

Abstract
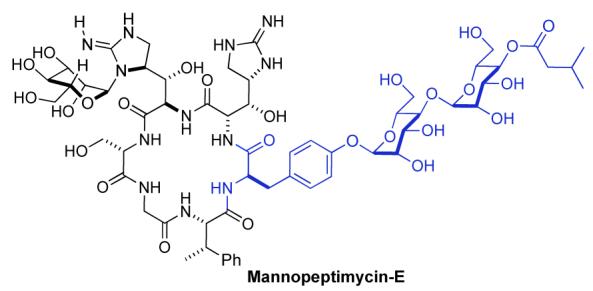
The enantioselective synthesis of the C-4′ acylated 1,4-α,α-manno,manno-disaccharide fragment of mannopeptimycin-E has been achieved in 7 steps from D-tyrosine. The route relies upon diastereoselective palladium-catalyzed glycosylation, diastereoselective reduction and diastereoselective bis-dihydroxylation. The efficiency of the synthesis is demonstrated by the high overall yield (37%) and the preparation of various analogues.
The continuing emergence of bacterial resistance to traditional antibiotics has inspired a never-ending search for new antibiotics.1 The five mannopeptimycins (1a-e) were isolated from the fermentation broths of Streptomyces hygroscopicus LL-AC98 and related mutant strains.2 The key structural features of the mannopeptimycins are a cyclic hexapeptide core with alternating D- and L-amino acids, three of which are rare. Two of the amino acids (β-D-hydroxyenuricididine and D-tyrosine) are glycosylated with mannose sugars. The glycosylated amino acids are an N-glycosylated β-hydroxyenuricididine with an α-mannose, and an O-glycosylated tyrosine with a α-(1,4-linked)-bis-manno-pyranosyl-pyranoside.
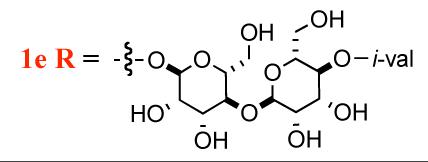
The unique structure and unprecedented biological activity have inspired both biological2,3 and synthetic studies4 from labs at Wyeth pharmaceuticals. Among the mannopeptimycins, mannopeptimycin-E (1e, Scheme 1) was reported as the most active member against methicillin-resistant staphylococci and vancomycin-resistant enterococci (Table 1).5
Scheme 1.
Structure of mannopeptimycin E 1e
Table 1.
Activities of the mannopeptimycins5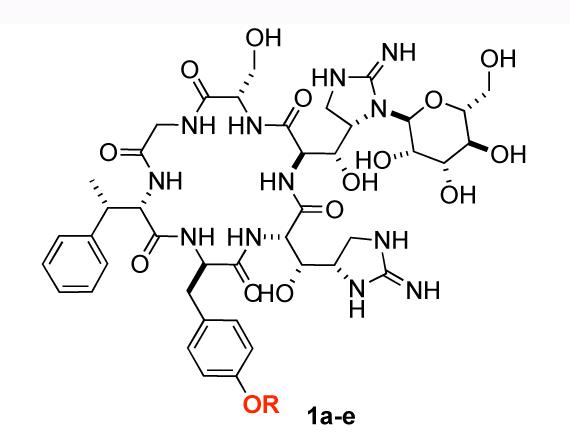
| mannopeptimycin A-E | MIC range (μg/mL) |
|
|---|---|---|
| MRSAa | Enterococus faeciumb |
|
 |
>128 | >128 |
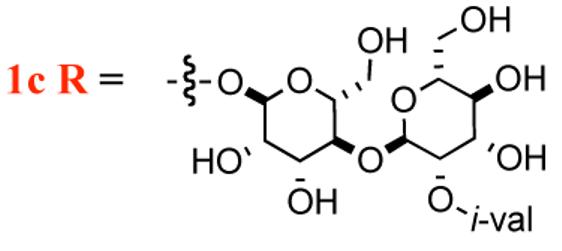
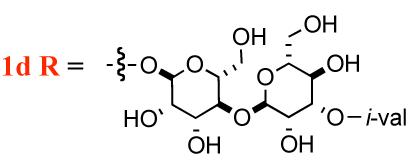
| 1b R= H | 64-128 | 32->128 |
 |
8 | 16-64 |
 |
8 | 8-64 |
 |
4 | 4-32 |
methicillin-resistant S. aureus.
a range of activities vs 4 lines of vancomycin-resistant.
i-val = i-valerate
A particularly interesting aspect of the SAR for the mannopeptimycins is how the specific placement of the isovalerate group on the bis-manno-disaccharide correlates with its antibacterial activity. It has been shown that C-4 isovalerate substitution on the terminal mannose leads to a substantial increase in antibacterial potency. For instance, mannopeptimycins-C and -D, which have C-2 and C-3 isovalerate groups respectively, have reduced activity, whereas mannnopeptimycins-A and -B, which lack isovalerate substitution have even lower activity (Table 1).5
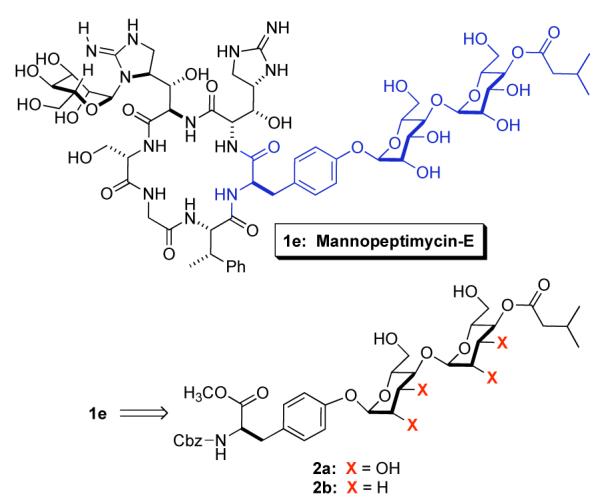
Although the total synthesis of mannopeptimycin has not been reported, Wang et al. have reported a synthesis of cyclic peptides related to mannopeptimycin having a C-4/C-6 acetal as an isovalerate substitute.4a This work also confirmed the importance of the C-4 isovaleryl group for antibiotic activity. The critical role isovalerate substitution has on the antibacterial activity of the mannopeptimycin-E inspired us to pursue a synthesis of an appropriate O-glycosylated D-tyrosine with C-4 isovalerate substitution (e.g. 2a and 2b, Scheme 1).6 In addition to our desire to synthesize and test the mannopeptimycin analogues 2a and 2b, we felt that the synthesis of 2a would serve as part of a model study for our synthesis of the natural product. In addition, the preparation of 3b, a fully protected bis-glycosylated tyrosine (Scheme 2), would be of use for the synthesis of mannopeptimycin E. Herein, we report the successful implementation of our palladium-catalyzed glycosylation reaction7,8 for the de novo installation of both a D,D- and an L,L-bis-mannodisaccharide fragment on a D-tyrosine. The flexibility of the approach is demonstrated by the syntheses of bis-2,3-dideoxy analogues in their D,D- and an L,L-forms.9
Scheme 2.
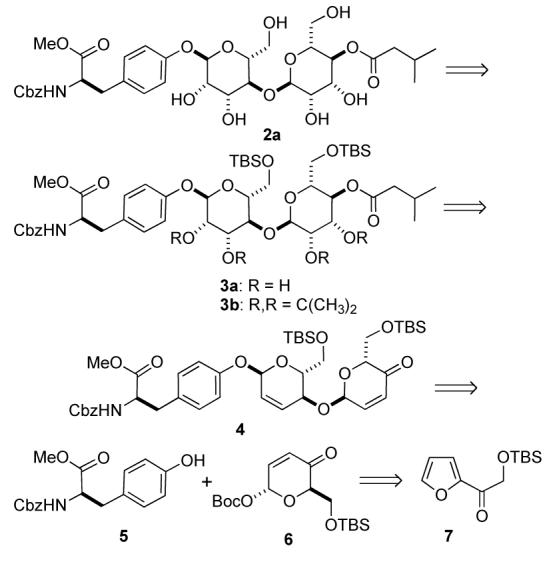
Retrosynthetic analysis of mannopeptimycins
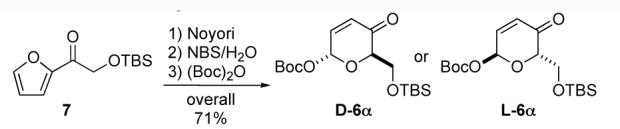
Our retrosynthetic analysis of the disaccharide fragment 2a and its fully protected variant 3b is outlined in Scheme 2. We envisioned that the manno-stereochemistry in both 2a and 3b could be installed by a diastereoselective ketone reduction and a bis-dihydroxylation of a 1,4-linked pyran/pyranone 4. Similarly, we believed that the pyran/pyranone 4 could be assembled using a diastereoselective palladium-catalyzed glycosylation of tyrosine 5.7 Recently, we reported a diastereoselective palladium-catalyzed glycosylation reaction that used alcohols as nucleophiles and pyranones like 6 as glycosyl donors. Thus, sequential application of our Pd(0)-glycosylation/NaBH4-reduction/Pd(0)-glycosylation sequence to tyrosine 5 and pyranone 6 was expected to allow for the rapid preparation of 4. Replacing the above-mentioned bis-dihydroxylation with a bis-diimide reduction might also allow for the preparation of the deoxy-analog 2b. Previously we have shown that pyranone 6 can be prepared in either enantiomeric form. Thus, this procedure was expected to allow the incorporation of either D- or L-sugars.10
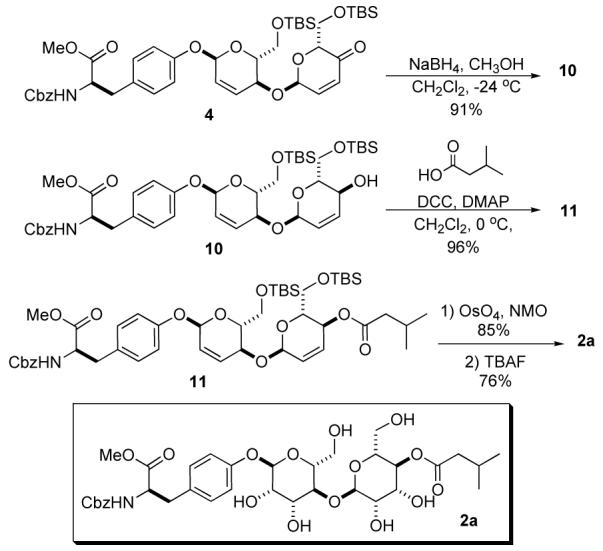
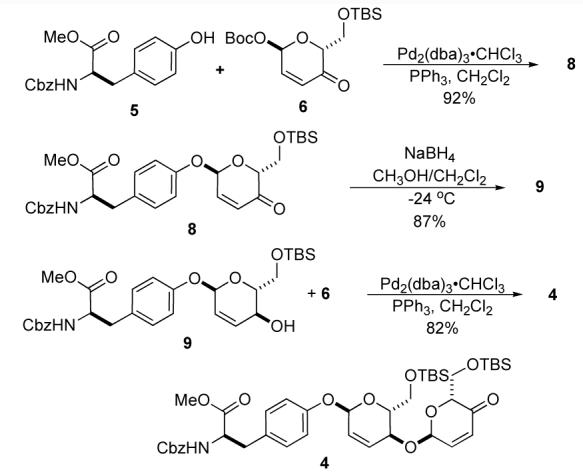
Our synthesis studies began with the protected D-tyrosine 5 and pyranone 6 which, exposed to 1 mol% Pd2(dba)3•CHCl3 and 2.5 mol% PPh3, underwent a diastereoselective glycosylation with complete α-selectivity to afford the pyranone 8 in 92% yield. A diastereoselective 1,2-reduction of the enone 8 , when subjected to NaBH4 in CH2Cl2/MeOH (1:1) at —24 °C, afforded allylic alcohol 9 as a single diastereomer (dr > 20:1). We next investigated the viability of the C-4 alcohol in the Pd-catalyzed glycosylation. Exposing allylic alcohol 9 to a second glycosylation using 1.2 equiv of pyranone 6 and 1 mol% Pd catalyst (1:2.5, Pd2(dba)3•CHCl3/PPh3) afforded the 1,4-linked-α-bis pyranone 4 in good yield (82%) and virtually complete stereocontrol.
The final post-glycosylation transformation of 4 is shown in Scheme 4. Treatment of 1,4-linked pyran/pyranone 4 under the same reduction conditions as before (8 to 9, Scheme 3) gave allylic alcohol 10 in excellent yield (91%) and diastereoselectivity (>20:1). The isovalerate group was installed by treating allylic alcohol 10 with isovaleric acid and DCC/DMAP in CH2Cl2, which provided the C-4 isovalerate disaccharide precursor 11 in excellent yield (96%). The manno-stereochemistry in 3a was diastereoselectively introduced11 upon exposure of 11 to the Upjohn conditions (OsO4/NMO, 85%).8 Removal of both TBS-ethers was accomplished with TBAF (0 °C in THF) affording the α-1,4-linked-bis-manno-disaccharide 2a in good yield (76%).
Scheme 4.
Synthesis of tyrosine-bis-manno-disaccharide
Scheme 3.
De novo synthesis of pyran/pyranone 4
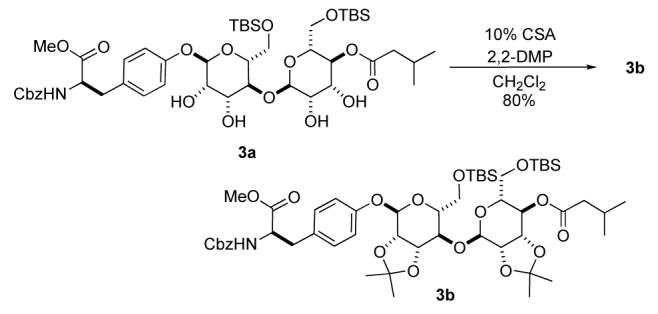
Finally the bis-manno-sugar 3a could also be converted to the fully protected α-1,4-linked-bis-manno-disaccharide 3b without any ester migration (Scheme 5). This was easily accomplished by treating a CH2Cl2solution of tetraol 3a with 2,2-dimethoxypropane and 10 mol% CSA; conditions which provided the bis-acetonide 3b in good yield (80%).
Scheme 5.
Synthesis of fully protected bis-manno-disaccharide
Replacing pyranone 6 with its L-enantiomer (ent)-6, resulted in an equally efficient synthesis of the L,L-bis-manno-sugar diastereomer of 2a, 14 (Scheme 6). Thus in three analogous steps, D-tyrosine was converted into pyran/pyranone 12 (69% overall yield). The L,L-1,4-linked pyran/pyranone 12 was stereoselectively reduced and acylated to form 13 in good overall yield (86%). Once again, two diastereoselective dihydroxylations occurred upon exposure of 13 to the Upjohn conditions. This bis-dihydroxylation occurred with near perfect stereocontrol, as with the diastereomeric series (c.f. Scheme 4).11 The tetraol product was converted to the unprotected bis-sugar 14 via a TBS group deprotection (TBAF, 78%) or to the fully protected diastereomer 15 by means of an acetonide protection (10 mol% CSA/2,2-DMP, 81%).
Scheme 6.
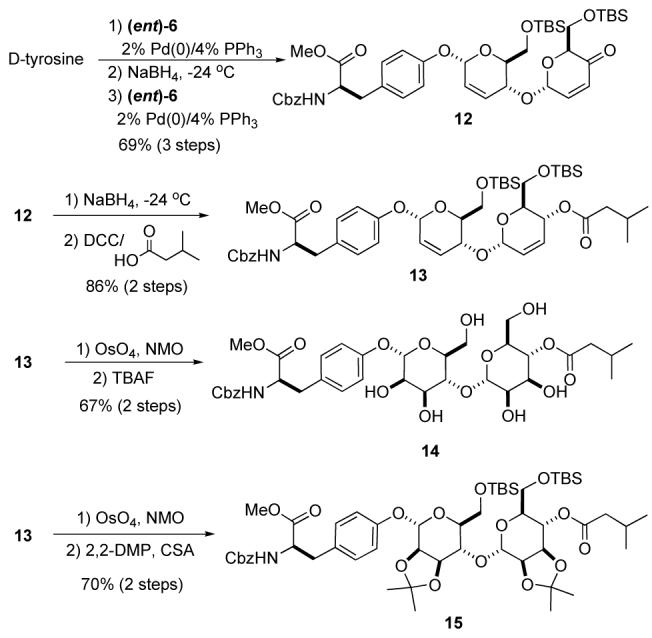
Synthesis of L,L-disaccharide analogs
Having synthesized the key disaccharide fragment of mannopeptimycin-E (2a and 3b) along with its L,L-diastereomers (14 and 15), we turned our attention to the preparation of deoxy analogues (Scheme 7). The simplest 2,3—deoxy analogue 16 was obtained by an exhaustive diimide reduction.12 Both double bonds of 11 were reduced using excess of the diimide precursor in CH2Cl2 affording the 2,3-deoxy-bis-pyranoside 16 in nearly quantitative yield (95%).13 Under identical conditions the diasteromeric L,L-1,4-linked bis-pyran 13 reduced to give an excellent yield of the bis-dideoxy analog 17 (97%).13
Scheme 7.
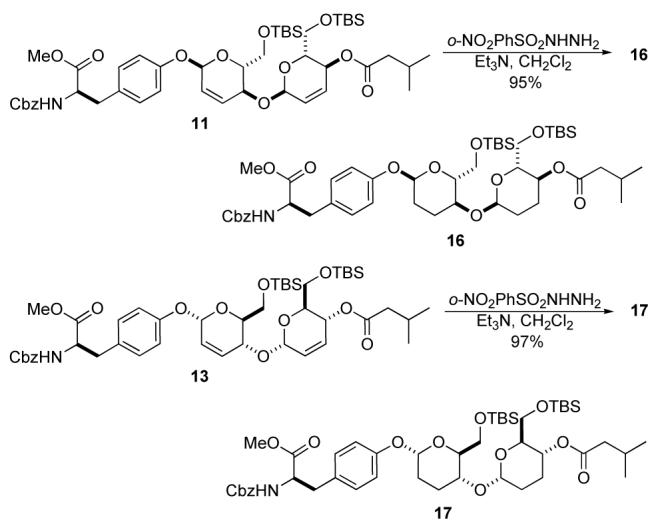
Synthesis of bis-2,3-dideoxy-disaccharide analogues
In conclusion, an enantioselective synthesis of the manno-disaccharide fragments of mannopeptimycin-E has been achieved in 7 steps and 37% overall yield from D-tyrosine via an iterative palladium-glycosylation strategy. Key to the success of this approach was the ease with which the C-4 isovalerate group was introduced, and the high diastereoselectivity of the palladium-catalyzed glycosylation and bis-dihydroxylation reactions. The use of this methodology for the synthesis of mannopeptimycin-E as well as various analogues is ongoing.
Supplementary Material
Acknowledgment
We thank the NIH (GM63150) and NSF (CHE-0415469) for their generous support of our research program. Funding for a 600 MHz NMR by the NSF-EPSCoR (#0314742) is also gratefully acknowledged.
Footnotes
Supporting Information Available: Complete experimental procedures and spectral data for all new compounds can be found in the Supporting Information (59 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Walsh CT. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- (2).He H, Williamson RT, Shen B, Grazaini EI, Yang HY, Sakya SM, Petersen PJ, Carter GT. J. Am. Chem. Soc. 2002;124:9729–9736. doi: 10.1021/ja020257s. [DOI] [PubMed] [Google Scholar]
- (3) (a).Petersen PJ, Wang TZ, Dushin RG, Bradford PA. Antimicrob. Agents Chemother. 2004;48:739–746. doi: 10.1128/AAC.48.3.739-746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sum PE, How D, Torres N, Petersen PJ, Lenoy EB, Weiss WJ, Mansour TS. Bioorg. Med. Chem. Lett. 2003;13:1151–1155. doi: 10.1016/s0960-894x(03)00045-3. [DOI] [PubMed] [Google Scholar]; (c) He H, Shen B, Petersen PJ, Weiss WJ, Yang HY, Wang T-Z, Dushin RG, Koehn FE, Carter GT. Bioorg. Med. Chem. Lett. 2004;14:279–282. doi: 10.1016/j.bmcl.2003.09.071. [DOI] [PubMed] [Google Scholar]
- (4) (a).Wang T-Z, Wheless KL, Sutherland AG, Dushin RG. Heterocycles. 2004;62:131–135. [Google Scholar]; (b) Dushin RG, Wang TZ, Sum PE, He H, Sutherland AG, Ashcroft JS, Graziani EI, Koehn FE, Bradford PA, Petersen PJ, Wheless KL, How D, Torres N, Lenoy EB, Weiss WJ, Lang SA, Projan SJ, Shlaes DM, Mansour TS. J. Med. Chem. 2004;47:3487–3490. doi: 10.1021/jm049765y. [DOI] [PubMed] [Google Scholar]
- (5).Singh MP, Petersen PJ, Weiss WJ, Janso JE, Luckman SW, Lenoy EB, Bradford PA, Testa RT, Greenstein M. Antimicrob. Agents Chemother. 2003;47:62–69. doi: 10.1128/AAC.47.1.62-69.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).We were mindful of Kahne’s discovery of simple disaccharide fragments of vancomycin with significant activity toward vancomycin resistance bacteria, see: Sun B, Chen Z, Eggert US, Shaw SJ, LaTour JV, Kahne D. J. Am. Chem. Soc. 2001;123:12722–12723. doi: 10.1021/ja0166693.
- (7).Babu RS, O’Doherty GA. J. Am. Chem. Soc. 2003;125:12406–12407. doi: 10.1021/ja037097k. [DOI] [PubMed] [Google Scholar]
- (8) (a).Babu RS, Zhou M, O’Doherty GA. J. Am. Chem. Soc. 2004;126:3428–3429. doi: 10.1021/ja039400n. [DOI] [PubMed] [Google Scholar]; (b) Babu RS, O’Doherty GA. J.Carbohydr. Chem. 2005;24:169–177. [Google Scholar]; (c) VanRheenen V, Cha DY, Hartley WM. Organic Syntheses. VI. Wiley & Sons; New York: 1988. p. 342. [Google Scholar]
- (9).Presumably, the L,L-diastereomer and the bis-2,3-dideoxy analogues of mannopeptimycin-E would have improved bioavailability.
- (10).Pyranones like 6 can be prepared in three steps from achiral acylfurans like 7 in either enantiomeric form (D/L). The pyranone asymmetry is derived from a Noyori reduction, see: ref. 7 and Li M, Scott JG, O’Doherty GA. Tetrahedron Lett. 2004;45:1005–1009.
- (11).The relative stereochemistry of 3a and 14a was determined by analysis of various coupling constants from their 1H NMR spectra, see supporting information.
- (12).We have found that o-nitrophenylsulfonylhydrazide/triethylamine to be an excellent diimide precursor, ideal for reducing pyrans of this type, see: ref. 8 and Haukaas MH, O’Doherty GA. Org. Lett. 2002;4:1771–1774. doi: 10.1021/ol025844x.
- (13).To achieve complete conversion, occasionally the crude reaction mixture may need to be re-subjected to the diimide conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.