

Abstract
Rapid-mix freeze-quench (RMFQ) methods and electron paramagnetic resonance (EPR) spectroscopy have been used to characterize the steady-state radical in the deamination of ethanolamine catalyzed by adenosylcobalamin (AdoCbl)-dependent ethanolamine ammonia-lyase (EAL). EPR spectra of the radical intermediates formed with the substrates, [1-13C]ethanolamine, [2-13C]ethanolamine, and unlabeled ethanolamine were acquired using RMFQ trapping methods from 10 ms to completion of the reaction. Resolved 13C hyperfine splitting in EPR spectra of samples prepared with [1-13C]ethanolamine and the absence of such splitting in spectra of samples prepared with [2-13C]ethanolamine show that the unpaired electron is localized on C1 (the carbinol carbon) of the substrate. The 13C splitting from C1 persists from 10 ms throughout the time course of substrate turnover, and there was no evidence for a detectable amount of a product like radical having unpaired spin on C2. These results correct an earlier assignment for this radical intermediate [Warncke et al. (1999) J. Am. Chem. Soc. 121, 10522-10528]. The EPR signals of the substrate radical intermediate are altered by electron spin coupling to the other paramagnetic species, cob(II)alamin, in the active site. The dipole-dipole and exchange interactions as well as the 1-13C hyperfine splitting tensor were analyzed through spectral simulations. The sign of the isotropic exchange interaction indicates a weak ferromagnetic coupling of the two unpaired electrons. A Co2+ to radical distance of 8.7Å was obtained from the magnitude of the dipole-dipole interaction. The orientation of the principal axes of the 13C hyperfine splitting tensor shows that the long axis of the spin-bearing p orbital on C1 of the substrate radical makes an angle of ∼98° with the unique axis of the dz2 orbital of Co2+.
Ethanolamine ammonia-lyase (EAL1, EC 4.3.1.7) is an AdoCbl dependent enzyme that catalyzes elimination of ammonia from the vicinal position of short chain amino-alcohols such as ethanolamine to give the corresponding oxo products. The functional protein is believed to be a hexamer of αβ-dimers ((αβ)6, α ∼50 kDa and β ∼31 kDa) (1, 2). EAL is proposed to be an important enzyme in the metabolism of some bacterial species such as Salmonella enterica, which can use ethanolamine, derived from the breakdown of phospholipids, as their sole source of carbon, nitrogen, and energy (3). Catalysis by EAL with various substrates and inactivation of EAL with substrate analogues have been studied extensively by kinetic methods, and radical states occurring during catalysis have been probed by EPR spectroscopy (4-9).
The putative catalytic cycle of EAL starts with the formation of a highly reactive 5′-deoxyadenosyl radical and cob(II)alamin after homolysis of the Co-carbon bond of the cofactor (Scheme 1) (10). The transient 5′-deoxyadenosyl radical abstracts the pro-S hydrogen atom from C1 of the substrate to create the initial substrate radical (11). Abstraction of the hydrogen atom from C1 is kinetically coupled to the Co-carbon bond homolysis and pulls the Co-carbon bond cleavage process forward (6). The substrate radical rearranges to a product-like cabinolamine radical, which abstracts a hydrogen atom from the 5′ carbon of 5′-deoxyadenosine to give a carbinolamine. Breakdown of the carbinolamine to ammonia and acetaldehyde and recombination of the 5′-deoxyadenosyl radical with cob(II)alamin complete the catalytic cycle. Details of the radical rearrangement step are not yet clear. The product radical was proposed to be a carbinolamine radical on the basis of theoretical energy calculations of possible intermediates and transition states (12-14).
Scheme 1.

EPR spectroscopy reveals the presence of organic radical intermediates spin coupled to cob(II)alamin whenever reaction mixtures with ethanolamine or with (R) or (S)-2-aminopropanol are frozen during turnover (15-18). EPR spectra of the paramagnetic species consist of the gx and gy signals of cob(II)alamin at g ∼ 2.3 and the signal of the organic radicals at g ∼ 2.003 (17). Cob(II)alamin and the organic radicals interact magnetically through exchange and dipole-dipole interactions, creating spectra in which signals for each paramagnetic species are perturbed by the electron-electron spin coupling with the partner (19).
The EPR spectrum of a radical intermediate in the reaction of EAL with ethanolamine was previously assigned as a product-like carbinolamine having unpaired spin on C2 (20). The distance between the radical intermediate and the cobalt center of cob(II)alamin was determined to be ∼ 9.7 Å (21). Theoretical hyperfine splitting values of different possible radical identities and conformations were obtained using electronic structure calculations (22). The structure of a radical intermediate of ethanolamine was further characterized by electron spin echo envelope modulation spectroscopy (13, 23, 24). However, conclusions from all of these studies were based on the original assignment of the radical intermediate as the carbinolamine product radical (20). In the earlier studies, concentrated solutions of enzyme were pre-mixed with a concentrated solution of ethanolamine (20). The reaction was initiated by addition of AdoCbl to the enzyme/substrate mixture prior to manual freezing of the reaction mixtures. There is a slow binding of AdoCbl to the enzyme that delays consumption of substrate using this order-of-addition (25, 26). AdoCbl binds to the enzyme with high affinity, and its binding and dissociation are probably not part of the normal catalytic cycle (27). Moreover, significant amounts of acetaldehyde and ammonia are generated during the several seconds required to mix and freeze the sample.
In this study, the radical intermediate observed during the steady-state regime in the reaction of EAL with ethanolamine is characterized by RMFQ trapping methods (28). These methods make it possible to use much lower concentrations of substrate and buffer and to use the physiological order-of-addition of substrate to the enzyme-cofactor complex. The radical intermediates are probed throughout the time-course of substrate turnover with C1 and C2 isotopomers of (13C)ethanolamine. Distance and geometrical information regarding the positions of the radical relative to cob(II)alamin are obtained from detailed analysis of electron spin-spin and nuclear spin electron spin interactions.
EXPERIMENTAL PROCEDURES
Materials
AdoCbl was purchased from Sigma-Aldrich. [1-13C]ethanolamine was purchased from Cambridge Isotopes, and [2-13C]ethanolamine was purchased from Isotec. The position of 13C in commercial samples of (13C)ethanolamine, was confirmed by 13C NMR (see Supplementary Material). The 13C chemical shift of C1 is more downfield (∼61 ppm) than the 13C chemical shift of C2 (∼45 ppm). The chemical shifts of the commercial samples of (13C)ethanolamine were further compared to the chemical shifts of samples of (13C)ethanolamine synthesized from (13C)glycine. The labeling patterns of the synthetic samples were verified by the one-bond or two-bond 1H-13C coupling constants of the starting materials, 2-13C-glycine and 1-13C-glycine.
Purification of EAL
EAL of Salmonella typhimurium (E. C. 4.3.1.7) was over-expressed in Escherichia coli and purified as previously described (4). All steps of purification were done at 4 °C. After ammonium sulfate precipitation, the enzyme was solubilized by dialyzing against 10 mM HEPES/NaOH (pH 7.5) for ∼48 hours, with 5-6 buffer changes. At the end of dialysis, the insoluble material was removed by centrifugation at 30000 g for 30 min. The final concentration of enzyme was 40-50 mg mL−1, and the specific activity was ∼30 – 45 μmols of ethanolamine min−1 mg−1 of EAL at 25 °C, as determined by the EAL-alcohol dehydrogenase coupled assay (27). Concentrations of apo-EAL were measured spectrophotometrically at 280 nm using an extinction coefficient of 0.69 mL mg−1 cm−1, molecular mass of 82 kD of an αβ unit of the (αβ)6 protomer, and six active sites per protomer (4).
Stopped Flow Spectrophotometry
The extent of Co-carbon bond cleavage in AdoCbl during the steady-state of the reaction of EAL with ethanolamine was assayed by stopped flow spectrophotometry. The measurements were carried out with an Applied Photophysics SX.18MV-R reaction analyzer. The transient decrease in absorption of AdoCbl at 525 nm was monitored upon mixing of a solution of the EAL-AdoCbl complex with ethanolamine. Calibration of Co-carbon bond cleavage was accomplished by mixing the EAL-AdoCbl complex with an excess of hydroxyethylhydrazine. The latter suicide inhibitor effects a stoichiometric conversion of enzyme-bound AdoCbl to its cleaved form, cob(II)alamin (4).
Preparation of Samples by RMFQ and Acquisition of EPR Spectra
RMFQ samples were prepared using an Update Instruments syringe RAM essentially in the same manner as described previously (28, 29). EAL and AdoCbl, both in 10 mM HEPES/NaOH, pH 7.5, were premixed at 0.46 mM enzyme (active sites) and 0.62 mM AdoCbl to form the holoenzyme. The holoenzyme solution was mixed with 8 mM ethanolamine, pH 7.5 using the rapid mixing apparatus. The reactions were freeze-quenched by injection into cold isopentane (−130 °C). Following packing at −130 °C, the samples were stored in liq. N2. X-band EPR spectra were obtained at 77 K using a Varian E-3 EPR spectrometer and a standard liquid N2 immersion Dewar. Data were acquired digitally through a custom interface and a PC using modified XEMR (v 0.7) software.
Spectral Simulations
Magnetic interaction parameters were obtained through simulation of EPR spectra using the following spin Hamiltonian (4):
| (1) |
The first two terms in eq 1 present the electronic Zeeman interaction of low spin Co2+ (S1) and the organic radical (S2). The third and fourth terms describe the exchange (J), and the dipole-dipole (D) interactions between the two electron spins. The final term describes the hyperfine interactions (HHFI) between the electron spins and nuclear spins. Orthogonal transformations, using Euler angles, are used to express the tensor quantities in eq 1 in a common reference frame that was chosen to be the g axis system of the low-spin Co2+. The Euler rotations used the “x-convention” as described by Goldstein (30). Additional details of the diagonalization of the energy matrix and generation of “field-swept” EPR spectra are provided in previous papers (4, 31). The fitting was done by trial and error.
In the EPR spectra the Co2+–radical pair, hyperfine splitting from 59Co (I=7/2) of cob(II)alamin and from the 1-13C (I=1/2) in the sample of labeled ethanolamine are the only resolved hyperfine features in the spectra. Hyperfine splitting from 14N (I=1) of the dimethylbenzimidazole lower axial ligand of Co2+ (29, 32, 33), from the α-1H (I=1/2), the β-14N (I=1), and two β-1H's are not resolved and contribute an inhomogeneous broadening in the signals. Estimates of hyperfine splitting parameters of these latter nuclear spins were derived from separate studies of selectively deuterated, 13C, and 15N isotopologues of ethanolamine (34). These parameters were included in the present simulations in order to account for the inhomogeneous line widths.
RESULTS
Extent of Cobalt-Carbon Bond Cleavage in the Steady-State
Cleavage of the Co–carbon bond of AdoCbl results in a decrease in the absorbance at 525 nm. The transient drop in absorbance upon mixing solutions of the EAL-AdoCbl complex with ethanolamine was compared to the absorbance change observed upon stoichiometric cleavage of enzyme-bound AdoCbl with hydroxyethylhydrazine (4). The transient ΔA525 for the reaction with ethanolamine is approximately (70 ± 5) % of the 100% cleavage achieved with hydroxyethylhydrazine (Figure S2 Supplementary Material).
Observation of the Earliest Paramagnetic Intermediate
EPR spectra of samples corresponding to 10 ms following mixing of substrate with the enzyme-cofactor complex exhibit signals at g values corresponding to low spin Co2+ and organic free radical, respectively (see Figure 1A, a). Signals for both the low spin Co2+ and organic free radical are perturbed by the electron spin-spin coupling interactions. In contrast to the “radical doublet” (see Figure 1A, b) observed in analogous spectra of the intermediate observed with the 2-aminopropanol's (17, 18, 29), the EPR signal of the organic radical component produced with ethanolamine gives a broad singlet.
Figure 1.
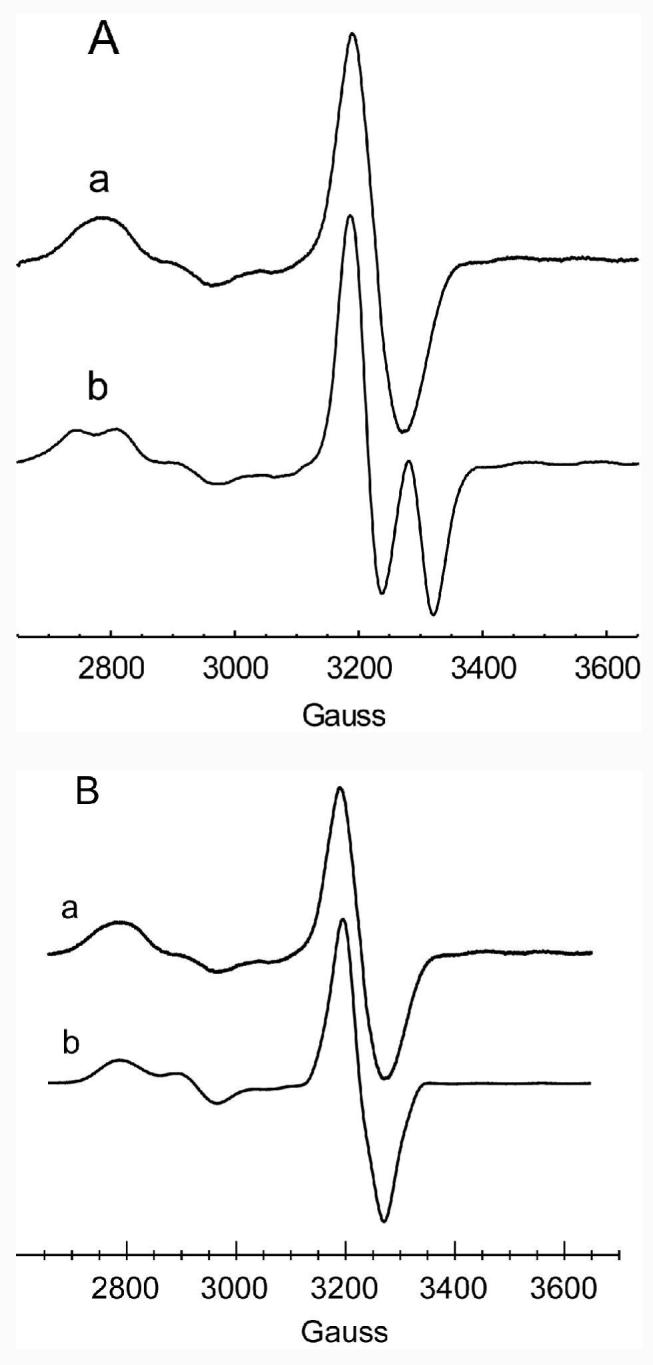
A: Comparison of the EPR spectra of the steady-state radical intermediates obtained with a ethanolamine by rapid mix freeze quench (RMFQ) at 10 ms, and b (S)-2-aminopropanol obtained by mixing and manually freezing the sample. For ethanolamine, a, the solution of EAL (0.46 mM active sites) and AdoCbl (0.62 mM) in 10 mM Hepes/NaOH, pH 7.5, was mixed with an equal volume of 8 mM ethanolamine in 10 mM Hepes/NaOH adjusted to pH 7.5. For the sample of (S)-2-aminopropanol, b, the sample contained initially 0.2 mM EAL (active sites), 0.4 mM AdoCbl, and 2 mM (S)-2-aminopropanol in 10 mM Hepes/NaOH adjusted to pH 7.5. (S)-2-Aminopropanol was added last and the sample was frozen in ∼ 20 s by dipping the EPR tube into a chilled slush of isopentane. The spectra were recorded at 77 K. Spectrum a is the average of 8 240 s scans and spectrum b is the average of 4 240 s scans. Spectra were recorded at 9.05 GHz (“free electron” g = 2.0023 at 3230 G) under non-saturating microwave power (4 mW) and a field modulation of 8 G.
B: Comparison of experimental, a, and simulated, b, EPR spectra of the EPR spectrum of the steady-state radical obtained with ethanolamine as the substrate. The experimental conditions are given in Figure 1A. The parameters used in the simulation are: J = −53 G; D = −43 G, E= −4 G; Euler angles for the D tensor 0°, 25°, 0°; g tensor of Co2+ [2.232, 2.205, 1.975]; A tensor of 59Co [7, 4, 109] G; line width of Co2+ transitions 25 G (isotropic); Aiso 14N lower axial ligand 15 G; substrate radical g tensor [2.005, 2.003, 2.002]; A tensor of α-proton [−35, −22, −4] G, Euler angles 140°, −110°, −130°; Aiso of β-Ha 12.5 G; Aiso of β-Hb 5.5 G; Aiso 14N (ethanolamine) 12 G; line width of radical transitions 12 G (isotropic). As noted in the text, the hyperfine splitting parameters representing unresolved hyperfine structure were included in the simulation to account for the inhomogeneously broadened lines. These parameters were derived from separate simulations of spectra of selectively deuterated samples in D2O (34).
The appearance of the radical component in the Co2+-radical spin coupled pair depends on the relative signs of the exchange coupling parameter, J, and the dipole-dipole coupling parameter, D (see eq 1) (29). The latter parameter is negative, by convention (35). Splitting from the effects of exchange and of dipole-dipole coupling add constructively in the central region of the radical spectrum whenever J and D are of opposite signs, and that situation occurs when the exchange coupling is antiferromagnetic (see Figure 1A, b) (29). On the other hand, for the case of ferromagnetic coupling (i. e., J negative using the sign convention in eq 1), splitting from the exchange and from the dipole-dipole interactions will tend to cancel one another in the middle of the pattern (29). Simulations of the present spectra (Figure 1B) as well as those obtained from samples made with perdeuterated ethanolamine indicate that the exchange interaction is ferromagnetic (34). The switch from a doublet signal to a broad singlet upon going from the slow substrates, (S) and (R)-2-aminopropanol, to ethanolamine can therefore be attributed to a change in the sign of the exchange coupling parameter, J, instead of a dramatic change in the inter-spin distance. Except for the sign of J, the magnitudes of the electron spin coupling parameters J = −53 ± 2 G and D = −43 ± 5 G are in the same range as those found for the substrate radical of (S)-2-aminopropanol (J= 70 G and D = −23 G) (29). Euler angles relating the D tensor to the g axis system of Co2+ are: 0°, (25 ± 10)°, 0°. The inter-spin vector and the z-axis of Co2+ therefore describe an angle of ∼ 25°. The small E term (− 4 G) may result from the slight off-axis positioning of the two spins (36). The |J| is close to that reported recently: however, Sun et al. (37) and Canfield & Warncke (24) use a model of antiferromagnetic coupling. It is also noteworthy that in the EPR spectra reported by Sun et al. (37), the radical signal obtained with unlabeled ethanolamine is split into a doublet–unlike the singlet spectrum reported here for the RMFQ sample with unlabeled ethanolamine. It is possible to generate splitting (data not shown) by combinations of high salt and glycerol used in the buffer system of Sun et al. (37). However, under these same conditions there is a noticeable amount of precipitation of the protein isolated by the procedure of Bandarian & Reed (4). This latter procedure avoids the use of Triton that was part of the purification originally described by Faust and Babior (2) and used by Sun et al. (37). Changes in the character of the EPR spectra with different buffer or salt conditions indicate that these solution components alter the active site in such a manner to influence the spin-spin coupling parameters. The activity of the enzyme is, however, virtually the same in the buffer used here and in that used by Sun et al. (37).
Adjustments to J and D allow simulation of the broad singlet spectrum for the radical observed with ethanolamine using an anitferromagnetic coupling scenario. However, when deuterated substrates and D2O are used, the improved resolution in the spectrum, wherein additional hyperfine structure is visible, makes it impossible to simulate the spectra with antiferromagnetic coupling (34).
Location of Unpaired Spin in the Radical Intermediate
Spectra obtained at 10 ms for samples of unlabeled ethanolamine, [2-13C]ethanolamine and [1-13C]ethanolamine are compared in Figure 2. Clearly the radical appearing at 10 ms has unpaired spin at C1 where it couples strongly to the 13C nuclear spin. The time courses for the reactions with the isotopomers of (13C)ethanolamine (Figure 3) show that the hyperfine splitting from [1-13C]ethanolamine persists throughout the course of substrate turnover whereas no hyperfine splitting appears for 13C at C2. These observations indicate that the substrate radical is the dominant paramagnetic species arising from the substrate during the entire time course of the reaction. Differences in the line shape of the spectrum of the radical with different buffer/salt compositions (noted above) do not, however, account for the position of 13C sensitivity, which is at C1 using either the present RMFQ protocol or the buffer system and manual mixing and freezing protocol of Warncke et al. (20). Radical intermediates that occur after rearrangement of the substrate radical do not achieve concentrations sufficient to allow detection by EPR. The results reported in Figures 2 and 3 have led to a correction to the original assignment of the radical signals to a rearranged or product radical species (38).
Figure 2.

EPR spectra of RMFQ samples with unlabeled ethanolamine and with isotopomers of (13C)ethanolamine. The sample compositions are given in the legend of Figure 1 except for substitution of: b [2-13C]ethanolamine ; c [1-13C]ethanolamine for a ethanolamine. The reactions were freeze-quenched at 10 ms. Each spectrum represents the average of 6 scans. Other acquisition parameters are given in the Legend of Figure 1.
Figure 3.

Time course of the EPR signal of the radical intermediate when either A [1-13C] ethanolamine or B [2-13C]ethanolamine as the substrate. The respective reaction times when the mixtures were freeze quenched are indicated on the figure. Acquisition parameters are as indicated in the Legend of Figure 1.
Analysis of the 13C Hyperfine Interaction
Central atom hyperfine splitting interactions in π radicals is typically strong, anisotropic, and approximately axially symmetric (39, 40). A 13C hyperfine interaction tensor was introduced into the spectral simulations to characterize this electron spin–nuclear spin interaction. Inclusion of the 13C hyperfine interaction in spectral simulations requires that the 13C hyperfine splitting tensor (together with the other tensors in eq 1) be expressed in a common frame of reference, which is taken to be the g axis system of the low spin Co2+. Results of the simulations are compared to the experimental spectrum in Figure 4. 13C hyperfine tensors ranging from [7,13,110] to [12,12,120] (values in Gauss) reproduce the observed splitting in the experimental spectra. Euler angles relating the 13C hyperfine tensor to the g axis were 0°, (98 ± 2)°, (20 ± 10)°. The simulations demonstrate that the line shape is particularly sensitive to the orientation (second Euler angle) of the z aixs of 13C hyperfine tensor to the gz axis of Co2+, and this second Euler angle shows that the unique axis of the spin-bearing p orbital of the substrate radical is oriented approximately perpendicular to the unique axis of the dz2 orbital of Co2+. The approximately axial symmetry of both tensors renders the direction of z 13C relative to gx and gy of Co2+ essentially arbitrary.
Figure 4.

Analysis of the 13C hyperfine splitting interaction in EPR spectra of the substrate radical during the reaction of 1-13C-ethanolamine. The experimental spectrum taken at t = 10 ms is reproduced in a. b: The spectrum calculated using a 13C hyperfine tensor [10, 10, 120] G and Euler angles 0°, 98°, 20°. c: The spectrum calculated using a 13C hyperfine tensor of [7, 13, 110] G and Euler angles 0°, 98°, 20°. d: The spectrum calculated using the same 13C tensor as in d but with Euler angles of 0°, 0°, 0°. The other simulation parameters are the same as given in the Legend of Figure 1.
DISCUSSION
Mechanistic Significance of the Substrate Radical
Optical spectra obtained by stopped flow, in which the enzyme–AdoCbl complex is mixed with ethanolamine, indicate that ∼ 70% of the enzyme-bound AdoCbl is cleaved to cob(II)alamin in the steady state of the reaction. The present EPR results indicate that virtually all of this cleaved cofactor is in a complex with the substrate radical of ethanolamine because the EPR spectra do not show detectable amounts of other radicals. This result in turn suggests that cleavage of the C–N bond in the substrate radical is a significant barrier in the overall reaction. This conclusion is in accord with the observation of a 15(V/K) KIE that was reported earlier from isotope ratio mass spectrometric measurements (18). The observation of a 15(V/K) KIE shows that the reaction is reversible in all steps up to and including the formation of the substrate radical. These steps include Co-carbon bond cleavage and abstraction of the pro S hydrogen atom from C1 of ethanolamine. The fact that the 15(V/K) KIE (1.0017) is significantly smaller than that expected from a full expression of a nitrogen IE (1.03) suggests that the forward commitment is high. The 15N KIE may be more fully expressed in Vmax. Preliminary results show that the 15V obtained by direct comparison is significantly larger than the measured 15(V/K) KIE (Poyner, R. R., Anderson, M. A., Cleland, W. W., and Reed, G. H. preliminary results).
The assignment of the substrate radical in the steady-state also brings ethanolamine into register with results for (S)-2-aminopropanol, wherein the substrate radical is present in the steady state (17, 29). The reports (13, 20, 23) of a rearranged-substrate (carbinolamine) radical were surprising from the viewpoint of predicted stability of free radicals. In addition to the intrinsic lability of hemiaminals, the carbinolamine radical would have hyperconjugation to β substituents as its sole means of spin delocalization. On the other hand, several resonance structures are possible for the substrate radical wherein the radical center is adjacent to an –OH group (41). The fact that radical intermediates following cleavage of the C–N bond of the substrate radical are too unstable, and thereby too dilute, to detect by EPR spectroscopy means that the rearrangement step of the catalytic cycle remains elusive in contrast to the earlier suggestions (13). The assignment of the radical center to C1 of ethanolamine also obviates the need to invoke two conformations of the radical in order to account for the duality of β-proton couplings seen in electron spin echo envelope modulations (24) because the substrate radical has two distinct β-protons.
Assignment of the substrate radical as the predominant intermediate in the reaction of EAL with ethanolamine simplifies interpretations of V and of V/K KIE's. If steps subsequent to the rearrangement were to be significantly rate limiting, then any paramagnetic species associated with these intermediates should be detectable in the steady-state. Thus, it is now clear that the 15N sensitive step assayed as 15(V/K) in the isotope ratio mass spectrometry measurements is the initial cleavage of the C2–N bond of the substrate radical (17). The steady-state mechanism fits the model shown in eq 3:
| (3) |
where EA is the enzyme-ethanolamine complex, EA• is the complex with the substrate radical, EP• is a product radical complex, and the “k7 arrow” includes all of the remaining steps leading to products. Because the concentrations of intermediates following C2–N bond cleavage are too low to be detected, the step represented by k5 is effectively the first irreversible step, and V/(KET) becomes:
| (4) |
The expression for V/ET is:
| (5) |
These equations can be used to derive expressions for the KIE's on V and on V/K in terms of the intrinsic rate constants (42, 43).
Structural Conclusions from the Spin-Spin Interaction Parameters
The magnitude and sign of the exchange interaction, J = − 0.005 cm−1, reflect a weak ferromagnetic exchange interaction between the unpaired electron on the radical and the unpaired electron in the dz2 orbital of Co2+. Because such weak interactions are likely mediated by spin polarization in intervening matter (super exchange) and the nature of intervening matter is not presently known, neither the magnitude nor the sign of J can be readily interpreted in structural terms (35). However, as noted above, both the sign and magnitude of J have significant influences on the appearance of the spectra. The approximately 98° angle between the respective unique axes of the two spin bearing orbitals versus an approximately co-linear orientation in the corresponding situation with the substrate radical of S-2-aminopropanol (29) is almost certainly a factor in the differences in signs and magnitudes of the exchange interactions observed for these two substrates (35). These differences in orbital alignment and distances between the radical centers and Co2+ observed with the substrate radicals of (S)-2-aminopropanol and ethanolamine could contribute to the 250-fold difference in kcat's of the two substrates.
An inter-spin distance, r, of 8.7Å is obtained using the point-dipole approximation eq 2 (44);
| (2) |
and D = −43 Gauss derived from the spectral simulations. This result places the spin-bearing carbon ∼ 2.3 Å closer to the Co2+ of cob(II)alamin than the ∼ 11Å in the corresponding substrate radical of (S)-2-aminopropanol (29). The structural results obtained from the spin-spin interaction parameters are summarized in the Scheme 2.
Scheme 2.

Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Steven O. Mansoorabadi for help with computations, Dr. Mark Anderson for acquiring the 13C NMR spectra, and Drs. Perry Frey and W. W. Cleland for helpful discussion.
Footnotes
This research was supported by NIH Grant R56-GM35752.
Abbreviations: EAL, ethanolamine ammonia lyase; AdoCbl, 5′-deoxyadenosylcobalamin; EPR, electron paramagnetic resonance; NMR, nuclear magnetic resonance; RMFQ, rapid mix freeze quench: HEPES, N-(2-hydroxyethyl)piperazine-N'-2-ethanesulfonic acid, KIE, kinetic isotope effect; IE isotope effect.
SUPPORTING INFORMATION AVAILABLE
13C NMR spectra of (13C)ethanolamines and 1H NMR spectra of (13C)glycine precursors. Stopped flow spectrophotometric assay for Co-carbon bond cleavage in the steady-state with ethanolamine. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Wallis OC, Johnson AW, Lappert MF. Studies on the subunit structure of the adenosylcobalamin-dependent enzyme ethanolamine ammonia-lyase. FEBS Lett. 1979;97:196–199. doi: 10.1016/0014-5793(79)80083-6. [DOI] [PubMed] [Google Scholar]
- 2.Faust LP, Babior BM. Overexpression, purification, and some properties of the AdoCbl-dependent ethanolamine ammonia-lyase from Salmonella typhimurium. Arch. Biochem. Biophys. 1992;294:50–54. doi: 10.1016/0003-9861(92)90135-j. [DOI] [PubMed] [Google Scholar]
- 3.Roof DM, Roth JR. Ethanolamine utilization in Salmonella-typhimurium. J. Bacteriol. 1988;170:3855–3863. doi: 10.1128/jb.170.9.3855-3863.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bandarian V, Reed GH. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry. 1999;38:12394–12402. doi: 10.1021/bi990620g. [DOI] [PubMed] [Google Scholar]
- 5.Babior BM. The mechanism of action of ethanolamine deaminase. VI. Ethylene glycol, a quasi-substrate for ethanolamine deaminase. J. Biol. Chem. 1970;245:1755–1766. [PubMed] [Google Scholar]
- 6.Bandarian V, Reed GH. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonia-lyase: determination of the number of exchangeable hydrogens in the enzyme-cofactor complex. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- 7.Graves SW, Babior BM. Interaction of N-substituted ethanolamine analogs with ethanolamine ammonia-lyase, an adenosylcobalamin-requiring enzyme. J. Biol. Chem. 1982;257:4102–4105. [PubMed] [Google Scholar]
- 8.Krouwer JS, Schultz RM, Babior BM. The mechanism of action of ethanolamine ammonia-lyase, an adenosylcobalamin-dependent enzyme. Reaction of the enzyme.cofactor complex with 2-aminoacetaldehyde. J. Biol. Chem. 1978;253:1041–1047. [PubMed] [Google Scholar]
- 9.Wang M, Warncke K. Kinetic and thermodynamic characterization of Co(II)-substrate radical pair formation in coenzyme B12-dependent ethanolamine ammonia-lyase in a cryosolvent system by using time-resolved, full-spectrum continuous-wave electron paramagnetic resonance spectroscopy. J. Am. Chem. Soc. 2008;130:4846–4858. doi: 10.1021/ja710069y. [DOI] [PubMed] [Google Scholar]
- 10.Bandarian V, Reed GH. Ethanolamine Ammonia-Lyase. In: Banerjee R, editor. Chemistry and Biochemistry of B12. Wiley-Interscience; 1999. pp. 811–834. [Google Scholar]
- 11.Yan S-J, McKinnie BG, Abacherli C, Hill RK, Babior BM. Stereochemistry of the ethanolamine ammonia lyase reaction with stereospecifically labeled [1-2H1]-2-aminoethanol. J. Am. Chem. Soc. 1984;106:2961–2964. [Google Scholar]
- 12.Semialjac M, Schwarz H. Computational exploration of rearrangements related to the vitamin B12-dependent ethanolamine ammonia lyase catalyzed transformation. J. Am. Chem. Soc. 2002;124:8974–8983. doi: 10.1021/ja020101s. [DOI] [PubMed] [Google Scholar]
- 13.Warncke K, Canfield JM. Direct determination of product radical structure reveals the radical rearrangement pathway in a coenzyme B12-dependent enzyme. J. Am. Chem. Soc. 2004;126:5930–5931. doi: 10.1021/ja031569d. [DOI] [PubMed] [Google Scholar]
- 14.Wetmore SD, Smith DM, Bennett JT, Radom L. Understanding the mechanism of action of B12-dependent ethanolamine ammonia-lyase: synergistic interactions at play. J. Am. Chem. Soc. 2002;124:14054–14065. doi: 10.1021/ja027579g. [DOI] [PubMed] [Google Scholar]
- 15.Babior B, Gould DC. An EPR signal generated by the ethanolamine deaminase-coenyzme B12 complex in the presence of substrate. Biochem. Biophys. Res. Commun. 1969;34:441–447. doi: 10.1016/0006-291x(69)90401-x. [DOI] [PubMed] [Google Scholar]
- 16.Babior BM, Moss TH, Gould DC. The mechanism of action of ethanolamine ammonia lyase, a B 12 -dependent enzyme. X. A study of the reaction by electron spin resonance spectrometry. J. Biol. Chem. 1972;247:4389–4392. [PubMed] [Google Scholar]
- 17.Babior BM, Moss TH, Orme-Johnson WH, Beinert H. The mechanism of action of ethanolamine ammonia-lyase, a B-12-dependent enzyme. The participation of paramagnetic species in the catalytic deamination of 2-aminopropanol. J. Biol. Chem. 1974;249:4537–4544. [PubMed] [Google Scholar]
- 18.Poyner RR, Anderson MA, Bandarian V, Cleland WW, Reed GH. Probing nitrogen-sensitive steps in the free-radical-mediated deamination of amino alcohols by ethanolamine ammonia-lyase. J. Am. Chem. Soc. 2006;128:7120–7121. doi: 10.1021/ja060710q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerfen GJ. EPR Spectroscopy of B12-Dependent Enzymes. In: Banerjee R, editor. Chemistry and Biochemistry of B12. Wiley-Interscience; New York: 1999. pp. 165–195. [Google Scholar]
- 20.Warncke K, Schmidt JC, Ke SC. Identification of a rearranged-substrate, product radical intermediate and the contribution of a product radical trap in vitamin B-12 coenzyme-dependent ethanolamine deaminase catalysis. J. Am. Chem. Soc. 1999;121:10522–10528. [Google Scholar]
- 21.Ke SC. Spin-spin interaction in ethanolamine deaminase. Biochim. Biophys. Acta. 2003;1620:267–272. doi: 10.1016/s0304-4165(03)00006-0. [DOI] [PubMed] [Google Scholar]
- 22.Sheu MJ, Ke SC. Molecular properties of the product radical in adenosylcobalamin-dependent ethanolamine deaminase. Physica a-Statistical Mechanics and Its Applications. 2005;350:131–143. [Google Scholar]
- 23.Warncke K. Characterization of the product radical structure in the Co(II)-product radical pair state of coenzyme B12-dependent ethanolamine deaminase by using three-pulse 2H ESEEM spectroscopy. Biochemistry. 2005;44:3184–3193. doi: 10.1021/bi048196t. [DOI] [PubMed] [Google Scholar]
- 24.Canfield JM, Warncke K. Active site reactant center geometry in the Co(II)-product radical pair state of coenzyme B12-dpendent ethanolamine deaminase determined by using orientation-selection electron spin-echo envelope modulation spectroscopy. J. Phys. Chem. B. 2005;109:3053–3064. doi: 10.1021/jp046167m. [DOI] [PubMed] [Google Scholar]
- 25.Hollaway MR, White HA, Joblin KN, Johnson AW, Lappert MF, Wallis OC. A spectrophotometric rapid kinetic study of reactions catalysed by coenzyme B12-dependent ethanolamine ammonia-lyase. Eur. J. Biochem. 1978;82:143–154. doi: 10.1111/j.1432-1033.1978.tb12005.x. [DOI] [PubMed] [Google Scholar]
- 26.Jones AR, Hay S, Woodward JR, Scrutton NS. Magnetic field effect studies indicate reduced geminate recombination of the radical pair in sbustrate-bound adenosylcobalamin-dependent ethanolamine ammonia-lyase. J. Am. Chem. Soc. 2007;129:15718–15727. doi: 10.1021/ja077124x. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan BH, Stadtman ER. Ethanolamine deaminase. a cobamide coenzyme-dependent enzyme. J. Biol. Chem. 1968;243:1787–1793. [PubMed] [Google Scholar]
- 28.Beinert H, Hansen RE, Hartzell CR. Kinetic studies on cytochrome c oxidase by combined epr and reflectance spectroscopy after rapid freezing. Biochim. Biophys. Acta. 1976;423:339–355. doi: 10.1016/0005-2728(76)90190-0. [DOI] [PubMed] [Google Scholar]
- 29.Bandarian V, Reed GH. Analysis of the electron paramagnetic resonance spectrum of a radical intermediate in the coenzyme B(12)-dependent ethanolamine ammonia-lyase catalyzed reaction of (S)-2-aminopropanol. Biochemistry. 2002;41:8580–8588. doi: 10.1021/bi0201217. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein H, Poole C, Safko JL. Classical Mechanics. 3rd ed. Addison-Wesley; San Francisco: 2002. pp. 150–154. [Google Scholar]
- 31.Mansoorabadi SO, Padmakumar R, Fazliddinova N, Vlasie M, Banerjee R, Reed GH. Characterization of a succinyl-CoA radical-cob(II)alamin spin triplet intermediate in the reaction catalyzed by adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry. 2005;44:3153–3158. doi: 10.1021/bi0482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abend A, Bandarian V, Nitsche R, Stupperich E, Retey J, Reed GH. Ethanolamine ammonia-lyase has a “base-on” binding mode for coenzyme B12. Arch. Biochem. Biophys. 1999;370:131–141. doi: 10.1006/abbi.1999.1382. [DOI] [PubMed] [Google Scholar]
- 33.Ke SC, Torrent M, G. MD, Morokuma K, Warncke K. Identification of dimethylbenzimidazole axial coordination and characterization of (14)N superhyperfine and nuclear quadrupole coupling on Cob(II)alamin bound to ethanolamine deaminase in a catalytically-engaged substrate radical-Cobal(II) biradical state. Biochemistry. 1999;38:12681–12689. doi: 10.1021/bi983067w. [DOI] [PubMed] [Google Scholar]
- 34.Bender G. Biophysics. University of Wisconsin-Madison; 2008. p. 135. Ph. D. Dissertation. [Google Scholar]
- 35.Bencini A, Gatteschi D. EPR of Exchange Coupled Systems. Springer-Verlag; Berlin: 1990. Exchange and Superexchange; pp. 1–19. [Google Scholar]
- 36.Mansoorabadi SO, Reed GH. Effects of electron spin delocalization on non-colinearity of interaction terms in EPR triplet powder patterns. In: Telser J, editor. Paramagnetic Resonance of Metallobiomolecules. ACS; Washington D. C.: 2003. pp. 82–96. [Google Scholar]
- 37.Sun L, Groover OA, Canfield JM, Warncke K. Critical role of arginine 160 of the EutB Protein subunit for active site structure and radical catalysis in coenzyme B12-dependent ethanolamine ammonia-lyase. Biochemistry. 2008;47:5523–5535. doi: 10.1021/bi702366e. [DOI] [PubMed] [Google Scholar]
- 38.Warncke K, Schmidt JC, Ke S-C. Identification of a rearranged-substrate, product radical intermediate and the contribution of a product radical trap in vitamin B12 coenzyme-dependent ethanolamine deaminase catalysis. J. Am. Chem. Soc. 2008;130:6055. [Google Scholar]
- 39.Wertz JE, Bolton JR. Electron Spin Resonance. Chapman and Hall; New York: 1986. pp. 170–171. [Google Scholar]
- 40.Gordy W. Theory and Applications of Electron Spin Resonance. Wiley; New York: 1980. pp. 244–247. [Google Scholar]
- 41.Frey PA. Radical mechanisms of enzymatic catalysis. Annu. Rev. Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 42.Frey PA, Hegeman AD. Enzymatic Reaction Mechanisms. Oxford University Press; Oxford: 2007. pp. 97–99. [Google Scholar]
- 43.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Garland Science; New York: 2007. pp. 253–295. [Google Scholar]
- 44.Luckhurst GR. Biradicals as spin probes. In: Berliner LJ, editor. Spin Labeling: Theory and Applications. Academic Press; New York: 1976. pp. 133–181. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.