin-mediated GSH increases was supported by the findings that overexpression of dominant negative Nrf2 suppressed insulin-induced as well as constitutive GSH synthesis. Additionally, insulin-mediated GCLc upregulation is kinetically faster in cells overexpressing wt-Nrf2 while being abrogated in dominant negative Nrf2 overexpressors. Furthermore, inhibition of insulin signaling with PI3K/Akt/mTOR kinase inhibitors effectively prevented insulin-induced GSH increases, in agreement with our previous studies [29]. Our suggestion that low glucose may be permissive for insulin-mediated GCLc induction and enhanced GSH synthesis is supported by the findings that (a) insulin increased GCLc expression on a significantly shorter time scale in cells cultured under low than normal glucose conditions, and (b) insulin administration under normal glucose conditions did not significantly elevate GCLc expression until glucose levels were markedly depleted.
The observations that insulin increases IHEC GCLc expression are in agreement with our recent findings in endothelial cells [29] and with previous studies in rat hepatocytes [16, 26]. However, the novel finding that insulin per se exerted no effect on GSH levels in IHECs is in contrast to hepatocytes, and suggests a critical post-transcriptional regulation of GSH at the level of the activity of GCL in endothelial cells. Importantly, the benefit of increased IHEC GCLc expression afforded by insulin is only apparent during an acute oxidative challenge wherein insulin treated cells exhibited an enhanced capacity to effectively recover their cellular GSH pool. It is notable that steady state cellular GSH levels was maintained at ~1 nmol/mg protein following tBH stress (Figures 2, 6); only in BSO-treated cells did cellular GSH fell below this level (Figure 2). One could speculate that when the free GSH pool reaches a minimal level during oxidative stress, the oxidation of an alternate thiol pool such as protein sulfhydryls could become important. This suggestion is consistent with our recent findings of a relationship between oxidation of protein thiols and susceptibility of IHECs and intestinal cells to hyperglycemia and menadione challenge, respectively [29, 32]. The current result that cells overexpressing dominant negative Nrf2 consistently maintained low GSH concentrations at ~1 nmol/mg protein that were unaltered by tBH treatment (Figure 6B) as compared to wild type cells (Figure 5B) supports the notion of a minimal threshold for GSH oxidation during oxidant exposure. The presence of a low and stable basal GSH pool in pEF-dnNrf2 cells (Figure 6B) in the absence of tBH stress could be the result of either incomplete inhibition of GCLc expression by the dominant negative mutant or greater competitive binding of endogenous Nrf2 or both. Alternatively this GSH pool could represent a GCL-independent pool derived from GSSG reduction and/or GSH transport.
A consistent observation in the current study is a two-fold increase in GCLc expression which supports the contention of a tight control of endothelial GSH synthesis. Our results are in agreement with published reports of two-fold induction of GCLc protein levels in various cell types regardless of whether the stimulus is endocrine [29, 33, 34], pharmacological [35], or genetic [5]. To our knowledge, only hepatocytes [26] exhibit GCLc increases of higher than 4-fold, consistent with a major role of hepatic cells in detoxication. Previous findings that GCLc knockout mice as well as systemic GCLc transgenic overexpressors are embryonic lethal [36, 37] supports a tight transcriptional control of GSH synthesis. Thus, while GSH concentrations are typically at millimolar levels in most cell types, cellular GSH homeostasis is a finely controlled process through rigorous regulation of GCLc expression.
As was previously demonstrated during hyperglycemic stress [29], PI3K/Akt/mTOR/Nrf2 signaling similarly mediates insulin-induced GCLc expression during hypoglycemic stress and GSH increases during tBH stress. Other observations that inhibition of PI3K/Akt/mTOR signaling abolished insulin-induced hepatocyte GCLc expression [26], and that PI3K signaling upregulates GCLc and/or GSH in response to growth factors [adrenomedullin, 38], T-type specific calcium channel inhibitor [flunarizine, 39], and carbon monoxide [22] suggest that PI3K/Nrf2 signaling represents a ubiquitous gene transcriptional pathway for GCLc induction in many cell types and under various stimuli. It is notable that a marked time lapse exists between activation of PI3K/Akt/mTOR (within minutes, Figure 3) and achievement of significant nuclear Nrf2 expression (in 24 hrs, Figure 5A). The reason for the slow response for Nrf2 nuclear expression is unclear, and could be related to mTOR signaling the translation of new Nrf2. The kinetic correspondence of a delayed 24-hr peak expression of nuclear Nrf2 with that of GCLc message that preceded expression of GCLc protein further suggests that achievement of threshold levels of nuclear Nrf2 may be necessary to activate GCLc transcription. Support for the interesting notion that blockade of PI3K/Akt/mTOR signaling prevented inducible but not basal GCLc expression comes from studies showing that an AP-1 sequence embedded within ARE4 of the GCLc promoter was responsible for constitutive enzyme activity but not for induction by β-naphthoflavone [40]. The findings of a lack of constitutive and insulin-induced GCLc expression in dominant negative Nrf2 cells confirm a role for Nrf2, in agreement with recent studies in IHECs [29]. Nrf2 involvement in constitutive and tert-butylhydroquinone (tBHQ) inducible expression of other antioxidant enzymes such as glutathione-S-transferase A2 (GST-A2) has been demonstrated [31]. Apart from PI3K, Nrf2 activity has also been shown to be regulated by other kinases including MAP kinases [41], protein kinase C (PKC) [42, 43], glycogen synthase kinase 3β (GSK3β) [44], and Fyn [45]. Moreover, its heterodimerization with different transcription factors represents another level of regulation [41, 46, 47].
Another novel observation is that low glucose promotes insulin induced GCLc expression and GSH increases. Taken together with our recent findings that hyperglycemia (25 mM glucose) modulates PI3K/Akt/mTOR and Nrf2 dependent transcriptional upregulation of GCLc [29], the current results suggest that altered glucose status contributed to insulin regulation of GCL activity. Hyper- and hypo-glycemia can induce oxidative stress via increased ROS generation [48, 49]. Hyperglycemia-stimulated H2O2 production was shown to enhance insulin-induced Akt phosphorylation [48], while diamide-induced GSH depletion and oxidative stress was demonstrated to inhibit the activity of Bach1, an ARE binding transcription factor that negatively regulates Nrf2 [50], thereby promoting Nrf2 signaling [51]. Interestingly, the effect of glucose on insulin signaling appears to be cell type specific; in rat hepatocytes insulin induction of GCLc and GSH was independent of media glucose concentration [16]. At present, the mechanism of glucose-induced effect on insulin-mediated GCLc expression is unclear and is a current focus in our laboratory. Preliminary evidence implicates ROS involvement and insulin induced GCLc promoter transactivation under conditions of hyper- and hypoglycemia.
In summary, we have demonstrated that insulin promoted GCLc expression and GSH recovery during oxidative challenge and that low glucose was permissive for the insulin effect. Our results are consistent with the activation of PI3K/Akt/mTOR signaling and Nrf2-dependent GCLc expression in insulin-mediated GSH synthesis. Given the existence of insulin receptors on endothelial cells, and that decreased glucose promoted insulin regulation of GCLc and GSH status, the current study will have important implications for understanding the role of insulin in endothelial protection under conditions of vascular oxidative stress and altered glycemic states, such as occurs in diabetes, hypertension, atherosclerosis, and coronary artery disease.
Acknowledgments
This study is supported by a grant from the National Institutes of Health, DK 44510.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maracine M, Aw TY. Redox Regulation of Cell Signaling. In: Matata BM, Elahi MM, editors. Oxidative Stress: Clinical and Biomedical Implications. Nova Science Publishers, Inc; 2007. pp. 103–132. [Google Scholar]
- 2.Usatyuk PV, Vepa S, Watkins T, He D, Parinandi NL, Natarajan V. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxid Redox Signal. 2003;5:723–730. doi: 10.1089/152308603770380025. [DOI] [PubMed] [Google Scholar]
- 3.Langston W, Chidlow JH, Jr, Booth BA, Barlow SC, Lefer DJ, Patel RP, Kevil CG. Regulation of endothelial glutathione by ICAM-1 governs VEGF-A-mediated eNOS activity and angiogenesis. Free Radic Biol Med. 2007;42:720–729. doi: 10.1016/j.freeradbiomed.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashino H, Shimamura M, Nakajima H, Dombou M, Kawanaka S, Oikawa T, Iwaguchi T, Kawashima S. Novel function of ascorbic acid as an angiostatic factor. Angiogenesis. 2003;6:259–269. doi: 10.1023/B:AGEN.0000029390.09354.f8. [DOI] [PubMed] [Google Scholar]
- 5.Kevil CG, Pruitt H, Kavanagh TJ, Wilkerson J, Farin F, Moellering D, Darley-Usmar VM, Bullard DC, Patel RP. Regulation of endothelial glutathione by ICAM-1: implications for inflammation. Faseb J. 2004;18:1321–1323. doi: 10.1096/fj.03-1401fje. [DOI] [PubMed] [Google Scholar]
- 6.Kugiyama K, Ohgushi M, Motoyama T, Hirashima O, Soejima H, Misumi K, Yoshimura M, Ogawa H, Sugiyama S, Yasue H. Intracoronary infusion of reduced glutathione improves endothelial vasomotor response to acetylcholine in human coronary circulation. Circulation. 1998;97:2299–2301. doi: 10.1161/01.cir.97.23.2299. [DOI] [PubMed] [Google Scholar]
- 7.Widder JD, Guzik TJ, Mueller CF, Clempus RE, Schmidt HH, Dikalov SI, Griendling KK, Jones DP, Harrison DG. Role of the multidrug resistance protein-1 in hypertension and vascular dysfunction caused by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:762–768. doi: 10.1161/01.ATV.0000259298.11129.a2. [DOI] [PubMed] [Google Scholar]
- 8.Biswas SK, Newby DE, Rahman I, Megson IL. Depressed glutathione synthesis precedes oxidative stress and atherogenesis in Apo-E(-/-) mice. Biochem Biophys Res Commun. 2005;338:1368–1373. doi: 10.1016/j.bbrc.2005.10.098. [DOI] [PubMed] [Google Scholar]
- 9.Tachi Y, Okuda Y, Bannai C, Bannai S, Shinohara M, Shimpuku H, Yamashita K, Ohura K. Hyperglycemia in diabetic rats reduces the glutathione content in the aortic tissue. Life Sci. 2001;69:1039–1047. doi: 10.1016/s0024-3205(01)01183-3. [DOI] [PubMed] [Google Scholar]
- 10.Prasad A, Andrews NP, Padder FA, Husain M, Quyyumi AA. Glutathione reverses endothelial dysfunction and improves nitric oxide bioavailability. J Am Coll Cardiol. 1999;34:507–514. doi: 10.1016/s0735-1097(99)00216-8. [DOI] [PubMed] [Google Scholar]
- 11.Kugiyama K, Miyao Y, Sakamoto T, Kawano H, Soejima H, Miyamoto S, Yoshimura M, Ogawa H, Sugiyama S, Yasue H. Glutathione attenuates coronary constriction to acetylcholine in patients with coronary spastic angina. Am J Physiol Heart Circ Physiol. 2001;280:H264–271. doi: 10.1152/ajpheart.2001.280.1.H264. [DOI] [PubMed] [Google Scholar]
- 12.Rouzer CA, Scott WA, Griffith OW, Hamill AL, Cohn ZA. Depletion of glutathione selectively inhibits synthesis of leukotriene C by macrophages. Proc Natl Acad Sci U S A. 1981;78:2532–2536. doi: 10.1073/pnas.78.4.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dethmers JK, Meister A. Glutathione export by human lymphoid cells: depletion of glutathione by inhibition of its synthesis decreases export and increases sensitivity to irradiation. Proc Natl Acad Sci U S A. 1981;78:7492–7496. doi: 10.1073/pnas.78.12.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J Biol Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- 15.Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257:13704–13712. [PubMed] [Google Scholar]
- 16.Lu SC, Ge JL, Kuhlenkamp J, Kaplowitz N. Insulin and glucocorticoid dependence of hepatic gamma-glutamylcysteine synthetase and glutathione synthesis in the rat. Studies in cultured hepatocytes and in vivo. J Clin Invest. 1992;90:524–532. doi: 10.1172/JCI115890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koide S, Kugiyama K, Sugiyama S, Nakamura S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. J Am Coll Cardiol. 2003;41:539–545. doi: 10.1016/s0735-1097(02)02866-8. [DOI] [PubMed] [Google Scholar]
- 18.Campolo J, Penco S, Bianchi E, Colombo L, Parolini M, Caruso R, Sedda V, Patrosso MC, Cighetti G, Marocchi A, Parodi O. Glutamate-cysteine ligase polymorphism, hypertension, and male sex are associated with cardiovascular events. Biochemical and genetic characterization of Italian subpopulation. Am Heart J. 2007;154:1123–1129. doi: 10.1016/j.ahj.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 19.Zuo HP, Xu WJ, Luo M, Zhu ZZ, Zhu GS. The glutamate-cysteine ligase catalytic subunit gene C-129T and modifier subunit gene G-23T polymorphisms and risk for coronary diseases. Zhonghua Xin Xue Guan Bing Za Zhi. 2007;35:637–640. [PubMed] [Google Scholar]
- 20.Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, Kunsch C. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J Biol Chem. 2003;278:703–711. doi: 10.1074/jbc.M203161200. [DOI] [PubMed] [Google Scholar]
- 21.Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 22.Li MH, Jang JH, Na HK, Cha YN, Surh YJ. Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress induces expression of glutamate-cysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J Biol Chem. 2007;282:28577–28586. doi: 10.1074/jbc.M701916200. [DOI] [PubMed] [Google Scholar]
- 23.Yang H, Magilnick N, Lee C, Kalmaz D, Ou X, Chan JY, Lu SC. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003;546:181–184. doi: 10.1016/s0014-5793(03)00517-9. [DOI] [PubMed] [Google Scholar]
- 25.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 26.Kim SK, Woodcroft KJ, Khodadadeh SS, Novak RF. Insulin signaling regulates gamma-glutamylcysteine ligase catalytic subunit expression in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 2004;311:99–108. doi: 10.1124/jpet.104.070375. [DOI] [PubMed] [Google Scholar]
- 27.Okouchi M, Okayama N, Aw TY. Differential susceptibility of naive and differentiated PC-12 cells to methylglyoxal-induced apoptosis: influence of cellular redox. Curr Neurovasc Res. 2005;2:13–22. doi: 10.2174/1567202052773535. [DOI] [PubMed] [Google Scholar]
- 28.Reed DJ, Babson JR, Beatty PW, Brodie AE, Ellis WW, Potter DW. High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Anal Biochem. 1980;106:55–62. doi: 10.1016/0003-2697(80)90118-9. [DOI] [PubMed] [Google Scholar]
- 29.Okouchi M, Okayama N, Alexander JS, Aw TY. NRF2-dependent glutamate-L-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia- induced brain endothelial cell apoptosis. Curr Neurovasc Res. 2006;3:249–261. doi: 10.2174/156720206778792876. [DOI] [PubMed] [Google Scholar]
- 30.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem. 2005;280:32485–32492. doi: 10.1074/jbc.M503074200. [DOI] [PubMed] [Google Scholar]
- 32.Circu ML, Rodriguez C, Maloney R, Moyer MP, Aw TY. Contribution of mitochondrial GSH transport to matrix GSH status and colonic epithelial cell apoptosis. Free Radic Biol Med. 2008;44:768–778. doi: 10.1016/j.freeradbiomed.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai-Turton M, Luderer U. Gonadotropin regulation of glutamate cysteine ligase catalytic and modifier subunit expression in rat ovary is subunit and follicle stage specific. Am J Physiol Endocrinol Metab. 2005;289:E391–402. doi: 10.1152/ajpendo.00531.2004. [DOI] [PubMed] [Google Scholar]
- 34.Takamura Y, Fatma N, Kubo E, Singh DP. Regulation of heavy subunit chain of gamma-glutamylcysteine synthetase by tumor necrosis factor-alpha in lens epithelial cells: role of LEDGF/p75. Am J Physiol Cell Physiol. 2006;290:C554–566. doi: 10.1152/ajpcell.00398.2005. [DOI] [PubMed] [Google Scholar]
- 35.Hoang YD, Avakian AP, Luderer U. Minimal ovarian upregulation of glutamate cysteine ligase expression in response to suppression of glutathione by buthionine sulfoximine. Reprod Toxicol. 2006;21:186–196. doi: 10.1016/j.reprotox.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279:324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- 37.Botta D, Shi S, White CC, Dabrowski MJ, Keener CL, Srinouanprachanh SL, Farin FM, Ware CB, Ladiges WC, Pierce RH, Fausto N, Kavanagh TJ. Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J Biol Chem. 2006;281:28865–28875. doi: 10.1074/jbc.M605143200. [DOI] [PubMed] [Google Scholar]
- 38.Kim JY, Yim JH, Cho JH, Kim JH, Ko JH, Kim SM, Park S, Park JH. Adrenomedullin regulates cellular glutathione content via modulation of gamma-glutamate-cysteine ligase catalytic subunit expression. Endocrinology. 2006;147:1357–1364. doi: 10.1210/en.2005-0895. [DOI] [PubMed] [Google Scholar]
- 39.So HS, Kim HJ, Lee JH, Park SY, Park C, Kim YH, Kim JK, Lee KM, Kim KS, Chung SY, Jang WC, Moon SK, Chung HT, Park RK. Flunarizine induces Nrf2-mediated transcriptional activation of heme oxygenase-1 in protection of auditory cells from cisplatin. Cell Death Differ. 2006;13:1763–1775. doi: 10.1038/sj.cdd.4401863. [DOI] [PubMed] [Google Scholar]
- 40.Wild AC, Gipp JJ, Mulcahy T. Overlapping antioxidant response element and PMA response element sequences mediate basal and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase catalytic subunit gene. Biochem J. 1998;332(Pt 2):373–381. doi: 10.1042/bj3320373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen G, Hebbar V, Nair S, Xu C, Li W, Lin W, Keum YS, Han J, Gallo MA, Kong AN. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem. 2004;279:23052–23060. doi: 10.1074/jbc.M401368200. [DOI] [PubMed] [Google Scholar]
- 42.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 43.Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol. 2003;285:C334–342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 44.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- 45.Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281:12132–12142. doi: 10.1074/jbc.M511198200. [DOI] [PubMed] [Google Scholar]
- 46.Marini MG, Chan K, Casula L, Kan YW, Cao A, Moi P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J Biol Chem. 1997;272:16490–16497. doi: 10.1074/jbc.272.26.16490. [DOI] [PubMed] [Google Scholar]
- 47.Dhakshinamoorthy S, Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 48.Wu X, Zhu L, Zilbering A, Mahadev K, Motoshima H, Yao J, Goldstein BJ. Hyperglycemia potentiates H(2)O(2) production in adipocytes and enhances insulin signal transduction: potential role for oxidative inhibition of thiol-sensitive protein-tyrosine phosphatases. Antioxid Redox Signal. 2005;7:526–537. doi: 10.1089/ars.2005.7.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGowan JE, Chen L, Gao D, Trush M, Wei C. Increased mitochondrial reactive oxygen species production in newborn brain during hypoglycemia. Neurosci Lett. 2006;399:111–114. doi: 10.1016/j.neulet.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 50.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 51.Ishikawa M, Numazawa S, Yoshida T. Redox regulation of the transcriptional repressor Bach1. Free Radic Biol Med. 2005;38:1344–1352. doi: 10.1016/j.freeradbiomed.2005.01.021. [DOI] [PubMed] [Google Scholar]