

Abstract
A well-characterized murine osteosarcoma model for metastasis and invasion was used in this study to determine the role of AP-1 in the progression of this disease. We analyzed K12 and K7M2 cells, two clonally related murine osteosarcoma cell lines that have been characterized as low metastatic or high metastatic, respectively, for AP-1 components and activity. AP-1 DNA binding was similar between the two cell lines; however AP-1 transcriptional activity was enhanced by 3- to 5-fold in K7M2 cells relative to that in K12 cells. The AP-1 complexes in K12 and K7M2 cells was composed primarily of cJun, JunD, FosB, Fra1, and Fra2, with the contribution of individual components in the complex varying between the two cell lines. In addition, an increase in phosphorylated cJun, JNK activity, and phosphorylated ERK1/2 was associated with the more metastatic osteosarcoma phenotype. The significance of AP-1 activation was confirmed by conditional expression of TAM67, a dominant negative mutant of cJun. Under conditions where TAM67 inhibited AP-1 activity in K7M2 cells, migration and invasion potential was significantly blocked. Tam67 expression in aggressive osteosarcoma cells decreased long-term in vivo experimental metastasis and increased survival of mice. This study shows that differences in metastatic activity can be due to AP-1 activation. The inhibition of AP-1 activity may serve as a therapeutic tool in the management of osteosarcoma.
Osteosarcomas are malignant tumors of the bone that frequently metastasize to the lung.1 During this process, a complex set of events is initiated that includes migration from the primary tumor location, intravasation, cellular arrest and adherence, extravasation at distant sites, cell proliferation, and angiogenesis.2 Each of these events is accompanied by changes in gene regulatory patterns, which are required for metastasis. Genetic profiling studies are beginning to decipher the global gene regulatory patterns associated with invasion.3 A murine model of osteosarcoma with differing metastatic potential has previously been developed and used to identify genes that associate with metastasis.4,5 In this model, two clonally related murine cell lines were used: K12 having low metastatic potential and K7M2 with high metastatic potential as observed by the aggressive behavior of K7M2 cells to establish experimental metastasis after tail-vein injection. Of the genes found differentially regulated between these clones, a number have a role in metastasis-associated events such as cytoskeleton rearrangement, motility, and proliferation. In particular, ezrin, a member of the ezrin-radixin-moesin family of proteins that function as cross-linkers between actin filaments and the plasma membrane, and galectin-3, a lectin binding protein that has a role in proliferation and apoptosis, were identified as genes that associate with K7M2 metastasis.4 Both ezrin and galectin-3 have been described as AP-1-regulated transcriptional targets.3,6,7 In addition to these two candidates, a number of other genes that were found to be differentially regulated between K12 and K7M2 are AP-1 targets.4
AP-1 is a transcription complex composed of members of the Jun, Fos, and activating transcription factor (ATF) family of proteins that bind as hetero- and/or homodimers to AP-1 binding sites in the promoters of various target genes.8,9,10 Transcriptional activation of AP-1 target genes requires phosphorylation of the individual components. The cJun component of AP-1 is phosphorylated in the transactivation domain at ser-63 and ser-73 by c-Jun NH (2)-terminal kinase (JNK) and ERK1/2.11,12,13,14 Over expression of Fos, the other major component of AP-1, has been shown to induce osteosarcoma formation in rodents.15,16 In addition, the co-expression of cJun and cFos in Fos-Jun double transgenic mice enhances Fos-induced osteosarcoma, suggesting that AP-1 is involved in osteosarcoma development in murine models.17 These studies and others identifying an association of AP-1-regulated genes with metastasis suggest that AP-1 may have a role in the aggressive phenotype observed in K7M2 osteosarcoma.
The objective of this study was therefore to establish if AP-1 had a role in the metastatic phenotypes observed in K12 and K7M2 murine osteosarcoma cells. To address this objective, we determined the activity and expression levels of the different components of the AP-1 complex and their upstream activators in K12 and K7M2. Our data show that AP-1 activity is increased in the highly aggressive K7M2 and that this phenotype could be suppressed by inhibition of AP-1 with dominant-negative cJun.
Materials and Methods
Cell Culture and Doxycycline Induction
K12 and K7M2 murine osteosarcoma cells were grown in Dulbecco’s minimal essential medium (DMEM; GIBCO/BRL, Burlington, ON, Canada) containing 10% fetal bovine serum and supplemented with penicillin-streptomycin (GIBCO/BRL). K7M2-Green Fluorescent Protein and K7M2-Tam67–3 clones were maintained in the presence of 5 μg/ml blastacidin.5 For each experiment, near-confluent cells were trypsinized with 0.5% trypsin/0.53 EDTA in Hanks-balanced salt solution (Tryp/EDTA; GIBCO/BRL) and plated in media containing 10% fetal bovine serum and penicillin-streptomycin with or without 2 μg/ml doxycycline to induce gene expression.
Preparation of Stable Clones
K7M2 osteosarcoma cells were infected with a Tam67- or GFP-containing pLRT retrovirus as previously described.7 The cells were split as necessary and treated with 8 μg/ml blastacidin. Clones resistant to blastacidin were selected and screened for inducible Tam67 expression using 2 μg/ml doxycycline (Sigma). The K7M2-Tam67 clone with the lowest background and highest inducible gene expression was used in subsequent experiments.
Total Protein and Nuclear Protein Preparation
Cells grown in 100-mm diameter tissue culture dishes were washed with cold PBS. Total protein was extracted from cells using RIPA lysis buffer (150 mmol/L NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, and 10 mmol/L Tris (pH 7.4) containing 100 μg/ml aprotinin, 100 μg/ml leupeptin, and 0.1 mmol/L Phenylmethanesulphonylfluoride. Samples were sonicated and centrifuged. The total protein concentrations were determined using a Biorad assay (Biorad). Nuclear proteins were harvested in cold PBS using a cell scraper. The cells were pelleted and resuspended in 1 ml of a buffer containing HEPES (pH 7.9), 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol, and allowed to swell on ice for 15 minutes. The nuclei were released by slow uptake and rapid ejection (five times) through a 25 gauge needle. The nuclei were pelleted by centrifugation and resuspended in 200 μl of a buffer containing HEPES (pH 7.9), 25% glycerol, 0.45 mol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, 100 μg/ml aprotinin, 1 μg/ml leupeptin, and 0.5 mmol/L Phenylmethanesulphonylfluoride. The samples were incubated at 4°C for 30 minutes with constant rotation. The samples were centrifuged and the supernatant was saved. Nuclear protein concentrations were determined using a Biorad assay. Both total and nuclear protein samples were stored at −80°C until needed.
Transfection and Dual Luciferase Assays
Transfection assays were performed using FuGene6 reagent. For each transfection and dual luciferase assay, 1 μg of pGL2-luciferase, TRE2-luciferase, −1048/+245 CyclinA-luciferase, 4×AP-1-luciferase, or mutated 4×AP-1-lucifase and 20 ng of thymidine kinase-Renilla luciferase (pRL-TK-Renilla) plasmid in Fugene6 reagent was added to cells in six-well plates. The transfected cells were incubated at 37°C for 24 hours. The proteins were harvested in passive lysis buffer (Promega, Madison, WI), and the luciferase activities of the lysates were analyzed using the dual luciferase reagents (Promega). All experiments were performed in triplicate.
Western Immunoblotting Analysis
Thirty to seventy μg of the protein sample was separated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The membranes were incubated in a 1:2000 dilution of primary antibody (Santa Cruz Biotechnology or Cell Signaling Technology) in Tris-buffered saline Tween 20, followed by incubation in a 1:4000 dilution of a secondary antibody linked to horseradish peroxidase (Amersham Biosciences) in Tris-buffered saline Tween 20. Proteins were identified using chemiluminescence (Amersham Biosciences) and autoradiography of X-ray films. Antibodies used include; c-Jun (sc-44), Jun B (sc-46), Jun D (sc-74), c Fos (sc-52), Fra 1 (sc-605), Fra 2 (sc-604), ATF 1 (sc-243), ATF 2 (sc-187), ATF 3 (sc-188), ATF 4 (sc-200), JAB-1 (sc-15353), JIP-1 (sc-9074), JNK1 (sc-571), and β-tubulin (sc-9404) obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-specific antibody (#92615) against Ser-63-c-Jun was obtained from New England Biolabs (Beverly, MA). Antibodies against Phospho-ERK1/2 (#9101) and ERK1/2 (#9102) were obtained from Cell Signaling Technology.
Gel Mobility and Gel Supershift Assay
Six μg of nuclear extract was incubated with 2 μl of poly dI/dC, 4 μl of 5× incubation buffer (100 mmol/L HEPES [pH 7.9], 250 mmol/L KCl, 2.5 mmol/L EDTA, 5 mmol/L MgCl2, 20% Ficoll 400), and 2 μl of cold AP-1 oligomer or cold mutated AP-1 oligomer, if necessary, in a total volume of 20 μl for 20 minutes on ice. AP-1 oligomers labeled with [γ-32P]ATP were then added and the mixtures were incubated on ice for 45 minutes. For supershift assays, 1 μl of supershift antibody (Santa Cruz Biotechnology) was added to the protein-probe mixture and allowed to incubate on ice for an additional 45 minutes. The samples were separated on a non-denaturing 5% polyacrylamide gel at 180 volts for 3 hours at 4° in 0.5×Tris/Borate/EDTA. Gels were dried and exposed to X-ray film.
JNK Assay
Cells grown in 100-mm diameter tissue culture dishes were washed with cold PBS. Non-denatured proteins were extracted from cells using cell lysis buffer (20 mmol/L Tris [pH 7.4], 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerolphosphate, 1 mmol/L Na3VO4, 1 μg/ml leupeptin, containing 1 mmol/L Phenylmethanesulphonylfluoride) obtained from Cell Signaling Technology. The lysed cells were sonicated, then centrifuged, and protein concentrations were determined using a Biorad assay. A 2-μg sample of c-Jun fusion beads (Cell Signaling Technology) was added to 250 μg total protein and rotated at 4°C for 24 hours. The sample was pelleted, washed twice with cell lysis buffer, and twice with kinase buffer (25 mmol/L Tris [pH 7.4], 5 mmol/L β-glycerolphosphate, 2 mmol/L DDT, 0.1 mmol/L Na3VO4, 10 mmol/L MgCl2) on ice. The sample was suspended in 50 μl kinase buffer supplemented with 100 μmol/L ATP and incubated at 30°C for 30 minutes. The reaction was terminated by the addition of 15 μl of 4×LDS sample buffer (NuPage). The sample was then boiled, and 20 μl loaded onto an SDS-polyacrylamide electrophoresis gel for Western immunoblotting analysis. A phospho-cJun antibody (Cell Signaling Technology) was used to detect the amount of cJun phosphorylation. Membranes were exposed to X-ray film.
Motility Assays
Motility assays were performed using transwell chambers (Costar, Cambridge MA) with 8-μm pore size. Cells in serum-free DMEM were added to an upper transwell chamber. The chamber was placed into lower chambers containing DMEM supplemented with 10% fetal bovine serum. Cells were incubated for 4, 8, or 24 hours at 37°C. On completion of the incubation period, the cells in the upper chambers were removed by gentle wiping with a cotton tip and the migrated cells on the bottom chamber stained using DiffQuick reagent (American Scientific Products, McGraw Park, IL). The transwell membranes were then viewed under a microscope for cell counting. At least six views per condition were counted and averaged. The percentage of motile cells was calculated related to the total number of cells plated as described in.4 Each experiment was performed in triplicate.
Invasion Assays
The in vitro invasive ability of K7M2-Tam67 cells grown in the absence and presence of doxycycline was determined using Transwell plates with 8 μm pore size coated with basement membrane matrix. The assay was performed using a fluorometry-based cell invasion assay according to the manufacturers protocols (Oncogene Research Products). Briefly, 300 μl of 0.5 × 106 cells/ml in serum-free media were added to the top chamber and incubated for 48 hours at 37°C. The bottom chamber contained 10% FBS containing DMEM. After incubation the cells on the underside of the chamber were removed by gently tapping the insert and incubated in a 500 μl 1:300 diluted calcein-AM solution for 30 minutes in the tissue culture incubator. The insert was removed and the dislodged cells incubated for an additional 30 minutes. Two hundred μl of the dislodged cells were then transferred to a 96-well plate in duplicate and fluorescence at excitation 485 nm and emission 520 nm measured. Experiments were done in triplicate.
Ex vivo Metastasis Videomicroscopy
Ex vivo metastasis videomicroscopy was performed as described by Khanna et al, 2000.5 Female, balb/c mice of 4 to 6 weeks of age were purchased from Charles Rivers Laboratories (NCI-Frederick Animal Production Area). Mice were divided into experiment groups consisting of three mice per condition per experiment and received either untreated water or water consisting doxycycline 2 mg/ml (Sigma Inc) in 5% sucrose solution (MP-Biomedicals, LLC) ad libitum for 3 days before each experiment. Cells were labeled with the Cell Tracker Green CMFDA (Invitrogen) at a final concentration of 25 μmol/L in serum-free DMEM. Subsequently, labeled cells were trypsinized, detached, and resuspended in 1×Hanks’ balanced salt solution. Using a 27-guage×1/2 inch hypodermic needle, in 0.1 ml volume of cells (1×106) was delivered by lateral tail vein injection. Mice were euthanized at either 1 hour or 6 hours postinjection. Three mice were injected per cell line per time point. On euthanization, lungs were removed and inflated using slow intratracheal injection of PBS. Lungs were then imaged ex vivo with a LEICA fluorescent inverted microscope as previously described.18 Images of 10 representative regions of each removed lung were captured using Openlab software. Experimental lung tissue was preserved in 10% formalin. The number of cells per image was counted using the Openlab software automatic feature counter and histographs were constructed in Microsoft Excel. Statistical significance was determined using non-parametric t-test and experiments were performed in quadruplicate.
In Vivo Experimental Metastasis
Mice were divided into four experiment groups (doxycycline induced K7M2-Tam67, uninduced K7M2-Tam67, doxycycline-induced K7M2-GFP, and uninduced K7M2-GFP) consisting of 10 mice per group per experiment and either received untreated water or water containing 2 mg/ml doxycycline (Sigma Inc) in 5% sucrose solution (MP-Biomedicals, LLC) ad libitum for 3 days before each experiment and then continuously through the duration of the experiment. Fresh doxycycline water was replaced twice weekly. Cells were resuspended in 1×Hanks’ balanced salt solution. Using a 27-guage×1/2 inch hypodermic needle, a 0.1 ml volume of cells (1 × 106) was delivered by lateral tail vein injection into 4- to 6-week-old BALB/c mice. Mice were monitored at least twice weekly for the development of metastasis related morbidity. Animals were sacrificed based on two different criteria. For experiment 1, animals were sacrificed in the premorbid state. Criterion for morbidity associated with metastases in mice included ill thrift, anorexia, dehydration, decreased activity and grooming behavior, and dyspnea. Sacrifice of mice with presumed pulmonary metastases was primarily based on the development of dyspnea. For experiment 2, animals were sacrificed at 37 days, a time determined to easily demonstrate the size and number of metastases in this model. All mice that were sacrificed due to presumed pulmonary metastases had necropsy confirmation of metastases. Their lungs were extracted, fixed in formalin, and examined for the presence of metastases by H&E light microscopy. Each experiment was repeated in triplicate. Statistical analysis was performed by and log-rank and log-rank trend statistics were used for survival curves (using Graphpad Prism v3.0a).
Results
Increased AP-1 Activity in Highly Metastatic Murine Osteosarcoma Cells
AP-1 target genes such as ezrin and galectin-3 have previously been shown to have increased expression in the more aggressive K7M2 osteosarcoma cell line when compared to the less aggressive K12 cell line.4 To determine whether differences in the biological phenotypes of these two cell lines could be attributed to alterations in AP-1 activity, we performed promoter activity assays. Transient transfections of two artificial promoter constructs containing either two (TRE2-luciferase) or four putative AP-1 binding sites (4×-AP-1-luciferase) were performed. These AP-1 regulated promoter constructs had enhanced activity in the K7M2 compared to K12 (Figure 1A, B). Promoter assays with mutated AP-1 binding sites confirmed that the differences in promoter regulation were a result of alterations in AP-1 activity (Figure 1B). Similar to artificial AP-1 promoter constructs, the promoter of a known AP-1 responsive gene, cyclin A had a significantly higher activity in K7M2 osteosarcoma (Figure 1C). These results suggest an increase in AP-1 activity in the more aggressive osteosarcoma cell type.
Figure 1.

AP-1 promoter activity in K12 and K7M2 osteosarcoma cell lines. AP-1 promoter activity was assayed by transient transfection of TRE2-Luciferase containing two AP-1 binding sites (A) and 4×AP-1-luciferase containing four AP-1 binding sites (B) and the −1048 to + 205 cyclin A promoter (C) fused to the luciferase reporter gene. Results are representative of the mean ± SD of experiments performed in triplicate. Renilla-luciferase under the control of the TK promoter was used as a control for transfection efficiency. Empty vectors and 4×mut AP-1-Luciferase containing four mutated AP-1 binding sites were used as negative controls (*P < 0.05, n = 4).
AP-1 Complex Components in Murine Osteosarcoma Cells
To further investigate what contributed to the differences in AP-1 activity between the two cell lines, we performed gel shift assays with an AP-1 consensus binding site. AP-1 binding was detected in K12 and K7M2 nuclear extracts and binding could be competed with unlabeled wild-type AP-1 oligomers but not the mutant oligomer (Figure 2). No difference in the relative amounts of AP-1 binding was observed between the two cell lines (Figure 2, lanes 1 and 11). Gel shift assays analysis with antibodies that have previously been shown to result in either the electrophoretic supershift or reduction of complexes containing individual AP-1 proteins19 identified supershift/reduction of cJun, JunD, FosB, Fra2, and to a lesser extent, Fra1 in K12 nuclear extracts (Figure 2, lanes 4, 6, 8, 9, 10). While the binding of the same AP-1 components was affected in K7M2 nuclear extracts, the supershift/reduction effect for JunD, Fra1, and Fra2 (Figure 2, lanes 16, 19, 20) appeared altered compared to K12. Western blot analysis for AP-1 components revealed no major differences in cJun, JunB, JunD, and cFos expression levels (Figure 3). An increase in Fra1 and ATF3 levels, and decrease in ATF2, was observed in K7M2 protein extracts (Figure 3). No difference in the upstream modulators of AP-1 activity, the Jun-activation domain binding protein (JAB1) and JNK interacting protein 1 (JIP1), was detected. Phosphorylation of cJun at ser-63 however, was approximately twofold higher in K7M2 cells (Figure 3). Thus, while total cJun amounts do not differ between K12 and K7M2 cells, increased phosphorylation of cJun is apparent in the more aggressive cell line, suggesting that JNK, and possibly other mitogen-activated protein kinases such as MEK, may have increased activity in K7M2 cells.
Figure 2.

Electrophoretic mobility shift assays using the AP-1 consensus oligomer. Five μg of nuclear protein extracted from K12 or K7M2 cells were incubated with a radioactively labeled AP-1 consensus oligomer. DNA-protein complexes were resolved by 4.5% non-denaturing polyacrylamide gel electrophoresis. Unlabeled wild-type and mutant AP-1 oligomers were used as competitors for complex formation. Antibodies against specific AP-1 components were used in supershift experiments to determine the nature of the DNA-protein complex. The asterisk shows the specific AP-1 DNA-binding complex and arrows show supershifted DNA-protein complexes.
Figure 3.
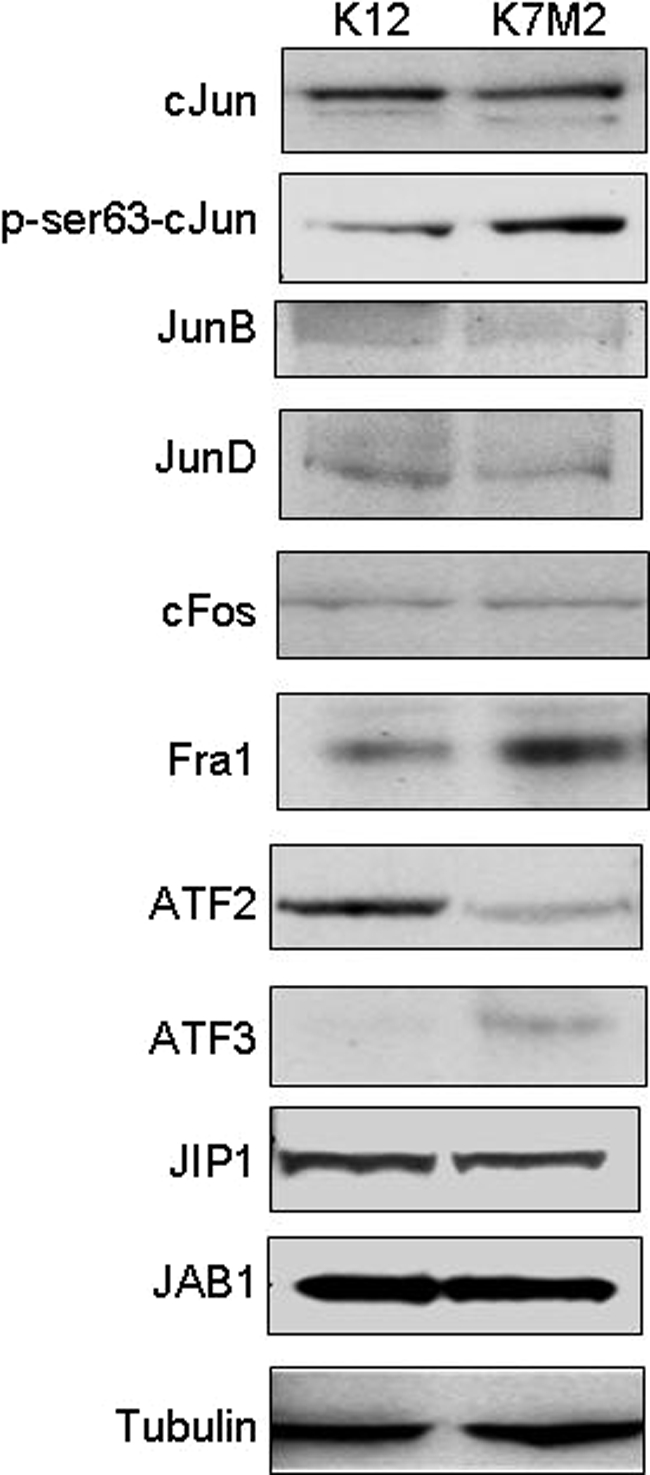
Western blot analysis for AP-1 components and upstream modulators. Western blot analyses were performed using 30 μg total cell protein extracted from K12 and K7M2 osteosarcoma cells separated by SDS-polyacrylamide gel electrophoresis. After transfer of the proteins to nitrocellulose membranes the blots were probed with antibodies against cJun, p-cJun, JunB, JunD, cFos, Fra1, ATF2, ATF3, JIP1, JAB1, and β-tubulin as a control for protein loading. All blots were generated using the same protein extract, with the p-cJun blots stripped to analyze total cJun levels. The results shown are representative of three independent experiments.
JNK and MEK1 Activity is Increased in Highly Metastatic Osteosarcoma
To determine the level of JNK activity in K12 and K7M2 cell lysates, JNK1/2 kinase assays were performed using GST-cJun-fusion beads. Phosphorylation of the GST-cJun fusion protein was achieved by incubation of K12 and K7M2 protein extracts with ATP in a kinase buffer. The phosphorylation of GST-cJun was then detected by Western blot analysis using a Ser63 phospho-specific antibody. An increase in JNK activity was observed in K7M2 compared to K12 cells (Figure 4A). This was not as a result of an increase in JNK1/2 levels since these remained largely unchanged between the two cell lines. Similarly, phosphorylation of ERK1/2 was considerably increased in K7M2 cells compared to K12 cells, suggesting an increase in MEK1/2 activity (Figure 4B). These results suggest that the increase in AP-1 activity in K7M2 may be associated with an increase in MAP Kinase activated signaling pathways.
Figure 4.

JNK and MEK1/2 kinase activity is increased in K7M2 cells. A: JNK assays were performed using protein extracted in a nondenaturing lysis buffer. Equal amounts of protein from K12 and K7M2 cells were incubated with a GST-cJun fusion protein linked to agarose beads to pull down JNK in the extracts. In vitro kinase reactions were performed in the presence of cold ATP and phosphorylation of the GST-cJun fusion protein at Ser63 were detected using a phospho-specific antibody in Western blot analysis. JNK1/2 and β-tubulin levels are shown, indicating that equal amounts of proteins were used in the assay. B: MEK1/2 activity was assayed by determining the phosphorylation status of ERK1/2. Equal amounts of K12 and K7M2 protein were used in Western blot analysis and p-ERK1/2 and total ERK1/2 levels are shown. The results shown are representative of three independent experiments.
Dominant Negative cJun Inhibits AP-1 Activity and the K7M2 Phenotype
The significance of increased AP-1 activity in K7M2 cells was determined by using a dominant negative mutant of cJun, Tam67, to inhibit endogenous AP-1 activity. K7M2 clones with doxycycline regulated Tam67 were prepared and screened for Tam67 by Western blot analysis. K7M2 cells with inducible GFP expression (K7M2-GFP) served as a control for the effect of doxycycline alone. A number of clones with varying conditional Tam67 expression were isolated (Figure 5A). The clone with the highest detectable doxycycline inducible Tam67 levels, K7M2-Tam67–3 was used in subsequent analysis. A time course of doxycycline treatment indicated that Tam67 was induced approximately 8 hours after treatment and was maximally expressed by 24 hours (Figure 5B). The expression of Tam67 occurred with an associated decrease in endogenous cJun levels (Figure 5B). This result confirmed that Tam67 inhibits AP-1 activity in K7M2 cells as c-jun is a known AP-1-regulated gene. The inhibition of AP-1 by Tam67 had no significant effect on the proliferation of K7M2 cells (Figure 5C). Cells grown in the absence of doxycycline had a doubling time of 22.09 ± 0.98 hours while that of those grown in the presence of doxycycline was 25.69 ± 1.77 hours. The decrease in cJun levels in Tam67 expressing K7M2 cells suggested that AP-1 activity should be decreased in these cells. Support for these observations was obtained by transient transfection of the TRE2-promoter-luciferase construct into K7M2-Tam67 cells grown in the absence and presence of doxycycline. Doxycycline regulated expression of Tam67 resulted in a significant inhibition in TRE2-promoter activity of approximately 63% in K7M2 Tam67 expressing cells (Figure 6A). This inhibition was not as a result of doxycycline alone, since promoter activity was not altered in the GFP expressing K7M2 control cells. Tam67 also had a nonspecific effect on the TK-Renilla promoter-reporter construct (Figure 6B). Taken together, these results show that dominant negative cJun can inhibit the expression of endogenous cJun expression as well as block activation of an AP-1 responsive promoter construct in aggressive osteosarcoma cells.
Figure 5.

Inducible dominant-negative cJun expression in K7M2 cells. A: K7M2 clones with stable inducible expression of GFP or a dominant-negative cJun, Tam67, were prepared as described in Materials and Methods. Inducible expression of Tam67 was determined by Western blot analysis using a cJun-specific antibody that recognizes Tam67 as a 29kD protein. Clone K7M2-Tam67–3 had the highest inducible Tam67 expression. B: Western blots showing a time course of doxycycline-inducible Tam67 expression in K7M2-Tam67 cells. Inducible Tam67 expression results in the inhibition of endogenous cJun expression. Results are representative of at least three independent clones. Tubulin is shown as a control for differences in loading. C: The effect of inducible Tam67 on the growth of K7M2 cells. Cell growth in the absence and presence of 2 μg/ml doxycycline (+Tam67) was performed in 96 well plates and measured by MTT assays (Promega) as described in materials and methods. Results are the mean ± SD of eight wells per condition. The doubling time for K7M2 cells was not significantly changed by TAM67.
Figure 6.

AP-1 promoter activity is inhibited by inducible Tam67 expression. K7M2-GFP and K7M2-Tam67 cells grown in the absence and presence of doxycycline for 24 hours were transiently transfected with the TRE2-promoter-luciferase construct containing two AP-1 binding sites and lysates extracted after 48 hour incubations. A: Luciferase activity was inhibited by inducible Tam67 expression in K7M2-Tam67 cells and not in K7M2-GFP control cells. (*P < 0.05, n = 3). B: Tam67 expression had an inhibitory effect Renilla-luciferase expression, although not to the same extent as that on the TRE2-Promoter construct. Results are the mean ± SD of experiments performed in triplicate.
Inhibition of AP-1 Activity Interferes with K7M2 Migration and Invasion
Biological phenotypes associated with metastasis include motility and invasion. K7M2 cells have previously been shown to be highly motile. To determine whether AP-1 activity is required for the movement of these cells, in vitro motility assays using transwell chambers were performed with control and Tam67 expressing K7M2 cells. These assays were performed after growing the cells in the absence and presence of doxycycline for 24 hours before setting up the experiment. Cell migration was allowed for 4, 8, and 24 hours in the absence and presence of doxycycline. Tam67 expression significantly inhibited the migration of K7M2 cells (Figure 7A). This inhibition was observed at 4, 8, and 24 hours and was significant in relation to the total cells assayed (Figure 7B). Doxycycline treatment of the K7M2-GFP control cells had no effect on cell migration while doxycycline-inducible Tam67 expression resulted in an approximately 80% reduction in K7M2 motility (Figure 7C). Taken together, these results suggest that AP-1 activation is essential for the motility associated with K7M2 osteosarcoma cells. To determine whether AP-1 activity associated with the invasive ability of K7M2 cells, we performed in vitro invasion assays using transwell chambers coated with basement membrane matrix and demonstrated that Tam67 inhibited the invasion of K7M2 cells (Figure 8A).
Figure 7.

K7M2 motility is inhibited by dominant-negative cJun. A: In vitro motility of K7M2-Tam67 cells grown in the absence and presence of doxycycline for 24 hours determined using Transwell culture chambers containing membranes with 8-μm pores as described in Materials and Methods. Motility of cells in the absence (−Tam67) and presence (+Tam67) of doxycycline were compared at 4, 8, and 24 hours. The total number of cells plated in each condition was stained on the membrane within 4 hours of plating. After incubation for 4, 8, and 24 hours the cells in the top on the chamber were removed and those that have migrated onto the bottom of the chamber were stained with DiffQuick solution and photographed at original magnification ×100. B: The percentage of motile cells scored as the number of cells at the bottom of the chamber relative to the total number of cells plated. Tam67 inhibited the motility of K7M2 cells relative to controls at 4, 8, and 24 hours. C: Motility of K7M2-GFP and K7M2-Tam67 at 24 hours. Motility in the presence of doxycycline is expressed relative to un-induced controls. Doxycycline-inducible GFP expression has no effect on the motility of K7M2 cells, while Tam67 expression significantly inhibits K7M2 motility. (*P < 0.05, n = 3).
Figure 8.

Inhibition of K7M2 in vitro and in vivo metastasis by Tam67. A: In vitro invasion assays of K7M2-Tam67 cells grown in the absence and presence of doxycycline. In vitro invasion was determined by a fluorometric-based cell invasion assay using transwell plates with 8-μm pore size that have been coated with basement membrane matrix. Induction of Tam67 expression with doxycycline significantly inhibited in vitro invasion. B: Western blot analysis showing Tam67 and cJun levels in a pool of K7M2-Tam67 cells prepared for lung colonization assays. Cells were grown in the absence and presence of doxycycline for 48 hours before harvesting for invasion assays. Overexpression of Tam67 results in a decrease in cJun levels as observed in Figure 5B. C: Short term in vivo experimental metastasis imaging of fluorescently labeled K7M2-Tam67 cells grown in the absence and presence of doxycycline for 48 hours before tail-vein injection of the cells into mice. Fluorescent cells located in the lungs of injected animals were photographed at 1 and 6 hours after tail-vein injection. Representative fields from lungs are shown at original magnification ×100. D: Quantification of fluorescence detected in the lungs of eight mice per group injected with control (−dox) and Tam67 expressing (+dox) K7M2 cells. Results shown are the mean ± SEM and are expressed as a percentage of cells in the lungs imaged at 1 hour after tail-vein injection. (*P < 0.05, n = 3).
Inhibition of AP-1 Activity Interferes with K7M2 Experimental Metastasis in Vivo and Prolongs Survival
To determine the effect of Tam67 expression on in vivo invasion and experimental metastasis, Tam67 expressing K7M2 cells were used to induce experimental metastasis in BalbC mice. A fraction of cell lysates from K7M2-Tam67 control (−dox) and Tam67-expressing K7M2 cells (+dox) to be injected into mice were first assayed by western blots to confirm the expression of Tam67 (Figure 8B). As observed previously, Tam67 expression resulted in an associated decrease in cJun levels confirming Tam67’s inhibitory effect on AP-1 activity. Using the single-cell fluorescent imaging of CMFDA-labeled cells as previously described,18 we examined the effects of Tam67 expression on the early steps that follow arrival of single metastatic cells in the lung. After tail vein injection of control (−dox) and Tam67 expressing (+dox) cells, the load of tumor cells was determined by imaging of fluorescent tumor cells in the lungs of injected animals at 1 hour and 6 hours post-experimental metastasis (Figure 8C). A decrease in the number of cells colonizing the lungs was observed within 6 hours of tail vein injection in the Tam67 expressing group; this change however, was not significant at this early time point (Figure 8D).
To determine the role of Tam67 expression on the consequences of osteosarcoma metastasis, mice were followed for the development of metastasis-associated morbidity. In this model, there is a predictable series of clinical changes in mice that occurs as a result of pulmonary metastases. Mice develop weight loss, poor hair coat, and eventually dyspnea (humane endpoint of survival; see Materials and Methods). Marked and diffuse pulmonary metastasis is confirmed by necropsy in all sacrificed mice. In the event that marked and diffuse metastasis is not seen, mice are censured from the data analysis. The biological differences in Tam67-expressing K7M2 cells became apparent during these longitudinal in vivo experiments. The suppression of AP-1 activity by both conditional (doxycycline-inducible) and constitutive expression of Tam67 (pcDNA-Tam67) significantly impacted the survival of the animals. Mice injected with Tam67-expressing cells showed longer survival compared to those injected with the untreated or control cell lines. Log-rank trend tests showed a significant increase in the survival of animals injected with Tam67 expressing osteosarcoma (Figure 9A and B). The survival of animals injected with doxycycline-inducible Tam67 expressing K7M2 cells was similarly increased (69 days, P = 0.015 under doxycycline induction) in comparison with that of animals injected with K7M2-GFP control cells (0 days, under doxycycline induction). In addition, mice treated with K7M2-GFP cells and doxycycline did not survive any longer than K7M2-Tam67 treated mice without doxycycline. The animals were followed to the same clinical endpoint (distress); however, the number of pulmonary metastases was decreased in animals with high Tam67 expression (25.2% decreased), although this was not statistically significant. While fewer pulmonary lesions were observed in the presence of doxycycline-inducible Tam67 expression, these lesions were large and showed a high indication of necrosis (Figure 10A–D). To confirm these results, we repeated the in vivo experiment but sacrificed all of the animals at a time when the animals were not yet showing distress (37 days), allowing for a more accurate assessment of differences in number and size of pulmonary metastases. Gross examination of the lungs demonstrated a decrease in pulmonary metastases in the animals with high TAM67 expression. Microscopic examination of the lungs revealed a 67% decrease in pulmonary metastases with TAM67 expression (P < 0.001) and a decrease in the size of those lesions (Figure 11). Our results suggest that the inhibition of AP-1 activity in aggressive osteosarcoma suppresses the formation of metastatic lung tumors and hence increases overall survival.
Figure 9.

Inhibition of AP-1 activity with Tam67 significantly prolongs survival of mice with experimental osteosarcoma metastasis. A: Doxycycline-induced Tam67 expression in K7M2 osteosarcoma cells resulted in an increase in survival (P < 0.0001 by log-rank test for K7M2-Tam67 −dox vs +dox). B: Constitutive Tam67 expression in K7M2 cells (K7M2-pCDNA-Tam67) results in a significant increase in survival compared to mice injected with K7M2 cells containing the empty vector (P < 0.005, n = 3).
Figure 10.

Pulmonary metastasis in animals with K7M2-Tam67 cells sacrificed at premorbid state. A: H&E staining of lung from an animal injected with K7M2-Tam67 cells without doxycycline treatment. B: H&E staining of lung from an animal injected with K7M2-Tam67 with doxycycline treatment (C) High power (original magnification × 20) image of lung (Lu) containing a metastatic lesion (tumor) from an animal without doxycycline treatment. D: High power (original magnification × 20) image of lung of lung containing metastasis (Tu) with necrosis (Ne) from an animal with doxycycline treatment. Images shown are representative of all experimental animals.
Figure 11.

Pulmonary metastasis in animals with K7M2-Tam67 cells sacrificed at 37 days. A: H&E staining of lung from an animal injected with K7M2-Tam67 with doxycycline treatment containing two lymph nodes (LN) and small metastasis (arrow). B: H&E staining of lung from an animal injected with K7M2-Tam67 cells without doxycycline treatment containing lymph node (LN) and many large metastases (arrows). Images shown are representative of all experimental animals.
Discussion
In this study, we identified an association between AP-1-dependent transcriptional activation and invasion with two clonally related murine osteosarcomas, K12 and K7M2. Enhanced AP-1 activity was observed in the highly aggressive K7M2 compared to K12. Similar results have been reported in breast cancer, where AP-1 overexpression associates with increased motility and invasion of MCF-7 breast cancer cells.20,21 To characterize the molecular basis for the difference in AP-1 activity in K12 and K7M2 osteosarcoma, we determined its composition and upstream activators. Supershift gel shift analysis identified slight variations in the major components of the AP-1 complex in K7M2 and K12 cells. This finding was supported by Western blot analysis showing that phospho-cJun and Fra1 levels were increased in K7M2. Previous studies have shown that phosphorylation of cJun in the transactivation domain enhances its activity as a regulator of transcription.12,22 Contrary to our expectations, ATF2 expression was significantly decreased in K7M2 relative to K12. ATF2 is a member of the ATF family of proteins and have been shown to have role in oncogenesis by interacting with v-Jun in primary chicken embryo fibroblasts.23 Our results suggest ATF2 is not required for the activation of AP-1 responsive promoters in K7M2 and that the involvement of cJun and possibly Fra1 may be the determining factors. This hypothesis is supported by our findings that JNK activity is significantly higher in K7M2 osteosarcoma. The activation of JNK signaling pathways have been demonstrated to have a role in osteoblast proliferation and apoptosis.24,25 JNK-activated AP-1 signaling pathways have also been associated with the progression of human osteosarcoma and the formation of osteosarcomas in transgenic mice.26,27 The increase in JNK and AP-1 activities in K7M2 may therefore be a prerequisite for the aggressive phenotype of these cells.
In addition to JNK activation, MEK activity, as determined by phosphorylation of ERK1/2, was significantly increased in K7M2. MEK has been reported to stimulate JNK activity in human leukemia cells.28 It is possible that MEK1 can stimulate JNK and hence AP-1 activity in these cells. A recent study showed that phosphorylation of cJun at ser-63 and ser-73 is mediated by ERK1/2 in response to TPA and EGF stimulation.14 The regulation of cFos expression is thought to involve ERK1/2.10 Interestingly, high levels of cFos have been detected in many osteosarcomas of human and murine origin.29,30,31 The overexpression of cFos in mice causes osteosarcoma by transforming chondroblasts and osteoblasts.32 We, however, did not observe an increase in cFos expression in K7M2 suggesting that cFos is not involved in AP-1 activity in K7M2. A study by Rupp et al., (1998) reported that while overexpression of AP-1 complex members such as c-fos and v-fos may be involved in tumorigenesis, the increased expression of these component proteins does not associate with murine osteosarcoma invasiveness and metastasis.33 It would thus appear that absolute levels of at least the Fos proteins are not essential for metastasis. Similarly, our results suggest that differences in metastatic potential reported for the two clonally related K12 and K7M2 murine osteosarcoma cell lines does not associate with differences in the expression levels of AP-1 components such as cFos, cJun, JunB, and JunD, but rather that an increase in AP-1 activity associated with an increase in MAP kinase-activated signaling pathways may be involved. Fra1 expression and DNA binding on the other hand was increased. The transactivation of Fra1 and resultant AP-1 transcriptional activation has been reported to occur in a ERK-dependent manner in a mouse epidermal model for oncogenesis.34 Fra1 overexpression has also been implicated in the transformation, motility and invasiveness of epithelial adenocarcinoma cells.35 The combination of JNK and ERK activation and the increase in phospho-cJun and Fra1 in K7M2 may therefore account for the increase in AP-1 activity and invasiveness observed in these cells.
The significance of AP-1 activation to the biological phenotype of K7M2 was determined by using Tam67, a dominant-negative mutant of cJun, to inhibit its activity. TAM67 can form blocking homodimers or inhibitory heterodimers with other Jun, Fos, and ATF members. In our study, Tam67 had a significant inhibitory effect on AP-1 activity, and in turn cellular motility and invasion. While TAM67 has been shown to inhibit AP-1 activity and cellular events such as motility and invasion in other cells such as keratinocytes, most of these studies have used in vitro systems and focused on early events in transformation such as tumor promotion.36 To our knowledge, this is the first study to report in vivo inhibition of AP-1 activity in an experimental metastasis model resulting in significant increase in survival of the animals. The survival of the animals in this model is the direct result of the number and size of the pulmonary metastases. The prolongation of survival by inhibition of AP-1 in our study associates with a decrease in experimental metastasis. We suspect that the decrease in the number of observed pulmonary metastases would have been greater if the animals were not allowed to progress till distress. Our data suggest that AP-1 plays an important role during the early stages of metastasis in this model. These early events would include cell motility and invasion, both shown to be inhibited by TAM67 in in vitro assays. Further, the number of cells in the lung after 6 hours was lower in the TAM67 expressing cells consistent, with a role early in the metastatic process. Of note, many of the animals that had decreased numbers of metastases (TAM67 expressors) had larger lesions than the control animals (sacrificed when premorbid). This observation is consistent with fewer cells initiating lesions in the lungs, but once established, growing at similar rates as those without TAM67. Presumably the larger size results from the greater available lung volume in animals with high TAM67 expression and less total pulmonary metastasis. This is supported by the observations of decrease in the number and size of metastases at 30 days.
The data presented in this study support the concept that AP-1 transcriptional activity and the expression of AP-1-regulated genes is important for the process of metastasis in osteosarcoma. Further, since AP-1 has an essential role in metastasis and invasion of osteosarcoma, it could be a potential therapeutic target for osteosarcoma.
Footnotes
Address reprint requests to Dr. Michael J. Birrer, National Institutes of Health, Building 37, Bethesda, MD, 20892, E-mail: birrerm@mail.nih.gov.
This work was prepared as part of our official duties. Title 17 U.S.C. §105 provide that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
References
- Bruland OS, Pihl A. On the current management of osteosarcoma: A critical evaluation and a proposal for a modified treatment strategy. Eur J Cancer. 1997;33:1725–1731. doi: 10.1016/s0959-8049(97)00252-9. [DOI] [PubMed] [Google Scholar]
- Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer. 1997;80:1529–1537. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1529::aid-cncr2>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- Ozanne BW, McGarry L, Spence HJ, Johnston I, Winnie J, Meagher L, Stapleton G. Transcriptional regulation of cell invasion: AP-1 regulation of a multigenic invasion programme. Eur J Cancer. 2000;36:1640–1648. doi: 10.1016/s0959-8049(00)00175-1. [DOI] [PubMed] [Google Scholar]
- Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, Trepel J, Meltzer P, Helman L. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res. 2001;61:3750–3759. [PubMed] [Google Scholar]
- Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin Exp Metastasis. 2000;18:261–271. doi: 10.1023/a:1006767007547. [DOI] [PubMed] [Google Scholar]
- Stapleton G, Malliri A, Ozanne BW. Downregulated AP-1 activity is associated with inhibition of protein-kinase-C-dependent CD44 and ezrin localisation and upregulation of PKC theta in A431 cells. J Cell Sci. 2002;115:2713–2724. doi: 10.1242/jcs.115.13.2713. [DOI] [PubMed] [Google Scholar]
- Kinoshita I, Leaner V, Katabami M, Manzano RG, Dent P, Sabichi A, Birrer MJ. Identification of cJun-responsive genes in Rat-1a cells using multiple techniques: Increased expression of stathmin is necessary for cJun-mediated anchorage-independent growth. Oncogene. 2003;22:2710–2722. doi: 10.1038/sj.onc.1206371. [DOI] [PubMed] [Google Scholar]
- Vogt PK. Jun, the oncoprotein. Oncogene. 2001;20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola D, Grover-Bardwick A, Heidecker G, Rapp UR, Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Mol Cell Biol. 1992;12:3507–3513. doi: 10.1128/mcb.12.8.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the c-Jun N-terminal kinase. Biochem Soc Symp. 1999;64:1–12. doi: 10.1515/9781400865048.1. [DOI] [PubMed] [Google Scholar]
- Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003;22:3876–3886. doi: 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Livne E, Erfle V, Gossner W, Silbermann M. Morphology and in vivo growth characteristics of an atypical murine proliferative osseous lesion induced in vitro. Cancer Res. 1986;46:3090–3098. [PubMed] [Google Scholar]
- Ward JM, Young DM. Histogenesis and morphology of periosteal sarcomas induced by FBJ virus in NIH Swiss mice. Cancer Res. 1976;36:3985–3992. [PubMed] [Google Scholar]
- Wang ZQ, Liang J, Schellander K, Wagner EF, Grigoriadis AE. c-fos-induced osteosarcoma formation in transgenic mice: cooperativity with c-jun and the role of endogenous c-fos. Cancer Res. 1995;55:6244–6251. [PubMed] [Google Scholar]
- Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, Helman LJ. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10:182–186. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- Leaner VD, Kinoshita I, Birrer MJ. AP-1 complexes containing cJun and JunB cause cellular transformation of Rat1a fibroblasts and share transcriptional targets. Oncogene. 2003;22:5619–5629. doi: 10.1038/sj.onc.1206644. [DOI] [PubMed] [Google Scholar]
- Briggs J, Chamboredon S, Castellazzi M, Kerry JA, Bos TJ. Transcriptional upregulation of SPARC, in response to c-Jun overexpression, contributes to increased motility and invasion of MCF7 breast cancer cells. Oncogene. 2002;21:7077–7091. doi: 10.1038/sj.onc.1205857. [DOI] [PubMed] [Google Scholar]
- Smith LM, Wise SC, Hendricks DT, Sabichi AL, Bos T, Reddy P, Brown PH, Birrer MJ. cJun overexpression in MCF-7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene. 1999;18:6063–6070. doi: 10.1038/sj.onc.1202989. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, Smeal T, Derijard B, Cobb M, Davis R, Karin M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguier S, Baguet J, Perez S, van Dam H, Castellazzi M. Transcription factor ATF2 cooperates with v-Jun to promote growth factor-independent proliferation in vitro and tumor formation in vivo. Mol Cell Biol. 1998;18:7020–7029. doi: 10.1128/mcb.18.12.7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Shuai K. Induction of apoptosis by protein inhibitor of activated Stat1 through c-Jun NH2-terminal kinase activation. J Biol Chem. 2001;276:36624–36631. doi: 10.1074/jbc.M101085200. [DOI] [PubMed] [Google Scholar]
- Hipskind RA, Bilbe G. MAP kinase signaling cascades and gene expression in osteoblasts. Front Biosci. 1998;3:D804–816. doi: 10.2741/a323. [DOI] [PubMed] [Google Scholar]
- Papachristou DJ, Batistatou A, Sykiotis GP, Varakis I, Papavassiliou AG. Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone. 2003;32:364–371. doi: 10.1016/s8756-3282(03)00026-7. [DOI] [PubMed] [Google Scholar]
- Behrens A, Jochum W, Sibilia M, Wagner EF. Oncogenic transformation by ras and fos is mediated by c-Jun N-terminal phosphorylation. Oncogene. 2000;19:2657–2663. doi: 10.1038/sj.onc.1203603. [DOI] [PubMed] [Google Scholar]
- Franklin CC, Kraft AS. Constitutively active MAP kinase kinase (MEK1) stimulates SAP kinase and c-Jun transcriptional activity in U937 human leukemic cells. Oncogene. 1995;11:2365–2374. [PubMed] [Google Scholar]
- Grigoriadis AE, Wang ZQ, Wagner EF. Fos and bone cell development: lessons from a nuclear oncogene. Trends Genet. 1995;11:436–441. doi: 10.1016/s0168-9525(00)89142-8. [DOI] [PubMed] [Google Scholar]
- Franchi A, Calzolari A, Zampi G. Immunohistochemical detection of c-fos and c-jun expression in osseous and cartilaginous tumours of the skeleton. Virchows Arch. 1998;432:515–519. doi: 10.1007/s004280050199. [DOI] [PubMed] [Google Scholar]
- Wu JX, Carpenter PM, Gresens C, Keh R, Niman H, Morris JW, Mercola D. The proto-oncogene c-fos is over-expressed in the majority of human osteosarcomas. Oncogene. 1990;5:989–1000. [PubMed] [Google Scholar]
- Grigoriadis AE, Schellander K, Wang ZQ, Wagner EF. Osteoblasts are target cells for transformation in c-fos transgenic mice. J Cell Biol. 1993;122:685–701. doi: 10.1083/jcb.122.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp B, Lorenz U, Schmidt J, Werenskiold AK. Discordant effects of activator protein-1 transcription factor on gene regulation, invasion, and metastasis in spontaneous, radiation-induced, and fos-induced osteosarcomas. Mol Carcinog. 1998;23:69–75. [PubMed] [Google Scholar]
- Young MR, Nair R, Bucheimer N, Tulsian P, Brown N, Chapp C, Hsu TC, Colburn NH. Transactivation of Fra-1 and consequent activation of AP-1 occur extracellular signal-regulated kinase dependently. Mol Cell Biol. 2002;22:587–598. doi: 10.1128/MCB.22.2.587-598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova O, Kramerov D, Grigorian M, Berezin V, Bock E, Lukanidin E, Tulchinsky E. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol Cell Biol. 1998;18:7095–7105. doi: 10.1128/mcb.18.12.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Crawford HC, Lavrovsky V, Taub D, Watts R, Matrisian LM, Colburn NH. A dominant negative mutant of jun blocking 12-O-tetradecanoylphorbol-13-acetate-induced invasion in mouse keratinocytes. Mol Carcinog. 1997;19:204–212. doi: 10.1002/(sici)1098-2744(199707)19:3<204::aid-mc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]