

Abstract
Genetic mutations affecting mitochondrial fission and fusion proteins cause human neurological disorders, but are assumed to be well tolerated in yeast. The conserved mitochondrial fission protein Dnm1/Drp1 is required for normal mitochondrial division, but also promotes cell death in mammals and yeast. Fis1, an outer mitochondrial membrane-anchored receptor for Dnm1/Drp1, also can promote cell death in mammals, but appears to have prosurvival activity in yeast. Here we report that deletion of the FIS1 gene in yeast consistently results in acquisition of a secondary mutation that confers sensitivity to cell death. In several independently derived FIS1 knockouts, tiling arrays and genomic sequencing identified the secondary mutation as a premature termination in the same stress-response gene, WHI2. The WHI2 mutation rescues the mitochondrial respiratory defect (petite formation) caused by FIS1 deficiency, but also causes a failure to suppress cell growth during amino-acid deprivation. Thus, loss of Fis1 drives the selection for specific compensatory mutations that confer defective growth control and cell death regulation, characteristic of human tumor cells. The important long-term survival function of Fis1 that is compensated by WHI2 mutation appears to be independent of fission factor Dnm1/Drp1 and its adaptor Mdv1, but may be mediated through a second adaptor Caf4, as WHI2 is also mutated in a CAF4 knockout.
Keywords: Fis1, WHI2, programmed cell death, mitochondrial fission, apoptosis, Drp1
Mitochondria are dynamic organelles that continuously undergo fission and fusion for organelle maintenance.1 Mitochondrial fission and fusion events are mediated by distinct molecular complexes that are characterized best in the budding yeast Saccharomyces cerevisiae.2 Deletion or depletion of the small, membrane-anchored fission protein Fis1 from yeast or mammalian cells results in a more connected mitochondrial network. Yeast Fis1 forms a complex with dynamin-related 1 (Dnm1), a dynamin-like GTPase conserved in animals (Drp1/DLP1) and plants (ADL2b) through either of two WD40-repeat adaptor proteins Mdv1 or its paralog Caf4.2-4 Mammalian Drp1 facilitates normal synaptic activity in neurons, and a mutation in human Drp1 is linked to severe neurological deficits.5-7 In addition to its critical role in healthy neurons, mammalian Drp1 also interacts with Bcl-2 family proteins and promotes the rapid mitochondrial fragmentation that is characteristic of apoptotic cells.6,8,9 Furthermore, Drp1/Dnm1 was shown to actively promote cell death in yeast, worms and flies, and a small molecule inhibitor of Drp1/Dnm1 inhibits cytochrome c release and delays mammalian cell death.10-13 However, the pro-death function of Drp1/Dnm1 may be separable from its fission function.
Consistent with the model that mitochondrial fission factors have an evolutionarily conserved pro-death function, mammalian Fis1 has been reported to promote mammalian cell apoptosis based on depletion of Fis1 by RNAi14,15 and overexpression of Fis1 by transfection.16-19 Thus, it was initially unexpected that deletion of FIS1 in yeast increases their sensitivity to multiple death stimuli, including acetic acid, H2O2, heat shock, ethanol and viruses.10,20,21 However, a protective function for Fis1 is supported by overexpression studies where yeast and human Fis1 rescue yeast FIS1-knockout strains from cell death, though Fis1 does not improve survival beyond that of wild-type.10 Furthermore, depletion of human Fis1 results in mammalian cell senescence.22,37
Here we report that yeast Fis1 has a long-term survival function, as de novo deletion of the FIS1 gene in S. cerevisiae selects for chromosome abnormalities and mitochondrial deficits, but does not immediately result in sensitivity to cell death. Rather, the acute sensitivity to cell death is due to secondary mutations in whiskey 2 (WHI2), a stress-response gene,23 that specifically arose in FIS1 knockouts but not in the mitochondrial fission-defective knockouts of DNM1 or MDV1. This process in yeast may share similarities to human tumorigenesis, where one mutation selects for additional mutations that confer loss of growth control despite the accompanying sensitivity to cell death induced by toxic chemicals such as anticancer agents.
Results
FIS1-knockout strains fail to suppress growth during amino-acid deprivation
Yeast deletion mutants lacking either FIS1 (fis1Δ), MDV1 (mdv1Δ) or DNM1 (dnm1Δ) all have netted mitochondrial morphology and, as a consequence, a mild mitochondrial segregation defect during sporulation, but lack obvious growth defects.24 However, FIS1 knockouts are uniquely sensitive to a variety of cell death stimuli, unlike DNM1 and MDV1 knockouts that are death resistant.10,20 The haploid fis1Δ strains in the yeast knockout (YKO) collection,25 designated here as fis1Δd1 and fis1Δd2 (Supplemental Table S1), were compared to their wild-type background strains WT1 (BY4741) and WT2 (BY4742), respectively. As previously reported,10 both of these haploid FIS1-knockout strains are sensitive to cell death induced by acetic acid, a mimic of overgrowth conditions (Figure 1a). This cell death phenotype was not caused by a general defect in cell health, because these strains grow normally on rich medium (yeast extract, peptone, dextrose, YPD) and on synthetic complete medium (SCDCSH, Cold Spring Harbor26).
Figure 1.

Fission 1 (FIS1) knockouts exhibit cell death sensitivity and growth control defects. (a) Growth of the indicated haploid yeast strains plated at the indicated dilutions on rich medium (YPD) with or without pretreatment with acetic acid. (b, c) Growth properties of two independent FIS1-knockout strains are compared to wild-type and a DNM1 knockout o on the three indicated o media. Quantified results are from 4 to 6 independent experiments presented as mean ±S.E.M. Growth of fis1Δ strains is ∼27-fold over WT, *P<10−5 for fis1Δd1; *P<0.0005 for fis1fis1Dd1; *Po0.0005d2. (d) Low amino-acid levels in SCDME explain reduced growth by wild-type strains (see text). Representative images from four independent experiments are shown
Serendipitously, we found that FIS1-knockout strains grow robustly compared to controls if the synthetic complete medium was prepared by a different recipe (SCDME, Methods in Enzymology27) (Figure 1b and c). Thus, the reduced growth of wild-type strains on SCDME medium is the default response to a signal that is apparently ignored by the FIS1-knockout strains, as fis1Δd1 and fis1Δd2 continue to grow robustly. Growth of DNM1 knockouts was similar to wild-type, suggesting that defective mitochondrial division is not the cause of the overgrowth phenotype. To identify which media components reveal the overgrowth control defect of FIS1 knockouts, two additional media were prepared to evaluate all media component differences. Reducing the amino-acid content of SCDCSH medium to match that of SCDME recapitulated the large growth differences between wild-type and FIS1-knockout strains (Figure 1d, right panels). In contrast, adjusting the four non-amino-acid ingredients, PIUA (PABA (para-aminobenzoic acid used in folate/vitamin B9 biosynthesis), inositol (incorporated into phospholipids), uracil and adenine), in the SCDCSH medium to the levels found in SCDME had no effect (Figure 1d, left panels). Therefore, we conclude that the wild-type control strains sense the reduced amino-acid supply in SCDME and deliberately repress their growth, whereas the FIS1-knockout strains fis1Δd1 and fis1Δd2 have a growth control defect allowing them to ignore these low-nutrient signals.
FIS1-knockout strains carry a previously unknown second mutation
To definitively determine whether FIS1 deficiency is responsible for the cell death and growth control defects, genetic linkage between these phenotypes and the FIS1 gene locus was assessed by tetrad analysis.27 FIS1-knockout strains fis1Δd1 and fis1Δd2 were mated to wild-type strains. The resulting heterozygous diploids were sporulated and the four haploid spores (a–d) in tetrads produced by each mating pair were analyzed for the segregation of genetic markers. As expected, FIS1 gene deletion (Δ, G418 resistant, G418R) co-segregated with netted (N) mitochondrial morphology (Figure 2a and b). If FIS1 deletion is directly responsible for the cell death and growth control defects, these phenotypes will co-segregate with FIS1-deletion (Δ, G418R). However, this was not the case. Instead, FIS1 deletion segregated independently, indicating that FIS1 deletion is not directly responsible for these phenotypes (Figure 2a). However, in all tetrads analyzed, the cell death and growth control phenotypes perfectly co-segregated with each other (2 : 2 segregation), suggesting that FIS1-knockout strains fis1Δd1 and fis1Δd2 each have a second previously unknown gene mutation, designated d (death), that is responsible for both the cell death and growth control defects (Figure 2c).
Figure 2.
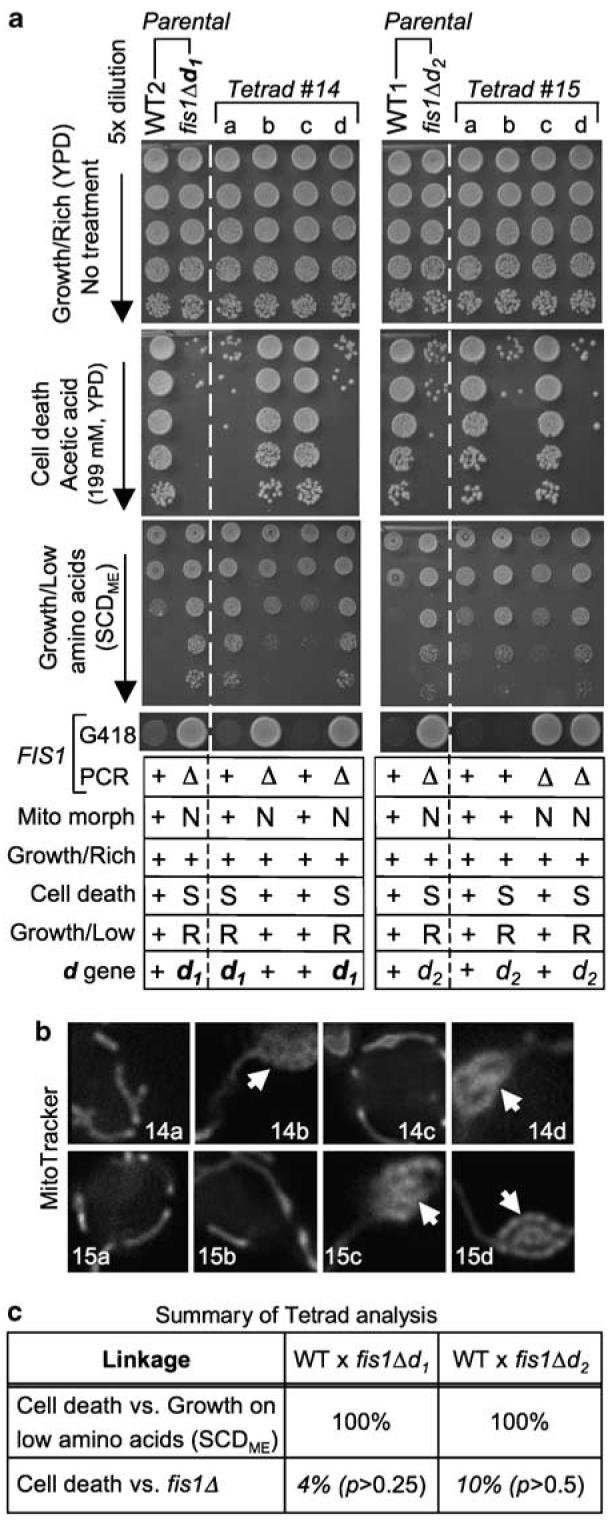
Tetrad analysis reveals a second mutation responsible for cell death and growth control defects. (a) Haploid parental strains and the four spores/strains of a representative tetrad derived from their cross was analyzed for growth on rich medium (YPD, top), tubular ( +/normal) or netted (N) mitochondrial morphology, sensitivity (S) to acetic acid-induced cell death, resistance (R) to growth inhibition due to low amino acids (synthetic complete dextrose medium, SCDME, Methods in Enzymology), and G418 resistance (indicating FIS1 gene replacement). Data are summarized for three or more independent experiments per condition in the table below; + indicates wild-type phenotype. (b) Mitochondrial morphology of each spore in tetrads no. 14 and no. 15 from (a) were visualized by MitoTracker staining.10 Arrows mark netted mitochondrial morphology. (c) Summary of genetic linkage determined by tetrad analyses as illustrated in (a). FIS1 deletion is unlinked to the cell death d gene, as the ratios of PD (parental ditype): NPD (nonparental ditype): TT (tetratype) within tetrads were 1 : 6 : 17 for fis1Δd1 (24 tetrads for cell death and/or overgrowth on SCDME) and 2 : 3 : 15 for fis1Δd2 (20 tetrads), which are close to random segregation (1 : 1 : 4, or 17% for PD (linked) versus 83% for NPD+TT (unlinked)). In contrast, cell death sensitivity was 100% linked to resistance to growth inhibition on low amino acids (SCDME), with ratios of 8 : 0 : 0 (fis1Δd1) and 20 : 0 : 0 (fis1Δd2). χ2 goodness-of-fit test was used to determine whether FIS1-deletion (fis1Δ) segregated independently from mutation d (death) at the expected ratio of 1 : 1 : 4 for PD : NPD : T. As none of the p values rejected the null hypothesis, FIS1-deletion segregated independently from mutation d
d1 and d2 are recessive mutations in the same gene
To determine if the secondary mutations d1 and d2 in the two FIS1-knockout strains are in the same or different genes, a complementation test was performed. However, this genetic test can only be applied if the mutations are recessive. To determine if d1 and d2 are recessive or dominant, we used FIS1+ strains containing only d1 or d2 (e.g. spores 14a and 15b, Figure 2a). These d1 and d2 haploid strains were mated to wild-type strains to create heterozygous diploids and tested for cell death sensitivity. The d mutations were complemented by the wild-type gene, as the heterozygous diploids were indistinguishable from the control diploid (WT×WT) when treated with acetic acid, indicating that the d mutations are recessive (Figure 3a, middle). Therefore, to determine if the d mutations are in the same complementation group, single mutants for d1 and d2 were mated together to generate double d1×d2 mutants. Because d1 (d2) failed to rescue the cell death phenotype of d2 (d1), based on similar cell death sensitivities for both heterozygous (d1×d2) and homozygous diploids (d1×d1 and d1×d2), d1 and d2 are in the same cell death complementation group (Figure 3b, middle). Although d1×d2 also failed to rescue the overgrowth phenotype on low amino-acid medium SCDME (Figure 3b, lower), both heterozygous diploids WT×d1 and WT×d2 had an intermediate phenotype indicating haploinsufficiency, compromising interpretation of the complementation test for this phenotype (Figure 3a). Nevertheless, because the cell death phenotype genetically co-segregates with the overgrowth phenotype, we conclude that d1 and d2 mutations are likely to be in the same gene.
Figure 3.
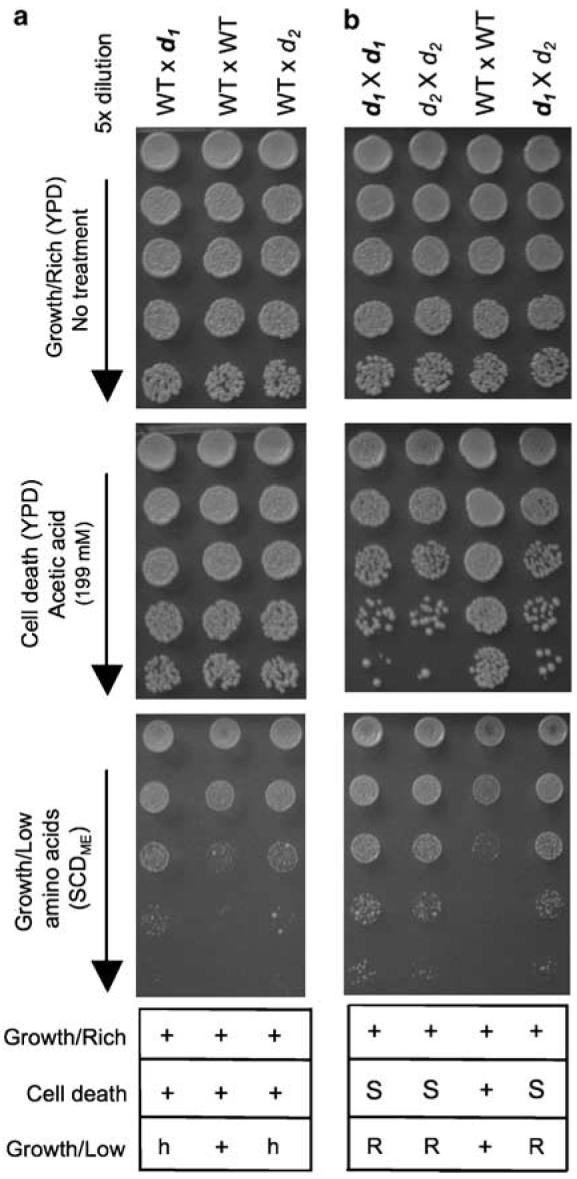
Mutations d1 and d2 are in the same complementation group. (a) d is a recessive mutation. Diploid strains heterozygous for d1 (WT×d1) or d2 (WT×d2) and a wild-type control diploid (WT×WT) were assayed for death sensitivity and the synthetic complete dextrose medium (SCDME, Methods in Enzymology) overgrowth phenotypes. (b) Complementation test. Growth of the indicated homozygous and heterozygous diploids generated from the indicated crosses was treated as in (a). Data for four independent experiments for each assay are summarized below representative images; + (WT-like), h (haploinsufficiency), S (sensitive), R (resistant)
Secondary mutations in WHI2 explain death sensitivity and growth control defects
To identify the gene responsible for both the cell death and growth control defects, a resequencing technique employing the tiling microarray was used to detect genome-wide sequence polymorphisms at single-nucleotide resolution.28,29 The tiling microarray predicted a mutation in the coding sequence of WHI2, a stress-response gene,23 in both d1 and d2 strains (FIS1+). Direct sequencing of genomic DNA (∼1600 nt) surrounding the polymorphism confirmed the predicted single nucleotide changes on chromosome 15 (G to T at 411326 (d1), T to G at 411075 (d2)) that introduce premature termination codons, implying loss-of-function (Figure 4a). The same mutations were confirmed in three d1 and two d2 mutant strains (including spores 14a, 14d, 15b and 15d, Figure 2a), and in their respective parental strains (fis1Δd1 and fis1Δd2) (Figure 4b and data not shown). Importantly, WHI2 mutations were not found in WT1, WT2 or four fis1Δ-only spores with no cell death or growth control phenotypes (14b, 14c, 15a and 15c, Figure 2a). Furthermore, the homozygous FIS1 knockout in the YKO collection (derived by mating the haploid knockouts) was heterozygous for both point mutations, indicating that these secondary mutations arose independently before mating (Supplemental Figure S1).
Figure 4.
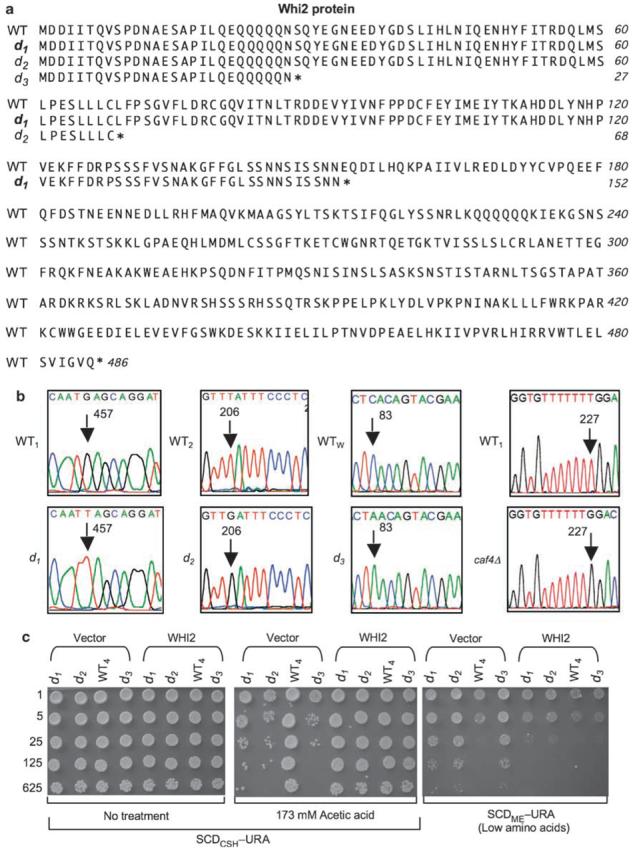
Secondary mutations in whiskey 2 (WHI2) are responsible for the cell death and overgrowth phenotypes. (a) Genomic sequencing identified premature terminations (*) in Whi2 from three independently derived FIS1-knockout strains. (b) DNA sequence chromatograms of WHI2 mutations in mutant strains d1, d2, d3, caf4Δ and their respective wild-type strains. Nucleotide position numbers in the WHI2 ORF mark the mutations (arrows). (c) A low-copy plasmid (CEN-URA) expressing WHI2 (by its own endogenous promoter) in d1, d2 and d3 strains, but not the empty plasmid vector, restores cell survival and reinstates the ability to reduce growth on low amino-acid medium. Note, a slightly lower dose of acetic acid is required when plating on synthetic medium. A representative of three independent experiments per condition is shown
Acquisition of WHI2 mutations in both FIS1-knockout strains raised the concern that this phenomenon could be restricted to the YKO collection. To address this point, we acquired a third independently constructed FIS1 knockout on the Winston background (fis1Δd3, RJ1365) in which the FIS1 ORF had been replaced with the HIS3 selection marker.30 This fis1Δd3 strain also has both the cell death10 and growth control phenotypes, and tetrad analysis verified that both phenotypes are caused by a secondary mutation, designated d3 (Figure 4c; W-C Cheng and JM Hardwick, unpublished data). Furthermore, sequence analysis of d3 revealed a premature stop codon after residue 27 in WHI2 that is distinct from d1 and d2 (Figure 4a and b). Remarkably, three independently derived FIS1-knockout strains all acquired a loss-of-function mutation in the same gene, WHI2.
To further verify that loss-of-function mutations in WHI2 are responsible for the observed death sensitivity and growth control defects of FIS1 knockouts (fis1Δd11–3), we reconstituted WHI2 into strains carrying only mutation d1, d2 or d3 (FIS1+). As expected, expression of WHI2 by its endogenous promoter on a low copy plasmid rescued both phenotypes, thereby restoring cell survival following a death stimulus and limiting cell growth during amino-acid deprivation (Figure 4c). Exogenous WHI2 also rescued both phenotypes in the parental strain fis1Δd1 as expected (Supplemental Figure S2).
FIS1 deficiency decreases respiratory competency independently of fission
To gain insight into the critical function of FIS1 that drives selection for a secondary mutation, we sequenced the WHI2 gene in the DNM1 and MDV1 knockouts from the YKO collection, which have netted mitochondrial morphology due to defective fission similar to FIS1 knockouts.1 No WHI2 mutations were present, consistent with their cell death (resistance) and normal growth control phenotypes (Figures 1b, c and 5a and data not shown). However, a screen of the yeast knockout collection for strains that exhibit overgrowth phenotypes in low amino acids identified CAF4, which encodes an Mdv1-like protein that also binds Fis1, though the knockout has nearly normal mitochondrial morphology.31 Direct sequencing revealed a frameshift (T deletion) following amino acid 75 of Whi2, resulting in a termination codon 10 residues later (Figure 4b, right). These findings suggest that a specific function of the Fis1 protein that is independent of Dnm1 and Mdv1, but potentially dependent on Caf4, is compensated by loss of Whi2 function.
Figure 5.
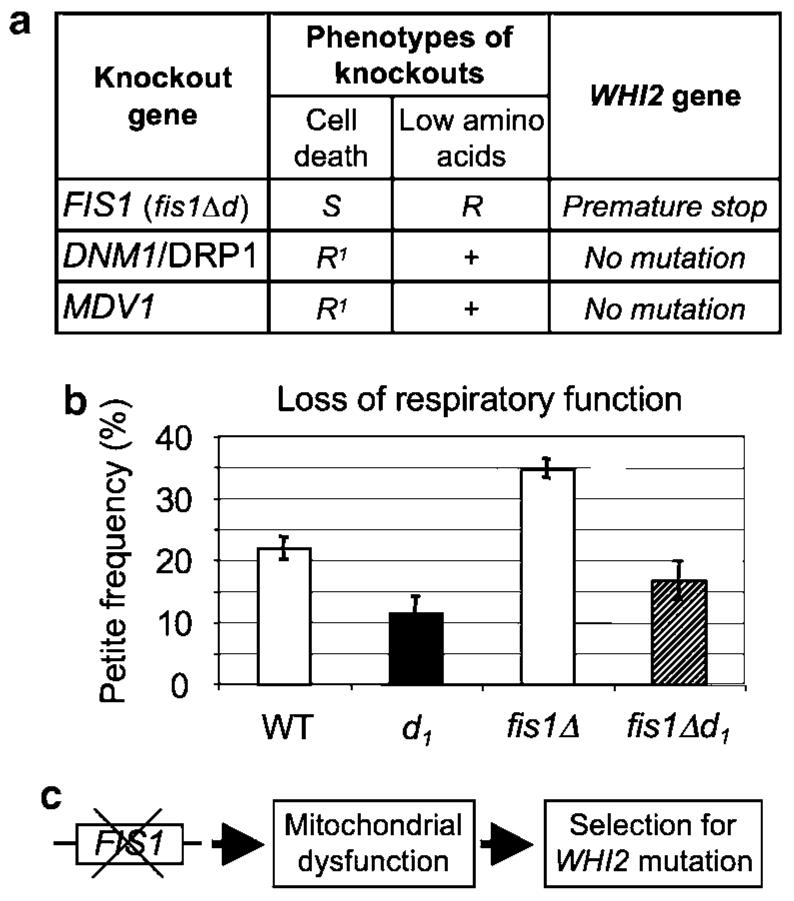
FIS1 deletion increases mitochondrial respiratory defects. (a) Results showing that whiskey 2 (WHI2) mutations do not arise in other knockout strains (dynamin-related 1, DNM1, or mitochondrial division 1, MDV1) that are defective for mitochondrial fission, or in another strain where secondary mutations are responsible for cell death sensitivity and overgrowth during amino-acid deprivation. (b) Loss of respiratory function measured as petite frequency (comparing strains with the indicated genotypes derived from eight spores of four tetrads) is caused by FIS1 deletion and is alleviated by WHI2 mutations. Means ± S.E. are presented for four independent experiments (9–10 determinations per genotype). P=0.02 for WT versus d1; P = 0.004 for WT versus FIS1Δ, P=0.006 for fis1Δ versus fis1Δd1. (c) Model summarizes data presented, indicating that loss of the FIS1 gene compromises mitochondrial function, which is compensated by selection of WHI2 loss-of-function mutations
Loss or mutation of mitochondrial DNA abolishes oxidative phosphorylation in yeast causing a petite (slow growth) phenotype on rich medium containing glucose (YPD) and a failure to grow on glucose-free medium containing glycerol as the carbon source (yeast extract, peptone, glycerol, YPG), where mitochondrial respiration is important for survival. Therefore, mitochondrial respiratory deficiency was assessed by replica plating strains derived from sets of tetrads (similar to those in Figure 2) from YPD onto YPG plates.24 FIS1-knockout strains with no WHI2 mutations had noticeably more petite colonies, consistent with an increased petite frequency when replica plated onto YPG (Figure 5b). Importantly, this mitochondrial defect in the FIS1 knockout was rescued by the d mutation in WHI2 (fis1Δd1), indicating that the WHI2 loss-of-function mutation protects the FIS1 knockout from mitochondrial damage. Taken together, these data suggest a model where deletion of FIS1 causes a mitochondrial defect that is compensated by a loss-of-function mutation in WHI2 (Figure 5c).
Gene duplication upon de novo deletion of FIS1
To further evaluate the importance of FIS1 to cell survival, we generated new FIS1-knockout strains by homologous recombination with a targeting cassette containing the auxotroph selection marker URA3 plus 200 bp arms of FIS1 ORF flanking sequences (Figure 6a). This targeting cassette was introduced into a wild-type strain (WT1) and transformants were selected on uracil-minus medium. Successful recombination at both 5′ and 3′ ends of the selection marker at the FIS1 locus of the resulting clones was confirmed by PCR using recommended primer pairs32 and by verifying netted mitochondrial morphology (Figure 6b, data not shown). These new FIS1-deleted strains lack a cell death phenotype (Supplemental Figure S3), and do not have WHI2 mutations (data not shown), confirming that WHI2 mutations do not develop immediately following deletion of FIS1.
Figure 6.

FIS1 deletion induces gene duplication. (a) Gene replacement strategy for generating knockouts of the FIS1 ORF (blue box) by recombination (dotted lines) with targeting cassettes consisting of the KanMX4 or URA3 selection marker flanked by TEF sequences for selection marker expression (red boxes). (b, c) Results of successful gene replacements (b) and unsuccessful attempts (c) resulting in strains with both FIS1 and the URA3/KanMX4 selection markers inserted in cis (dashed line) or in trans (solid arrows). (d) Presumed rare event of gene (dashed line) or segmental/chromosome duplication upon replacement of FIS1 with KanMX4 for G418R in the YKO collection, resulting in two copies of KanMX4 cassettes per cell. (e) Results obtained by marker swap (MS) of the KanMX4 cassette in the YKO collection for a URA3 cassette resulting in 0–1 copies of KanMX4 and 1–2 copies of URA3. (f) Examples of growth on synthetic complete dextrose medium (SCDCSH, Cold Spring Harbor) minus uracil plates, with or without G418, confirm PCR genotype analysis from strains as shown in (e). A colour version of this figure is available online
In contrast to the recombination strategy above, attempts to delete FIS1 were unsuccessful with the strategy used for the YKO collection with only 40 bp homologous arms.25 All 14 putative knockouts analyzed from 3 independent experiments had normal tubular mitochondria, rather than the netted morphology characteristic of fis1Δ strains, indicating retention of the FIS1 gene despite the presence of the selection marker used to replace FIS1 (Figure 6c, data not shown). The presence of FIS1 was confirmed by CGH analysis, and by three different PCR primer pairs covering ∼200 to 500 bp flanking sequences (data not shown). These results raise the possibility that attempted deletion of FIS1 may have selected for duplication of the FIS1 locus or entire chromosome, and that only one copy of duplicated FIS1 was replaced by the selection marker (Figure 6c). Similar to our observation, aneuploidy has been observed in the YKO haploid collection during initial construction.32,33
Given these difficulties, we considered the possibility that haploid FIS1Δ strains in the YKO collection could also have undergone gene/chromosome duplications. Results from our marker-swap (MS) experiment are consistent with this possibility. In this experiment, we replaced the KanMX4 cassette at the FIS1 locus in the fis1Δd1 strain from the YKO collection with a URA3 cassette by homologous recombination as described34 (Figure 6d and e). Only two clones were obtained from several independent experiments and both retained their ability to grow on uracil-minus medium when retested (Figure 6f, right). Strikingly, one of them retained resistance to G418, confirming the presence of KanMX4 in addition to the URA3 cassette (Figure 6e and f left). This finding suggests a prior duplication of FIS1, one of which is now occupied by the URA3 cassette. PCR analysis confirmed that both the KanMX4 and URA3 cassettes are associated with at least ∼500 bp of FIS1 flanking sequences (not shown).
Duplication of FIS1 can be mediated by segmental or chromosomal duplication
The evidence presented above suggests that gene or chromosome duplication may occur with relative frequency when attempting to delete the FIS1 gene. Hughes et al.33 examined gene duplication using whole-genome DNA microarray, and found that 8% of ∼300 homozygous and haploid YKO strains have either chromosomal or segmental duplication, and suggested that duplication was selected for by dosage/functional compensation of the deleted gene.
To determine if FIS1 duplication is mediated by chromosomal or segmental duplication, the genome copy number was analyzed by Southern blot analysis using intergenic probes both proximal and distal to the FIS1 locus on chromosome 9 (Figure 7a). All results from three different colonies each for fis1Δd1 and FIS1Δd2 strains confirmed the absence of a FIS1 gene as expected based on fragment sizes detected with the proximal probe (Figure 7b and c). However, a comparison of signal intensities for proximal and distal probes relative to the actin probe suggests one of three events, no detectable duplication (fis1Δd2 colonies 1 and 3), segmental duplication (fis1Δd1 colonies 1 and 3, fis1Δd2 colony 2) or chromosome duplication (fis1Δd2 colony 2). These variants are presumably a reflection of an unstable rare preexisting event (before FIS1 deletion) that was selected as a consequence of Fis1 deficiency and then subsequently evolved, perhaps more as a reflection of instability, but potentially also a consequence of FIS1 deletion. Taken together, the evidence suggests negative selection for de novo deletions of FIS1 and indicates that WHI2 mutations are not preexisting, but arise by selection when FIS1 is deleted.
Figure 7.

Evidence for segmental and chromosome duplication in FIS1 knockouts from the YKO collection. (a) Chromosome 9 for parental and FIS1-deleted strains shows approximate locations of the intergenic probe proximal to the FIS1 gene (blue bar), distal probe near end of opposite arm on chromosome 9 (orange bar), centromere (circle), BamHI cut sites (black triangles), BglII sites (tall red triangles), fragment sizes detected by probes (brackets), Ty LTRs flanking 50 kb region encoding FIS1 (pink triangles). (b) Representative Southern blots of genomic DNA hybridized with the indicated probes. (c) Southern blot analyses for colonies of each of the indicated strains tested in independent experiments. A colour version of this figure is available online
Discussion
Loss of FIS1 imposes a selective pressure for genetic alterations in yeast
We report here that three independently constructed FIS1-knockout strains of S. cerevisiae acquired a loss-of-function mutation in the same gene, WHI2. Although FIS1 is not classified as an essential gene, our findings suggest that depletion of FIS1 is detrimental. However, the long-term survival function of FIS1 may be independent of a fission function mediated through Dnm1 and MDV1, as deletion of FIS1-interacting genes DNM1 or MDV1 does not select for cell death sensitivity, overgrowth on low amino acids or for secondary mutations that alter these functions. Thus, FIS1 may have additional functions not shared with DNM1 or MDV1. Consistent with this possibility, FIS1 appears to be required for maintenance of mitochondrial DNA-encoded function, as FIS1 knockouts (with wild-type WHI2) exhibit increased petite frequency (loss of mitochondrial respiratory function). Although mitochondrial fusion factors are known to be required for maintenance of mitochondrial DNA in yeast and mammals,2 this is unexpected upon loss of a fission factor. Importantly, loss-of-function mutations in WHI2 specifically suppress increased petite frequency caused by FIS1 deletion, although the mechanism is not known.
WHI2 is a fungi-specific gene that was first identified for its involvement in cell size and cell cycle, but was not known to have an involvement in programmed cell death. Others found that deletion of WHI2 causes a failure to undergo cell-cycle arrest in G1 when nutrients become depleted, resulting in continuous division/growth, reduced cell size and eventual arrest at random stages of the cell cycle upon exhaustion of nutrients.35,36 In addition, WHI2 is important for mounting stress responses against H2O2, heat shock and high salt,23 consistent with our observation that loss of WHI2 provides a growth advantage during amino-acid deprivation and confers sensitivity to cell death when stressed. By binding to the protein phosphatase Psr1, WHI2 was suggested to stimulate expression of stress-response genes through transcription factors Msn2/Msn4.23 Thus, although WHI2 mutants can outgrow wild-type cells during nutrient limitation, any growth advantage could potentially be offset by the propensity of FIS1-knockout strains to undergo cell death.
A model of tumorigenesis?
Attempts to generate new FIS1 knockouts can also select for FIS1 gene or chromosome duplication, presumably because the cells that contain these preexisting events have an advantage when FIS1 is deleted, and because these events are more common than acquisition of inactivating point mutations in WHI2. Because WHI2 mutations did not arise immediately after losing FIS1, it is also possible that a step-wise progression of mutations eventually allows fixation of WHI2 loss-of-function. This situation may be analogous to the process of tumorigenesis in mammals, where the selection for cell growth and survival functions may be governed by some of the same evolutionarily conserved mechanisms operating in yeast. Although secondary mutations are confounding for genetic studies in both yeast and mammals, these events are likely to be far more common than currently appreciated, but will be much easier to dissect in yeast than in mammals. We anticipate that identification of such mutations in the YKO collection will reveal new genetic interactions relevant to cell death and survival pathways.
Cryptic secondary mutations
Several additional studies have identified a genetic linkage between FIS1 and WHI2. Two independent genome-wide screens for mutants (homozygous diploids) with altered sporulation efficiencies identified both FIS1 and WHI2 knockouts.38,39 A third genome-wide screen for synthetic lethal interactions with the Hsp82 chaperone identified both FIS1 and WHI2.40 These screens may reflect FIS1–WHI2 genetic interactions, but a cryptic WHI2 mutation in the FIS1 knockout could also explain these results. However, the mere presence of a cryptic WHI2 mutation in FIS1 knockouts implies a genetic interaction, even though the phenotypes being scored in these screens may directly reflect only the function of WHI2, rather than FIS1. The presence of a WHI2 mutation in the CAF4 knockout, but not in other knockout strains, including DNM1 and MDV1, further fuels the hypothesis that the secondary mutations are specifically selected to compensate for an undetermined function of FIS1. FIS1 can bind Caf4 to promote mitochondrial fission in the absence of MDV1,31 but it is not evident how this would occur independently of Dnm1/Drp1, suggesting additional functions for FIS1.
The cryptic secondary mutation conundrum is a major inconvenience to yeast geneticists, as it compromises applications of the YKO collection to epistasis analyses and other strategies, though case reports are generally lacking. The prevalence of cryptic mutations is also underappreciated because these mutations lack phenotypes detected by the assays being applied. Secondary mutations may account for many examples of inconsistent phenotypes, but the underlying mutation is rarely pursued, in part because these mutations are generally thought to be more artifact than of biological significance. To the contrary, these secondary mutations will likely yield insightful new genetic interactions. Application of tiling microarray techniques developed by Gresham et al.28 makes it feasible to rapidly identify these cryptic mutations.
Materials and Methods
Yeast strains, plasmids and media
Genotypes of all yeast strains used in this study are listed in Supplemental Table S1. The 3 kb SalI/KasI fragment containing the WHI2 gene and regulatory sequences was subcloned from plasmid pWhim3, provided by PE Sudbery, into vector YCplac33, a CEN-URA3 plasmid (accession number X75456) provided by J Heitman. Synthetic media SCDCSH (or SCDCSH–URA, minus uracil)26 and SCDME (or SCDME–URA),27 and rich media (2% peptone, 1% yeast extract)27 with 2% dextrose (YPD) or 3% glycerol (YPG), and 2% agar for solid media, were prepared as described.
Cell death assay
Yeast strains were seeded at low density into 5 ml liquid YPD or uracil dropout media (SCDCSH–URA), incubated overnight at 30°C with rotation until reaching 0.5. OD600, and 2 ml of culture were treated 4 h with the indicated concentrations of acetic acid. Treated and untreated cultures were serially diluted fivefold in media and 5 μl of each dilution were plated on solid media starting with undiluted sample. Plates were incubated at 30°C in a humidified chamber for 2–3 days.
Quantification of growth on SCDCSH and SCDME media
Quantification of yeast growth in Figure 1 was performed using ImageJ (Rasband, W.S., ImageJ, US National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2007). TIFF images of plates were imported into ImageJ and converted from RGB color to 8-bit. Using the tool functions ‘subtract background’, ‘selection’ and ‘measure’, the ratios of measured area intensities were calculated for each dilution spot. Dilutions with the most similar intensities between strains/treatments were used to calculate a ratio, and subsequently multiplied by the dilution factor to represent fold differences relative to WT (y axis).
CGH and yeast tiling microarray
Yeast CGH and tiling microarrays were processed and analyzed as described29 to identify single nucleotide differences between WT and fis1Δ-derived spores, including spore 2a, derived from sporulation of a heterozygous FIS1-knockout strain that was created by mating WT2 and fis1Δd1, and spore 3b, derived by sporulation of a heterozygous FIS1-knockout strain created by mating WT1 and fis1Δd2.
Mitochondrial function (petite frequency)
Overnight cultures (YPD) started from single colonies were diluted to 0.2 OD600, incubated (∼16 h) at 30°C until mid-log phase (∼0.5 OD600), diluted 8000-fold, and 200 μl was spread onto duplicate/triplicate YPD plates. Colonies were counted after 3 days of incubation, replica plated onto YPG plates and incubated for 3 days at 30°C. The percent of respiration-incompetent colonies, defined as petites, were calculated as the percent of total colonies (YPD) that failed to grow on YPG.24
Southern blotting
Genomic DNA from each strain was purified as described,26 and 10 μg was digested with BamHI and BglII, separated on a 0.7% agarose gel and hybridized with [32P]radiolabeled probes using the High Prime DNA Labeling Kit (Roche). Signal intensities on autoradiographs were quantified from multiple exposures using the ImageGauge Version 4.22 software (Fuji Photo Film). Probes correspond to chromosome 9 nucleotide numbering for proximal probe (243 013–242 416) and distal probe (424 383–424 999), and chromosome 6 for the act1 probe (52 937–53 520).
Supplementary Material
Acknowledgements
We thank Jef D Boeke and Xuewen Pan for helpful discussions and reagents. This work was supported by National Institutes of Health grants RO1 GM077875 (JMH) and GM071508 (MJD).
Abbreviations
- CAF4
CCR4 associated factor
- CSH
Cold Spring Harbor Laboratory Press
- G418R
G418 resistant
- ME
Methods in Enzymology
- SCD
synthetic complete dextrose medium
- WHI2
whiskey 2
- YKO
yeast knockout
References
- 1.Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- 2.Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- 3.Wells RC, Picton LK, Williams SC, Tan FJ, Hill RB. Direct binding of the dynamin-like Gtpase, Dnm1, to mitochondrial dynamics protein Fis1 is negatively regulated by the Fis1 N-terminal Arm. J Biol Chem. 2007;282:33769–33775. doi: 10.1074/jbc.M700807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Chan DC. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc NatlAcad Sci USA. 2007;104:18526–18530. doi: 10.1073/pnas.0706441104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, et al. Bcl-Xl Induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 8.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 9.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of Bap31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome C release to the cytosol. J Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fannjiang Y, Cheng WC, Lee SJ, Qi B, Pevsner J, McCaffery JM, et al. Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 2004;18:2785–2797. doi: 10.1101/gad.1247904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagasia R, Grote P, Westermann B, Conradt B. Drp-1-mediated mitochondrial fragmentation during Egl-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 12.Goyal G, Fell B, Sarin A, Youle RJ, Sriram V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev Cell. 2007;12:807–816. doi: 10.1016/j.devcel.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bras M, Yuste VJ, Roue G, Barbier S, Sancho P, Virely C, et al. Drp1 mediates caspase-independent type Iii cell death in normal and leukemic cells. Mol Cell Biol. 2007;27:7073–7088. doi: 10.1128/MCB.02116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.James DI, Parone PA, Mattenberger Y, Martinou JC. Hfis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 17.Yu T, Fox RJ, Burwell LS, Yoon Y. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein Hfis1. J Cell Sci. 2005;118:4141–4151. doi: 10.1242/jcs.02537. [DOI] [PubMed] [Google Scholar]
- 18.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for Ced-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 19.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related Gtpases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanovska I, Hardwick JM. Viruses activate a genetically conserved cell death pathway in a unicellular organism. J Cell Biol. 2005;170:391–399. doi: 10.1083/jcb.200503069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitagaki H, Araki Y, Funato K, Shimoi H. Ethanol-induced death in yeast exhibits features of apoptosis mediated by mitochondrial fission pathway. FEBS Letts. 2007;581:2935–2942. doi: 10.1016/j.febslet.2007.05.048. [DOI] [PubMed] [Google Scholar]
- 22.Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, et al. Formation of elongated giant mitochondria in Dfo-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- 23.Kaida D, Yashiroda H, Toh-e A, Kikuchi Y. Yeast Whi2 and Psr1-phosphatase form a complex and regulate Stre-mediated gene expression. Genes Cells. 2002;7:543–552. doi: 10.1046/j.1365-2443.2002.00538.x. [DOI] [PubMed] [Google Scholar]
- 24.Gorsich SW, Shaw JM. Importance of mitochondrial dynamics during meiosis and sporulation. Mol Biol Cell. 2004;15:4369–4381. doi: 10.1091/mbc.E03-12-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet. 1996;14:450–456. doi: 10.1038/ng1296-450. [DOI] [PubMed] [Google Scholar]
- 26.Burke D, Dawson D, Stearns T. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. 2000 edn. xvii. Cold Spring Harbor Press; Plainview, NY: 2000. [Google Scholar]
- 27.Sherman F. Guide to yeast genetics and molecular biology, part B. In: Guthrie C, Fink GR, editors. Methods in Enzymology. Vol. 350. Elsevier; London and New York: 2002. pp. 3–41. [Google Scholar]
- 28.Gresham D, Ruderfer DM, Pratt SC, Schacherer J, Dunham MJ, Botstein D, et al. Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science. 2006;311:1932–1936. doi: 10.1126/science.1123726. [DOI] [PubMed] [Google Scholar]
- 29.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 30.Cerveny KL, Jensen RE. The Wd-repeats of Net2p interact with Dnm1p and Fis1p to regulate division of mitochondria. Mol Biol Cell. 2003;14:4126–4139. doi: 10.1091/mbc.E03-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffin EE, Graumann J, Chan DC. The Wd40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 33.Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet. 2000;25:333–337. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- 34.Pan X, Yuan DS, Ooi SL, Wang X, Sookhai-Mahadeo S, Meluh P, et al. Dslam analysis of genome-wide genetic interactions in Saccharomyces cerevisiae. Methods (San Diego, Calif) 2007;41:206–221. doi: 10.1016/j.ymeth.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sudbery PE, Goodey AR, Carter BL. Genes which control cell proliferation in the yeast Saccharomyces cerevisiae. Nature. 1980;288:401–404. doi: 10.1038/288401a0. [DOI] [PubMed] [Google Scholar]
- 36.Radcliffe P, Trevethick J, Tyers M, Sudbery P. Deregulation of Cln1 and Cln2 in the Saccharomyces cerevisiae Whi2 mutant. Yeast (Chichester, England) 1997;13:707–715. doi: 10.1002/(SICI)1097-0061(19970630)13:8<707::AID-YEA130>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, et al. Mitochondrial fission and fusion mediators, Hfis1 and Opa1, modulate cellular senescence. J Biol Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- 38.Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:15530–15535. doi: 10.1073/pnas.202604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marston AL, Tham WH, Shah H, Amon A. A genome-wide screen identifies genes required for centromeric cohesion. Science. 2004;303:1367–1370. doi: 10.1126/science.1094220. [DOI] [PubMed] [Google Scholar]
- 40.Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.