

Abstract
The regulation of TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) in cancer chemotherapy is not fully understood. Here, we show that the HDACIs (inhibitors of histone deacetylases) induce TRAIL in human breast cancer cells. Induction of TRAIL by the HDACI MS275 can be enhanced by adriamycin. Using different reporter constructs in conjunction with transcription activity assays and chromatin immunoprecipitation assays, we provide evidence that the transcription factor Sp1 is responsible for TRAIL induction by MS275 alone or in combination with adriamycin. Further, we show that the combined treatment of breast cancer cells with MS275 and adriamycin significantly increases apoptotic cell death via the activation of both death receptor and mitochondrial apoptotic pathways. Down-regulation of TRAIL by small interfering RNA (siRNA) silencing decreased MS275-mediated adriamycin-induced caspase activation and apoptosis, thus conferring adriamycin resistance. More importantly, breast cancer cell T47D in which Sp1 was knocked down or Sp1 knockout mouse embryonic stem cells were resistant to the combined treatments. Taken together, our results indicate that induction of TRAIL by the combined treatments with MS275 and adriamycin is mediated by Sp1 and suggest that transcription factor Sp1 is an important target for the development of novel anticancer agents.
Keywords: TRAIL, apoptosis, Sp1, chemosensitivity
Introduction
TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) is a member of the tumor necrosis factor family (1, 2). TRAIL selectively induces apoptosis of transformed or tumor cells but not normal cells, making it a promising new agent for cancer therapy (1, 2). There are four membrane-bound receptors for TRAIL, including DR4 (3), DR5 (4-8), TRAIL-R3 (6, 9-11), TRAIL-R4 (12, 13). DR4 and DR5 both contain a conserved death domain (DD) motif and are proapoptotic receptors (14), whereas TRAIL-R3 lacks an intracellular domain and TRAIL-R4 has a truncated DD. Thus, both TRAIL-R3 and TRAIL-R4 act as decoy receptors to antagonize TRAIL-induced apoptosis by competing for ligand binding (14, 15). Binding of TRAIL to DR4 or DR5 triggers formation of the death-inducing signaling complex (DISC) by recruiting FADD and caspase-8 or -10, resulting in the activation of caspase-8 or -10, which in turn activates caspase-3, -6 and -7 to cleave death substrates and cause cell death. Activated caspase-8 can cleave the proapoptotic Bcl2 family member Bid to generate truncated Bid. Truncated Bid translocates to the mitochondria to cause cytochrome c release which amplifies the apoptotic signal from the TRAIL pathway.
It has been shown that DR5 could be transcriptionally induced by some anticancer drugs (4, 16), thus sensitizing cancer cells to TRAIL (16, 17). However, the regulation of TRAIL ligand expression is much less understood. The TRAIL promoter contains a number of transcription regulatory elements including ISRE, NF-κB, and Sp1 (18, 19). It has been shown that IFNs directly induce TRAIL in both human leukemia Jurkat and colon cancer cell line HT29 (18, 19). IFN-γ also acts as a mediator to induce TRAIL in response to retinoid treatment (20). The induction of TRAIL via an IFN-stimulated response element results in apoptosis (20, 21). In addition, the TAX oncoprotein encoded by human T-cell leukemia virus induces TRAIL through the NF-κB-dependent pathway (22). TRAIL is also induced by T cell receptor (TCR) mimetics in human T cells and such induction involves a c-Rel binding site in the proximal TRAIL promoter (23). However, the regulation of TRAIL by cancer chemotherapy is not fully understood.
The histone-deacetylase inhibitors (HDACIs) are novel anticancer agents that can activate transcription of target genes via histone acetylation (24). By activation of gene expression, HDACIs may induce cell differentiation, growth arrest and apoptosis. The ability of HDACIs to induce apoptosis is attributed to the activation of both extrinsic and intrinsic apoptotic pathways (25). Several HDACIs, including valproic acid (VPA), suberoylanilide hydroxamic acid (SAHA) and the benzamide derivative MS275, exhibit anti-tumor activity with little toxicity to normal cells both in vitro and in vivo (24). Recent studies have shown that HDACIs induce leukemia-selective apoptosis through the TRAIL apoptotic pathway (26, 27) and sensitize leukemia cells to anticancer agents (28). In addition, several studies also indicated that HDACIs induce DR5 and subsequently sensitize cancer cells to TRAIL-mediated cell death (29). How activation of TRAIL pathway contributes to HDACI-induced apoptosis in solid tumors is of critical importance and needs to be determined.
In this paper, we show that the HDACI MS275 induces TRAIL via an Sp1-dependent pathway. MS275-mediated TRAIL induction was enhanced by adriamycin at both the RNA and protein levels. The induction of TRAIL by MS275 enhanced adriamycin-induced apoptosis in human breast cancer cells. Down-regulation of TRAIL by siRNA decreased adriamycin-mediated cell death induced by MS275. Importantly, T47D cells in which Sp1 was knocked down or Sp1 null ES (embryonic stem) cells were more resistant to adriamycin, MS275 or combination treatment as compared to their counterparts with intact Sp1. Thus, our data indicate that Sp1-mediated TRAIL induction plays a critical role in chemosensitivity and suggest that Sp1 is a therapeutic target for the development of novel anticancer therapeutics.
Materials and Methods
Reagents
MS275 was purchased from Alexis Biochemicals (San Diego, CA). SAHA was purchased from Cayman (Ann Arbor, MI). Adriamycin was obtained from the Oncology Outpatient Pharmacy at the Karmanos Cancer Institute. Monoclonal anti-human TRAIL, DR4, and polyclonal DR5 antibodies were purchased from Imgenex (San Diego, CA). Rabbit anti-caspase-9, -8, -3, and anti-poly (ADP-ribose) polymerase (PARP) polyclonal antibodies were purchased from Cell Signaling Technology (Beverly, MA). Monoclonal p21 antibody was purchased from Calbiochem (San Diego, CA). Anti-actin antibody was purchased from Sigma (St. Louis, MO). Sp1 antibody was purchased from Upstate Biotechnology (Lake Placid, NY).
Cell lines, culture conditions and treatment
The human breast cancer MCF7 cells were obtained from Karmanos Cancer Institute and maintained in DMEM/F12. The human breast cancer MDA231 and T47D cells were obtained from ATCC (Manassas, VA) and maintained in DMEM. Cells were supplemented with either 10% fetal bovine serum (FBS) for MDA231 and T47D or 5% FBS for MCF7 and antibiotics at 37 °C in a humidified atmosphere consisting of 5% CO2 and 95% air. Sp1 knockout mouse embryonic stem cells (Sp1-/-) and their normal control ES cells (Sp1+/+) were described previously (30) and maintained in GMEM medium (Sigma) supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 1× nonessential amino acids, 10% FBS, beta-mercaptoethanol and leukemia inhibitory factor (Chemicon, Temecula, CA). ES cells were grown in bovine gelatin coated dishes.
Isolation of RNA and Northern blot analysis
The procedures for preparation of total RNA and Northern blot analysis were described previously (31).
siRNA transfection for knockdown of TRAIL and Sp1
On-TARGETplus SMARTpool siRNAs for TRAIL, Sp1, and corresponding control siRNA were purchased from Dharmacon Research (Lafayette, CO). The transfection was performed as suggested by Dharmacon with slight modifications, as described previously (32). Briefly, T47D cells were plated at 6 × 105 cells per well in 6-well plates. The next day, cells were transfected with TRAIL, Sp1 or non-target control oligonucleotides using Oligofectamine (Invitrogen). After 3 days, transfected cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml) or in combination for 48 hr and then harvested for assessing the expression of TRAIL, Sp1 and activation of the caspase cascade by Western blot analysis. To determine chemosensitivity, cells with or without transfections were placed at 8,000 cells per well in 96-well plates and then treated with MS275, adriamycin or in combination for 48 hr, and cell viability was determined by MTT assays.
MTT assays
MTT assays were described previously (33).
Western blot analysis
The procedures for preparation of whole cell protein lysates and Western blot analysis were described previously (33)
Assay of caspase-3 activity
The enzymatic activity of caspase-3 was assayed using the caspase-3 colorimetric assay kit (R & D Systems, Minneapolis, MN) according to the manufacturer's protocol. Cells were left untreated or treated with MS275, adriamycin, or in combination for 48 hr and then lysed in lysis buffer for 10 min on ice. The lysed cells were centrifuged at 14,000 rpm for 5 min, and 150 μg protein was incubated with 50 μl of reaction buffer and 5 μl of caspase-3 substrate at 37°C for 2 hr, and the absorbance was measured at a wavelength of 405 nm on a plate reader.
Construction of reporter vectors
TRAIL reporter constructs pGL3-TRAIL2, pGL3-TRAIL5 and pGL3-TRAIL6 were described previously (33) and shown in Fig. 3A. TRAIL5mu in which the second Sp1 binding site was mutated was amplified from pGL3-TRAIL2 using GC-Rich PCR system (Roche Molecular Biochemicals, Indianapolis, IN) and the following primers: 5′-CCGCTCGAGAGGAAATTTTCTTTACAGTT-3′ and 5′-CCCAAGCTTGATCCTGTCAGAGTCTGACTGCTG-3′. The PCR conditions were as follows: 95°C/3 min; 30 cycles at 95°C/30 sec, 45°C /30 sec and 68°C/2 min, followed by 68°C/7 min for the final extension. The amplified fragment was isolated from 1% agarose gel, digested with XhoI and Hind III, and subcloned into pGL3-Basic (Promega). The insert was verified by DNA sequencing.
Figure 3.

Effect of MS275, adriamycin, or in combination on the TRAIL promoter activity. A, schematic depiction of luciferase reporter constructs. The translation start site is indicated by an arrow and designated at + 1 and both Sp1 binding sites were indicated. B, activation of the TRAIL promoter by MS275, adriamycin, or in combination. T47D cells were transfected with pGL3-TRAIL2, pGL3-TRAIL5, pGL3-TRAIL5mu, pGL3-TRAIL6, and pGL3-Basic. pRLSV40 was added in transfections for normalization. The next day, cells were left untreated or treated with 5 μM MS275, 0.1 μg/ml adriamycin, or in combination. Luciferase activity was determined 24 hr later. Data are shown as relative firefly luciferase activities, normalized to Renilla luciferase activities. C, induction of TRAIL protein. T47D cells were left untreated or treated with 5 μM MS275, 0.1 μg/ml adriamycin, or in combination for 24 hr. Induction of TRAIL, DR4, and DR5 proteins were determined by Western blot analysis. Actin was used as a loading control.
Luciferase reporter assays
Transfections for luciferase assays were carried out as described previously (33). Briefly, T47D cells were plated at 8 × 105 per well in 6-well plates. The next day, the cells were transfected with 5 μg of reporter constructs and 5 ng of pRLSV40 (Promega) using Lipofectamine 2000 reagent (Invitrogen). After 24 hr, transfected cells were treated with or without 5 μM MS275, 0.1 μg/ml adriamycin or in combination for 24 hr. Firefly luciferase activities were assayed using the dual-luciferase reporter assay system (Promega) in a Turner TD20/20 luminometer and normalized to Renilla luciferase activity.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the ChIP Assay Kit (Upstate Biotechnology) as described previously (33). The PCR primers used in ChIP were 5′-AATGGGCTTGAGGTGAGTGCAGAT-3′ and 5′-ATGAGTTGTTTTTCTGGGTTCTGT-3′.
ELISA for Sp1 transcription activity
T47D cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml), or in combination for 24 hr and nuclear protein was extracted using a Nuclear Extraction kit (Panomics, Fremont, CA). Nuclear protein was then quantified using the Bio-Rad Protein Assay kit (Bio-Rad). Total 15 μg of nuclear protein from each treatment was analyzed for Sp1 activity using the Transcription Factor ELISA kit (Panomics). Sp1 antibody was used as primary antibody and anti-rabbit IgG HRP was used as secondary antibody. The absorbance was measured at a wavelength of 450 nm on the spectrophotometer.
Statistical analysis
Statistical analysis was performed using Student's t test. The data were presented as the mean ± SD, and p≤ 0.05 was considered significant.
Results
HDACIs induce TRAIL expression in human breast cancer cells
The regulation of TRAIL by cancer chemotherapy in solid tumor cell lines is not fully understood. We treated three breast cancer cell lines; MDA231, T47D and MCF7, with various doses of MS275 for 24 hr, and induction of TRAIL was determined by Western blot analysis. Fig. 1 shows that TRAIL protein was induced in all three lines at different doses of MS275 and that such treatments appear to have no effect on the levels of DR4 and DR5 protein except for an increase in DR5 protein in MCF7 cells treated with 5 μM MS275. Consistent with previous studies (26, 27), p21 was induced by MS275, which served as a positive control. TRAIL mRNA was also increased by MS275 treatment (Fig. 1B). Further, we found that SAHA, another HDACI, can also induce TRAIL expression (Fig. 1A). These data suggest that TRAIL induction by HDACIs is a common event in breast cancer cells.
Figure 1.
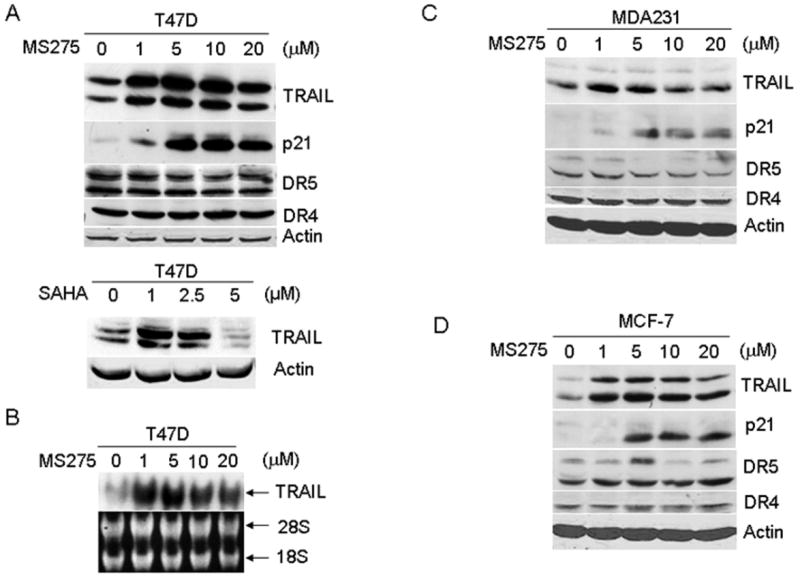
Induction of TRAIL by HDACIs. A, C and D, the effects of MS275 or SAHA on TRAIL, DR4, DR5, and p21 proteins. T47D (A), MDA231 (C), and MCF7 (D) cells were left untreated or treated with various doses of MS275 or SAHA for 24 hr, and total protein was then extracted for assaying the expression of TRAIL, DR4, DR5 and p21 by Western blot analysis. β-actin was used as a loading control. B, induction of TRAIL mRNA by MS275 in T47D cells. Cells were treated as in (A), and total RNA was extracted to examine for TRAIL mRNA expression by Northern blot analysis. rRNA was used as loading controls.
MS275 sensitizes human breast cancer cells to adriamycin
We previously showed that induction of TRAIL by TNFα and 5-aza-2′-deoxycytidine plays a critical role in sensitizing breast cancer cells to chemotherapy (33, 34). To determine the role of TRAIL induction in breast cancer cell death induced by adriamycin, we first tested the effect of MS275 treatment alone on the growth of breast cancer cells. As shown in Fig. 2A, MS275 inhibited the growth of all three cell lines in a dose-dependent manner. We next asked whether the effect of MS275 on growth inhibition could be enhanced by anticancer agents. Fig. 2B shows that the growth inhibition was ∼ 86% in cells treated with MS275 in combination with adriamycin, as compared to ∼50% and ∼ 33% in cells treated with MS275 and adriamycin, respectively. These data suggest that MS275 could sensitize T47D cells to adriamycin.
Figure 2.

Effect of MS275, adriamycin or in combination on cell death. A, role of MS275 in growth inhibition. The human breast cancer cell lines T47D, MDA231, and MCF7 were left untreated or treated with various doses of MS275 for 48 hr. Cell viability was determined by MTT assays. Data are representative of three independent experiments. B, role of MS275 in T47D cell death induced by adriamycin. T47D cells were treated with 5 μM MS275, 0.1 μg/ml adriamycin or in combination for 48 hr. Cell viability was determined by MTT assays. Data are representative of three independent experiments. Asterisks indicate statistical significance (**p<0.01). C, activation of the apoptotic pathways by MS275, adriamycin or in combination. T47D cells were left untreated or treated with 5 μM MS275, 0.1 μg/ml adriamycin or in combination, and total protein was extracted at 48 hr. Cleavage of caspase-9, -8, -3 and PARP was determined by Western blot analysis. β-actin was used as a loading control. D, role of the treatments with MS275, adriamycin or in combination in caspase 3 activity. T47D cells were treated as in (C) and then lysed in lysis buffer. Cell protein was extracted and used for measuring caspase-3 activity. Data are representative of three independent experiments. Asterisks indicate statistical significance (**p<0.01).
To determine the underlying mechanisms by which MS275 sensitizes T47D cells to adriamycin, we tested the activation of the apoptotic pathways since HDACIs can kill cancer cells by apoptosis (25). Fig. 2C shows that adriamycin or MS275 treatment alone causes modest cleavage of caspase-9 and PARP. In contrast, the combination of 5 μM MS275 with 0.1 μg/ml adriamycin significantly enhanced cleavage of caspase-9, -3 and PARP (Fig. 2C). Importantly, the combined treatments resulted in a significant increase in cleavage of caspase-8, which was not obvious in cells treated with either agent alone (Fig. 2C). In addition, the combined treatment also enhanced caspase-3 activity (Fig. 2D) relative to the treatments with either agent alone. Collectively, these data suggest that the enhanced cell killing by the combined treatments is attributable to the augmented induction of apoptosis.
Sp1 is responsible for TRAIL induction by MS275 alone or in combination with adriamycin
To define the mechanisms of TRAIL regulation by MS275, we transfected T47D cells with either the TRAIL luciferase reporter construct pGL3-TRAIL2 or empty vector pGL3-basic, followed by the treatment with MS275 (5 μM). Cells were harvested after 24 hr, and luciferase activity was assayed using the dual-luciferase reporter assay system. Fig. 3B shows that transfections of pGL3-TRAIL2 containing a 504 bp fragment upstream of translational start site results in an approximately 2.5-fold increase in luciferase activity in response to MS275 treatment, as compared to untreated cells. To understand TRAIL regulation by MS275 in detail, we tested the effects of MS275 on luciferase activity using several deletion constructs (Fig. 3A). As shown in Fig. 3B, pGL3-TRAIL5 containing two Sp1 sites was still activated by MS275 whereas pGL3-TRAIL6 with one Sp1 binding site was inert. This suggests that the second Sp1 is critical for MS275-mediated TRAIL induction. To further confirm this, we mutated the second Sp1 site in pGL3-TRAIL5mu and found that pGL3-TRAILmu is no longer responsive to MS275 treatment, suggesting that the second Sp1 binding site is responsible for transactivation of the TRAIL promoter by MS275.
Since MS275 could sensitize T47D cells to adriamycin (Fig. 2), we asked if the combination of MS275 with adriamycin has any effects on the TRAIL promoter. To answer this question, T47D cells transfected with TRAIL reporter constructs were treated with either agent alone or in combination and then assayed for the TRAIL promoter activity. As shown in Fig. 3B, although adriamycin alone had no effects on the TRAIL promoter activity, the combination of MS275 with adriamycin resulted in a more pronounced increase in the activation of the TRAIL promoter as compared to cells treated with MS275 alone. Similar to the results obtained with MS275, loss of the second Sp1 binding site abolished the activation of the TRAIL promoter by the combined treatments (Fig. 3B). Importantly, we showed that the combined treatments increase the level of TRAIL protein, as compared to cells treated with either agent alone (Fig. 3C). Interestingly, in spite of the fact that adriamycin alone had no effect on the activation of the TRAIL promoter, adriamycin could augment MS275-mediated activation of the TRAIL promoter, suggesting that this adriamycin-mediated effect may be indirect. Collectively, these results indicate that the second Sp1 site is critical for the activation of the TRAIL promoter by MS275 alone or in combination with adriamycin.
To determine whether Sp1 binds directly to the TRAIL promoter in response to MS275 or the combined treatment, we performed ChIP analysis of the TRAIL promoter with anti-Sp1 antibody. T47D cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml) or in combination and then harvested 24 hr later for the ChIP experiments. Fig. 4A and B show that PCR amplification of the immunoprecipitated chromatin with Sp1 antibody results in single bands of a size expected for the TRAIL promoter. Importantly, there were higher levels of amplified DNA in MS275 treated cells than observed in untreated cells. Moreover, the combined treatment resulted in more robust DNA amplification than that of cells treated with MS275 alone. In addition, although adriamycin did not activate TRAIL promoter constructs (Fig. 3B), we observed a slight increase in DNA amplification in adriamycin treated cells as compared to untreated cells. To further investigate the activation of Sp1 transcription, we extracted nuclear proteins from cells treated with MS275, adriamycin or in combination and used ELISA kit to assay the Sp1 transcription activity. As shown in Fig. 4C, either MS275 or adriamycin treatment resulted in a modest increase in Sp1 transcription activity as compared to untreated cells. In contrast, the combination of MS275 and adriamycin resulted in ∼ 4-fold increase in Sp1 transcription activity. Taken together, these results suggest that the induction of TRAIL by MS275 or in combination with adriamycin is mediated by the transcription factor Sp1.
Figure 4.
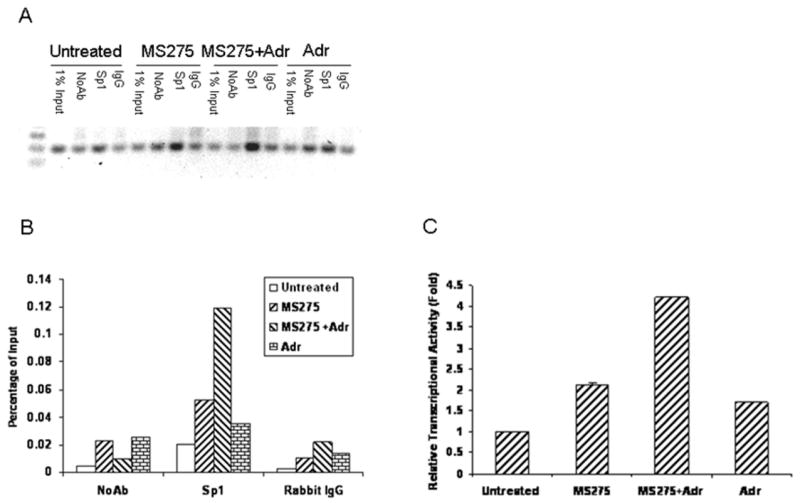
Role of Sp1 in TRAIL induction by MS275, adriamycin, or in combination. A and B, in vivo binding of Sp1 to the TRAIL promoter by ChIP assays. T47D cells were treated with MS275, adriamycin or in combination for 24 hr. ChIP was performed using the ChIP Assay Kit as described in “Materials and Methods”. Immunoprecipitations were performed using rabbit polyclonal antibodies against Sp1. Immunoprecipitations with rabbit IgG used as negative controls. The amplified DNA fragments for Sp1 sites were shown in (A). Quantification of amplified DNA was shown in (B). The results are presented as percentages of DNA immunoprecipitated relative to the input. C, ELISA assay for Sp1 transcription activity. T47D cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml), in combination for 24 hr and nuclear protein was extracted using a Nuclear Extraction kit. Total 15 μg of nuclear protein from each treatment was analyzed for Sp1 activity using the Transcription Factor ELISA kit. Absorbance was obtained using the spectrophotometer at 450 nm. Relative transcriptional activity in untreated cells was arbitrarily given as 1.
Induction of TRAIL by MS275 is required for sensitization of T47D cells to adriamycin-induced apoptosis
We have shown that MS275 induces TRAIL and that the combination of MS275 and adriamycin enhances cell death relative to either agent alone. We asked whether TRAIL induction by MS275 is required for this enhanced killing effect. To this end, we transfected T47D cells with either control siRNA or siRNA against TRAIL, and then tested the effect of siRNA-mediated TRAIL down-regulation on cell death. As shown in Fig. 5A, the basal levels of TRAIL protein in cells transfected with TRAIL siRNA were decreased as compared to cells tranfected with control siRNA. Further, the induction of TRAIL by MS275 was also decreased in cells transfected with TRAIL siRNA as compared with cells transfected with control siRNA. Importantly, we found that cells transfected with TRAIL siRNA were more resistant to MS275, adriamycin, or in combination, as compared with cells transfected with control siRNA (Fig. 5C), suggesting that TRAIL is important for cell death induced by such treatments. Because TRAIL is a potent apoptosis inducer, we investigated the effects of down-regulation of TRAIL on MS275-mediated adriamycin-induced apoptosis. Cells transfected with TRAIL or control siRNA were treated with either MS275 (5 μM), adriamycin (0.1 μg/ml) or in combination for 48 hr, and the activation of the apoptotic pathway was then examined. As shown in Fig. 5B, cleavage of caspase-9, -8, -3 and PARP was significant in cells treated with both MS275 and adriamycin, as compared to untreated or cells treated with MS275 alone, while such changes were minimal in adriamycin treated cells. In contrast, cleavage of caspase-9, -8, -3 and PARP was decreased in cells transfected with TRAIL siRNA following the combined treatment (Fig. 5B). Additionally, we found that such treatments increased caspase-3 activity, which was abolished in cells transfected with TRAIL siRNA (data not shown), indicating that down-regulation of TRAIL impairs the activation of the caspase cascade induced by the combined treatment, thereby enhancing cell survival. Collectively, these results suggest that TRAIL plays an important role in MS275-mediated adriamycin-induced apoptosis.
Figure 5.
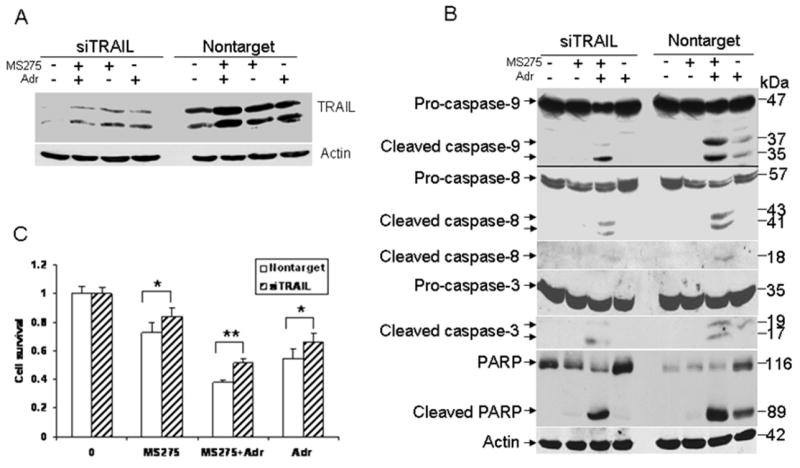
Effect of TRAIL down-regulation by siRNA on cell death induced by MS275, adriamycin or in combination. A, down-regulation of TRAIL by siRNA silencing. T47D cells were plated at 6 ×105 cells per well in 6-well plates. Cells were transfected with TRAIL or control siRNA. After 3 days, cells were left untreated or treated with MS275 (5 μM), adriamycin 0.1 μg/ml) or in combination for 48 hr, and induction of TRAIL was determined by Western blot analysis. B, effect of down-regulation of TRAIL by siRNA on the caspase cascade. T47D cells were transfected with TRAIL or control siRNA and treated with MS275 (5 μM) with and without adriamycin (0.1 μg/ml) for 48 hr as described in (A). Total protein was extracted for assaying cleavage of caspase-9, -8, -3 and PARP by Western blot analysis. β-actin was included as a loading control. C, effect of silencing TRAIL expression on cell survival. T47D cells were transfected with TRAIL or control siRNA as described in (B). After 3 days, cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml), or in combination for 48 hr. Cell viability was determined by MTT assays. Cell survival data are expressed as percentage of untreated cells. Data are representative of three independent experiments. Asterisks indicate statistical significance (**p<0.01; *p<0.05).
The role of Sp1 in chemosensitivity
We have shown that TRAIL expression is regulated by Sp1. Since TRAIL induction plays an important role in apoptosis (26, 27), we asked whether Sp1 could substitute TRAIL to induce apoptosis by MS275 in the presence or absence of adriamycin. To answer this question, we transfected T47D cells with Sp1 or control siRNA and then treated transfected cells with MS275 (5 μM), adriamycin (0.1 μg/ml) or in combination for 48 hr, and the growth inhibition was then examined. As shown in Fig. 6A, the levels of Sp1 in cells transfected with Sp1 siRNA were significantly decreased as compared to cells transfected with control siRNA. Interestingly, we did not detect the changes in the levels of Sp1 protein in response to the treatments (Fig. 6A), suggesting that increased Sp1 transcription activity (Fig. 4) may not be due to an increase in the total level of Sp1 protein. However, we found that induction of TRAIL by MS275 was abolished in Sp1 siRNA transfected cells as compared to control siRNA transfected cells. Furthermore, the induction of TRAIL by the combination was also decreased when Sp1 was down regulated (Fig. 6A). These data indicate that induction of TRAIL by MS275 or in the combination with adriamycin is dependent on the presence of Sp1.
Figure 6.

Role of Sp1 in cell death induced by MS275, adriamycin or in combination. A, knockdown of Sp1 by siRNA silencing. T47D cells were plated at 6 ×105 cells per well in 6-well plates. The next day, cells were transfected with Sp1 or control siRNA. Cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml) or in combination for 48 hr, and induction of TRAIL and Sp1 was determined by Western blot analysis. B, effect of down-regulation of Sp1 by siRNA on the caspase cascade. T47D cells were transfected with Sp1 or control siRNA and treated with MS275 (5 μM) in the presence or absence of adriamycin (0.1 μg/ml) for 48 hr. Total protein was extracted for assaying cleavage of caspase-9, -8, -3 and PARP by Western blot analysis. β-actin was included as a loading control. C, effect of silencing Sp1 expression on cell survival. T47D cells were transfected with Sp1 or control siRNA. Cells were left untreated or treated with MS275 (5 μM), adriamycin (0.1 μg/ml), or in combination for 48 hr and cell viability was determined by MTT assays. Cell survival data are expressed as percentage of untreated cells. Data are representative of three independent experiments. Asterisks indicate statistical significance (**p<0.01; *p<0.05). D, effect of Sp1 depletion on chemosensitivity. In upper panel, Sp1+/+ and Sp1-/- ES cells were treated with both MS275 (5 μM) and adriamycin (0.1 μg/ml) for 24 hr. Total protein was extracted and then assayed for the levels of TRAIL and Sp1 by Western blot analysis. β-actin was used as a loading control. In lower panel, Sp1+/+ and Sp1-/- ES cells were treated with MS275 (3 μM), adriamycin (0.01 μg/ml), or in combination for 48 hr, and cell viability was determined by MTT assays. Cell viability data are expressed as percent of untreated cells. Data are representative of three independent experiments. Asterisks indicate statistical significance (**p<0.01; *p<0.05).
To determine the effect of Sp1 knockdown on cell viability, T47D cells transfected with either Sp1 or control siRNA were treated with MS275, adriamycin or in combination for 48 hr, and cell viability was then determined. Fig. 6C shows that cells transfected with Sp1 siRNA are more resistant to MS275, adriamycin or in combination, as compared to cells transfected with control siRNA, which is similar to the results obtained with cells in which TRAIL was down regulated (Fig. 5C). These data suggest that Sp1-dependent TRAIL expression is critical for cell death induced by MS275, adriamycin or in combination.
To investigate the effects of down-regulation of Sp1 on MS275-mediated adriamycin-induced apoptosis, cells transfected with Sp1 or control siRNA were treated with either MS275 (5 μM), adriamycin (0.1 μg/ml) or in combination for 48 hr, and activation of caspases and cleavage of PARP were examined. As expected, in cells transfected with control siRNA, cleavage of caspase-9, -8, -3, and PARP was significantly increased in cells that received the combined treatments, as compared to untreated cells, while such changes were minimal in either agent treated cells (Fig. 6B). In contrast, such changes were decreased in cells transfected with Sp1 siRNA following the combined treatment (Fig. 6B), indicating that down-regulation of Sp1 decreases activation of the caspase cascade induced by the combined treatment, leading to improved cell survival. Taken together, these results suggest that Sp1 plays an important role in MS275-mediated adriamycin-induced caspase activation and apoptosis.
Role of Sp1 in chemosensitivity in Sp1 knockout mouse embryonic stem (ES) cells
We have shown that down-regulation of Sp1 by siRNA decreases T47D cell death induced by the combined treatments with MS275 and adriamycin (Fig. 6C). Since Sp1 expression could not be completely eliminated by siRNA silencing (Fig. 6A), the results obtained with an siRNA approach may not completely reflect the role of Sp1 in chemosensitivity. To overcome this difficulty, we examined the role of Sp1 in chemosensitivity using Sp1 knockout mouse ES cells. As expected, full length Sp1 was expressed in Sp1+/+ ES cells but not in Sp1-/- ES cells (Fig. 6D, upper panel), confirming Sp1 deletion in Sp1-/- cells. We then treated these cells with MS275, adriamycin or in combination, and the effects of such treatments on cell death were assessed. As shown in Fig. 6D (lower panel), Sp1-/- cells were more resistant than Sp1+/+ cells to MS275 or adriamycin; there were ∼57% and ∼74% of surviving Sp1-/- cells as compared to ∼43% and ∼42% of surviving Sp1+/+ cells following the treatments with MS275 and adriamycin, respectively. More importantly, we showed that the combined treatment results in a significant increase in cell survival of Sp1-/- cells as compared to Sp1+/+ cells (42% versus 18%). In addition, we showed that TRAIL is induced in Sp1+/+ but not in Sp1-/- cells by the combined treatments (Fig. 6D, upper panel), further confirming the requirement of Sp1 for TRAIL induction by the combined treatment. Thus, these results suggest that Sp1 plays a critical role in cell death induced by combined treatment with MS275 and adriamycin, which may be through the induction of TRAIL and activation of the TRAIL apoptotic pathway.
Discussion
In this study, we show that MS275 induces TRAIL, which sensitizes human breast cancer T47D cells and mouse ES cells to adriamycin-induced death. We also show that the underlying mechanism of such sensitization is mediated by the transcription factor Sp1 because Sp1 knockdown or deletion abolishes TRAIL induction and subsequently renders cells resistant to MS275, adriamycin or their combination. Thus, our findings indicate for the first time that the transcription factor Sp1 plays a critical role in chemosensitivity and thereby is a potential therapeutic target for the development of novel anticancer agents.
Sp1 was the first mammalian transcription factor to be identified (35). It binds to GC-rich sequences to regulate gene expression (36). Although Sp1 is a basal transcription factor, recent studies suggested that it plays an important role in tumor growth and metastasis. For example, Sp1 is over expressed in both gastric and pancreatic cancers, and overexpression of Sp1 enhances the expression of vascular endothelial growth factor (VEGF), which promotes cancer angiogenesis (37, 38). However, it is not known whether Sp1 plays a role in the responses of cancer cells to chemotherapy. In this study, we showed that knockdown of Sp1 in T47D cells abolishes cell death induced by MS275, adriamycin, or their combination. We also showed that knockdown of Sp1 in T47D cells substantially decreases the activation of the caspase cascade induced by the combined treatments, suggesting that Sp1 knockdown impairs caspase-mediated cell death induced by chemotherapeutic agents. Furthermore, we showed that Sp1 knockout decreased mouse ES cell death induced by MS275, adriamycin, or in combination, particularly for the combined treatments. However, it is not clear whether Sp1 plays a role in sensitizing other cancer cells to additional anticancer agents, which requires further investigation. Additionally, although Sp1 is a basal transcription factor and has many target genes, it is possible that in some cells after chemotherapeutic drug treatment, Sp1 might preferentially activate some apoptosis-related genes (e.g. TRAIL) to induce cell death. Because Sp1 is over expressed in some cancers (37, 39), anticancer drugs that can activate Sp1-dependent TRAIL expression might be effective against these tumors and this requires for further investigation. Nevertheless, this study provides the first proof-of-principal to target Sp1 for cancer therapy.
What are the underlying mechanisms by which Sp1 favors cell death induced by test agents? We believe that Sp1 could transcriptionally activate TRAIL, leading to the activation of the TRAIL apoptotic pathway because knockdown of Sp1 decreased TRAIL induction and subsequently increased chemoresistance. Although we showed that knockdown of Sp1 could decrease chemosensitivity of the breast cancer T47D cells to MS275, adriamycin or in combination (Fig. 6C), the effect of Sp1 knockdown on decreased chemosensitivity in T47D was not as evident as observed in Sp1 knockout ES cells (Fig. 6D). This difference may be due to the level of Sp1 protein. Because Sp1 is highly expressed in T47D cells, siRNA silencing can substantially down-regulate but not completely eliminate Sp1 expression (Fig. 6A). The remaining Sp1 may contribute to the observed cell death in Sp1 knockdown T47D cells. Consistent with this, we showed that more surviving cells were observed in Sp1-/- as compared to Sp1+/+ ES cells presumably because Sp1 was completely eliminated in Sp1-/- cells (Fig. 6D).
It has been shown that protein modifications including phosphorylation by anticancer agents increase Sp1 transcription activity (40). Furthermore, Sp1 can be cleaved and cleaved Sp1 has a higher activity (41). Since treatment of either agent alone or in combination had no effects on the levels of Sp1 protein, we believe that TRAIL expression induced by the treatments may not be due to the alteration of the level of Sp1 protein. Our preliminary data indicated that adriamycin, MS275 or in combination could cause Sp1 cleavage (data not shown), suggesting that Sp1 cleavage by the treatments may enhance Sp1 activity, leading to increased TRAIL expression, which is under investigation. In addition, it has been shown that HDAC1 could interact with Sp1 to regulate its activity (42). Therefore, we tested the effect of MS275 on Sp1 and HDAC1 interaction by IP-western. Although HDAC1 activity was inhibited, MS275 had no effect on the interaction of Sp1 with HDAC1 (data not shown). Since there are several HDAC family members, it is possible that MS275 could affect other HDAC members to regulate Sp1 activity. Regardless, we showed that MS275 alone or in combination with adriamycin increased Sp1 transcription activity.
Currently, there are few agents that are truly cancer cell specific in terms of efficacy and induction of cell death. TRAIL is an example of a molecule that selectively kills transformed and cancer cells but not most normal cells (14). Although TRAIL can selectively kill tumor or transformed cells without harming normal cells, its regulation in tumors, particularly in solid tumors, is not fully understood. Previous studies identified several regulatory elements in TRAIL promoter, which include ISRE, NF-κB, and Sp1 (18-20). We previously showed that TRAIL is induced by TNFα and 5-aza-2′-deoxycytidine through distinct mechanisms (33, 34). In this study, we showed that deletion of the second Sp1 binding site results in a complete loss of TRAIL promoter activity induced by MS275 or in combination with adriamycin, indicating that this Sp1 site is required for transactivation of the TRAIL promoter by our test agents. Furthermore, we have shown by ChIP assays that Sp1 can bind to the TRAIL promoter upon the treatments. Taken together, we conclude that the second Sp1 site is important for the activation of the TRAIL promoter.
We have shown that adriamycin can induce TRAIL at the protein level but had no effect on the TRAIL promoter activity. We have also shown that adriamycin can enhance the TRAIL promoter activity induced by MS275. This suggests that adriamycin induces other factors that indirectly affect the activity of the TRAIL promoter. Consistent with this, we have shown that there is a slight increase in TRAIL promoter activity induced by adriamycin treatment alone using ELISA (Fig. 4C).
Defective apoptotic responses are hallmarks of cancer cells, and apoptotic pathways are therefore attractive therapeutic targets. In addition to the use of TRAIL and agonistic antibodies alone or in combination with clinical chemotherapeutic agents, several new compounds including HDACIs that target the apoptotic pathway are under development (26, 27). Although HDACIs can activate transcription of target genes via histone acetylation to kill cancer cells by cell cycle arrest and apoptosis (24), recent studies indicated that induction of TRAIL plays a critical role in leukemia cell death (26, 27). Similarly, we showed here that HDACIs induce TRAIL in breast cancer cells (Fig. 1) and that such induction is critical for T47D and mouse ES cell death induced by adriamycin (Fig. 6C and D). Thus, our study suggests that induction of endogenous TRAIL sensitizes cancer cells to chemotherapy.
In summary, we demonstrate that HDACIs induce TRAIL via the second Sp1 binding site in the promoter of the TRAIL gene. We also demonstrate that the treatment with MS275 sensitizes breast cancer cells or mouse ES cells to adriamycin-induced death. More importantly, we demonstrate that Sp1-dependent TRAIL induction plays a critical role in cell death induced by combined treatments with MS275 and adriamycin because down-regulation of TRAIL by siRNA in T47D or deletion of Sp1 in Sp1 knockout mouse ES cells abolishes such sensitization. Therefore, our findings suggest that Sp1 is a new target in human cancer therapy.
Acknowledgments
Grant support: in part by NIH grant R01 CA100073 (GS Wu) and Department of Defense Breast Cancer Program W81XWH-07-1-05-21 (WZ Wei and GS Wu).
Abbreviations
- TRAIL
Tumor necrosis factor-related apoptosis-inducing ligand
- Adr
adriamycin
- siRNA
small interfering RNA
- HDACI
inhibitors of histone deacetylases
- FBS
fetal bovine serum
- ES cells
embryonic stem cells
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2-3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 2.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–90. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 3.Pan G, O'Rourke K, Chinnaiyan AM, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–3. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 4.Wu GS, Burns TF, McDonald ER, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–3. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 5.MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272:25417–20. doi: 10.1074/jbc.272.41.25417. [DOI] [PubMed] [Google Scholar]
- 6.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 7.Walczak H, Degli-Esposti MA, Johnson RS, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–97. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Screaton GR, Mongkolsapaya J, Xu XN, Cowper AE, McMichael AJ, Bell JI. TRICK2, a new alternatively spliced receptor that transduces the cytotoxic signal from TRAIL. Curr Biol. 1997;7:693–6. doi: 10.1016/s0960-9822(06)00297-1. [DOI] [PubMed] [Google Scholar]
- 9.Degli-Esposti MA, Smolak PJ, Walczak H, et al. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med. 1997;186:1165–70. doi: 10.1084/jem.186.7.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheridan JP, Marsters SA, Pitti RM, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–21. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 11.Mongkolsapaya J, Cowper AE, Xu XN, et al. Lymphocyte inhibitor of TRAIL (TNF-related apoptosis-inducing ligand): a new receptor protecting lymphocytes from the death ligand TRAIL. J Immunol. 1998;160:3–6. [PubMed] [Google Scholar]
- 12.Marsters SA, Sheridan JP, Pitti RM, et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7:1003–6. doi: 10.1016/s0960-9822(06)00422-2. [DOI] [PubMed] [Google Scholar]
- 13.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–20. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 14.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 15.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14(34):337–48. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 16.Sheikh MS, Burns TF, Huang Y, et al. p53-dependent and -independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor alpha. Cancer Research. 1998;58(8):1593–8. [PubMed] [Google Scholar]
- 17.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4(2):139–63. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 18.Gong B, Almasan A. Genomic organization and transcriptional regulation of human Apo2/TRAIL gene. Biochem Biophys Res Commun. 2000;278(3):747–52. doi: 10.1006/bbrc.2000.3872. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Ji Y, Wang X, Evers BM. Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem Biophys Res Commun. 2000;276(2):466–71. doi: 10.1006/bbrc.2000.3512. [DOI] [PubMed] [Google Scholar]
- 20.Clarke N, Jimenez-Lara AM, Voltz E, Gronemeyer H. Tumor suppressor IRF-1 mediates retinoid and interferon anticancer signaling to death ligand TRAIL. Embo J. 2004;23(15):3051–60. doi: 10.1038/sj.emboj.7600302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirshner JR, Karpova AY, Kops M, Howley PM. Identification of TRAIL as an interferon regulatory factor 3 transcriptional target. J Virol. 2005;79(14):9320–4. doi: 10.1128/JVI.79.14.9320-9324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivera-Walsh I, Waterfield M, Xiao G, Fong A, Sun SC. NF-kappaB signaling pathway governs TRAIL gene expression and human T-cell leukemia virus-I Tax-induced T-cell death. J Biol Chem. 2001;276(44):40385–8. doi: 10.1074/jbc.C100501200. [DOI] [PubMed] [Google Scholar]
- 23.Baetu TM, Kwon H, Sharma S, Grandvaux N, Hiscott J. Disruption of NF-kappaB signaling reveals a novel role for NF-kappaB in the regulation of TNF-related apoptosis-inducing ligand expression. J Immunol. 2001;167(6):3164–73. doi: 10.4049/jimmunol.167.6.3164. [DOI] [PubMed] [Google Scholar]
- 24.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 25.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–99. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 26.Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 27.Nebbioso A, Clarke N, Voltz E, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 28.Rahmani M, Yu C, Reese E, et al. Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21(CIP1/WAF1) induction rather than AKT inhibition. Oncogene. 2003;22(40):6231–42. doi: 10.1038/sj.onc.1206646. [DOI] [PubMed] [Google Scholar]
- 29.Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23(37):6261–71. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 30.Marin M, Karis A, Visser P, Grosveld F, Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 1997;89(4):619–28. doi: 10.1016/s0092-8674(00)80243-3. [DOI] [PubMed] [Google Scholar]
- 31.Sun SY, Yue P, Zhou JY, et al. Overexpression of bcl2 blocks TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in human lung cancer cells. BBRC. 2001;280:788–97. doi: 10.1006/bbrc.2000.4218. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Xu J, Zhou JY, Liu Y, Wu GS. Mitogen-Activated Protein Kinase Phosphatase-1 Is Required for Cisplatin Resistance. Cancer Res. 2006;66(17):8870–7. doi: 10.1158/0008-5472.CAN-06-1280. [DOI] [PubMed] [Google Scholar]
- 33.Xu J, Zhou JY, Wu GS. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Is Required for Tumor Necrosis Factor {alpha}-Mediated Sensitization of Human Breast Cancer Cells to Chemotherapy. Cancer Res. 2006;66(20):10092–9. doi: 10.1158/0008-5472.CAN-06-1633. [DOI] [PubMed] [Google Scholar]
- 34.Xu J, Zhou JY, Tainsky MA, Wu GS. Evidence that tumor necrosis factor-related apoptosis-inducing ligand induction by 5-aza-2′-deoxycytidine sensitizes human breast cancer cells to adriamycin. Cancer Res. 2007;67(3):1203–11. doi: 10.1158/0008-5472.CAN-06-2310. [DOI] [PubMed] [Google Scholar]
- 35.Kadonaga JT, Carner KR, Masiarz FR, Tjian R. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell. 1987;51(6):1079–90. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- 36.Kadonaga JT, Courey AJ, Ladika J, Tjian R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science. 1988;242(4885):1566–70. doi: 10.1126/science.3059495. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Wei D, Huang S, et al. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin Cancer Res. 2003;9(17):6371–80. [PubMed] [Google Scholar]
- 38.Shi Q, Le X, Abbruzzese JL, et al. Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res. 2001;61(10):4143–54. [PubMed] [Google Scholar]
- 39.Lou Z, O'Reilly S, Liang H, Maher VM, Sleight SD, McCormick JJ. Down-regulation of overexpressed sp1 protein in human fibrosarcoma cell lines inhibits tumor formation. Cancer Res. 2005;65(3):1007–17. [PubMed] [Google Scholar]
- 40.Niina I, Uchiumi T, Izumi H, et al. DNA topoisomerase inhibitor, etoposide, enhances GC-box-dependent promoter activity via Sp1 phosphorylation. Cancer Sci. 2007;98(6):858–63. doi: 10.1111/j.1349-7006.2007.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spengler ML, Brattain MG. Sumoylation inhibits cleavage of Sp1 N-terminal negative regulatory domain and inhibits Sp1-dependent transcription. J Biol Chem. 2006;281(9):5567–74. doi: 10.1074/jbc.M600035200. [DOI] [PubMed] [Google Scholar]
- 42.Doetzlhofer A, Rotheneder H, Lagger G, et al. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol. 1999;19(8):5504–11. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]