

Abstract
C-1027 is a potent antitumor antibiotic composed of an apo-protein and a reactive enediyne chromophore. The chromophore consists of four different chemical subunits including an (S)-3-chloro-4,5-dihydroxy-β-phenylalanine moiety, the biosynthesis of which from L-α-tyrosine is catalyzed by six proteins, SgcC, SgcC1, SgcC2, SgcC3, SgcC4, and SgcC5. Biochemical characterization of SgcC3 unveiled that: (i) SgcC3 is a flavin adenine dinucleotide (FAD)-dependent halogenase, (ii) SgcC3 acts only on the SgcC2 peptidyl carrier protein-tethered substrates, (iii) SgcC3-catalyzed halogenation requires O2 and reduced FAD and either the C-1027 pathway-specific flavin reductase SgcE6 or E. coli flavin reductase (Fre) can support the SgcC3 activity, (iv) SgcC3 also efficiently catalyzes bromination but not fluorination or iodination, and (v) SgcC3 can utilize both (S)- and (R)- β-tyrosyl-S-SgcC2 but not 3-hydroxy-β-tyrosyl-S-SgcC2 as a substrate. These results establish that SgcC3 catalyzes the third enzymatic transformation during the biosynthesis of the (S)-3-chloro-4,5-dihydroxy-β-phenylalanine moiety moiety of C-1027 from L-α-tyrosine. SgcC3 now represents the second biochemically characterized flavin-dependent halogenase that acts on a carrier protein-tethered substrate. These findings will facilitate the engineering of new C-1027 analogs by combinatorial biosynthesis methods.
Keywords: C-1027, chlorination, enediyne, halogenase, SgcC3, Streptomyces globisporus
Introduction
C-1027 is a chromoprotein antitumor antibiotic isolated from the fermentation broth of Streptomyces globisporus.1 Similar to other nine-membered enediynes, which includes neocarzinostatin2 and maduropeptin,3 C-1027 is composed of an apo-protein (CagA) and a reactive enediyne chromophore that is essential for bioactivity.4 Upon release from CagA, the C-1027 chromophore readily undergoes a Bergman cycloaromatization to form a benzenoid diradical intermediate (Figure 1). These free radicals are capable of abstracting hydrogen atoms from the DNA backbone, ultimately resulting in DNA double-stranded breaks (DSB) in the presence of molecular oxygen.5–7 The induction of DSB correlates well with cytotoxicity, and because of this novel mechanism of DNA damage, the enediynes have attracted interest as potential cancer chemotherapeutic agents.8,9
Figure 1.

The Bergman cycloaromatization exemplified by the C-1027 chromophore. Upon release from the apo-protein CagA, the enediyne core (shown in red) undergoes an electronic rearrangement to form a 3, 6-diradical species that can abstract hydrogen atoms from DNA. DSB, double-stranded break.
The nine-membered enediynes all have an enediyne core consisting of an unsaturated nine-membered carbocycle containing two acetylenic groups conjugated to a recipient or incipient double bond. The C-1027 chromophore (1) has three additional chemical components that are covalently appended to the enediyne core, namely a deoxy aminosugar, a benzoxazolinate, and a β-amino acid moiety (Figure 1). The C-1027 biosynthetic gene cluster was previously identified and, based on bioinformatics analysis, the biosynthetic pathway for each moiety was predicted and a convergent pathway was hypothesized to yield 1. Of note, the β-amino acid moiety (S)-3-chloro-4,5-dihydroxy-β-phenylalanine was predicted to be biosynthesized from L-α-tyrosine (2) mediated by five proteins: SgcC, SgcC1, SgcC2, SgcC3 and SgcC4, with the condensation enzyme SgcC5 catalyzing the final incorporation of 2 into 1 via a β-aminoacyl-S-SgcC2 intermediate (3) (Figure 2).8 We have subsequently confirmed in vitro using purified recombinant enzymes that (i) SgcC4 catalyzes the conversion of 2 to (S)-β-tyrosine (4) as the first step10–12 and (ii) SgcC1 activates 4 as an (S)-β-tyrosyl adenylate (5) and subsequently loads 4 to SgcC2, a peptidyl carrier protein (PCP), to yield (S)-β-tyrosyl-S-SgcC2 (6) as the second step.13,14 SgcC3 and SgcC were also identified as the C-3 halogenase and C-5 hydroxylase, respectively, by gene inactivation: the ΔsgcC3 mutant produced 20-deschloro-C-1027 (7) and 20-deschloro-22-deshydroxy-C-1027 (8),13 while the ΔsgcC mutant accumulated 22-deshydroxy-C-1027 (9).8 Thus, the requirement of SgcC3 for chlorination was clearly established, although these results provided little insight into the substrate specificities and mechanistic details of this enzyme.
Figure 2.
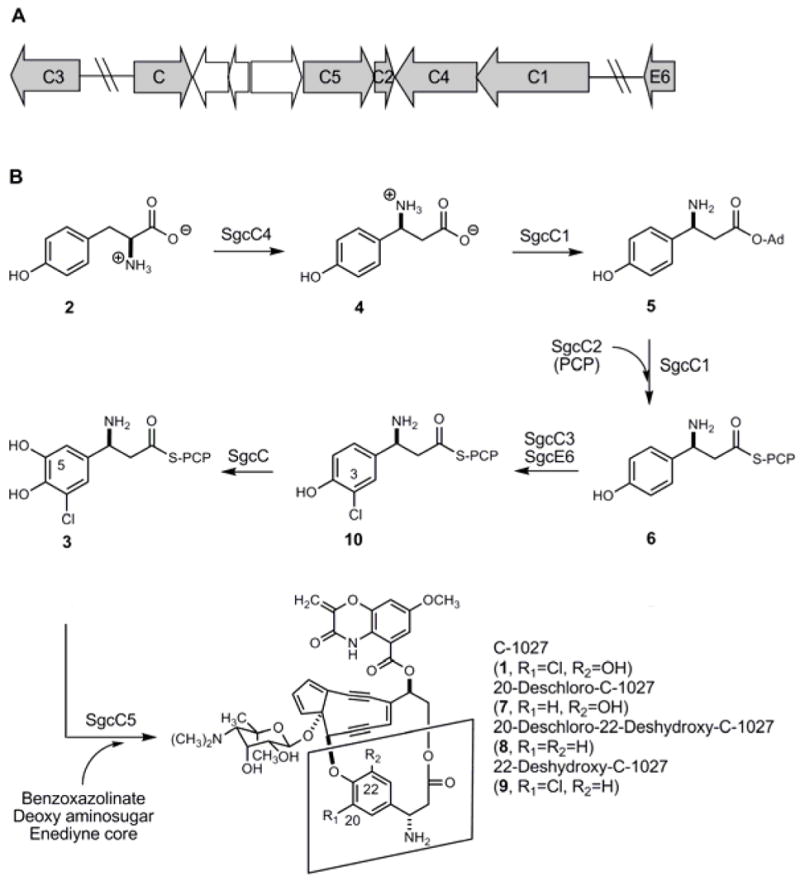
Biosynthesis of (S)-3-chloro-4,5-dihydroxy-β-phenylalanine moiety (3) of C-1027 (1, boxed). (A) A sub-cluster of genes within the C-1027 gene cluster that encode biosynthetic proteins for 3 production and (B) proposed biosynthetic pathway for 3 from L-α-tyrosine (2) and structures of 1 and engineered analogs 7, 8, and 9.
In general, incorporation of a halogen into natural products plays an important role in increasing the diversity and biological activity of natural products. Indeed, 7 was recently shown to be less proficient at inducing DSB, less cytotoxic, as well as have an altered mechanism of action compared to the typical cellular response induced by treatment with 1.9 Prior to 1997, halogenation was believed to occur only by the action of haloperoxidases – enzymes that utilize a heme or vanadium cofactor and hydrogen peroxide as a co-substrate.15 Since this time, several O2-dependent halogenases have been described and their activity biochemically confirmed. Currently, the O2-dependent halogenases fall into one of two families: α-ketoglutarate-dependent halogenases that act on non-activated aliphatic substrates16–21 and flavin-dependent halogenases that catalyze the regioselective halogenation of primarily aromatic compounds.16,22–31 SgcC3 has sequence homology to flavin-dependent halogenases, and combined with the in vivo data, SgcC3 was predicted to incorporate chloride using flavin as a redox cofactor and O2 as an oxidizing agent.
Given the importance of halogen atoms in natural products and the intriguing variety and complexity of halogenases,16 we set out to characterize SgcC3 in vitro as part of our continuous efforts to investigate the biosynthetic pathway of 1. In this study, we report the functional characterization of recombinant SgcC3 as a flavin adenine dinucleotide (FAD)-dependent halogenase that catalyzes the regiospecific chlorination of 6, requiring O2 and reduced FAD (FADH2), to afford (S)-3-chloro-β-tyrosyl-S-SgcC2 (10) as the third step for the biosynthesis of 3 from 2 (Figure 2). The general catalytic properties of SgcC3 are reported, and SgcC3 now is the second biochemically characterized flavin-dependent halogenase that acts on a carrier protein-tethered substrate, which likely represents a general strategy for oxidative halogenation of secondary metabolites that are assembled via carrier protein-dependent biosynthetic machinery. The results established herein, along with the evidence that SgcC5 has relaxed specificity as implied by the isolation of 7, 8, and 9, affords the opportunity to generate new C-1027 analogs by combinatorial biosynthesis methods.
Experimental Procedures
Synthesis of the β-Tyrosine Analogues
Syntheses of the β-tyrosine analogues [i.e., 3-chloro-β-tyrosine, 3-bromo-β-tyrosine, 3-hydroxy-β-tyrosine, and 3-chloro-5-hydroxy-β-tyrosine] were achieved by following literature procedure32 (see Supporting Information for details).
Cloning of sgcC2, sgcC3, sgcE6, and E. coli fre Genes for Heterologous Expression
The genes encoding SgcC2, SgcC3, SgcE6 and E. coli flavin reductase (Fre) were amplified by PCR using Platinum Pfx DNA polymerase following the program and conditions provided by Invitrogen (see Table S1 for primers used in Supporting Information). Templates utilized for PCR were pBS103413 (for sgcC2), pBS10058 (for sgcC3), pBS10068 (for sgcE6), and E. coli DH5α genomic DNA (for fre), respectively. Purified PCR product of sgcC2 was cloned into the pCDF-2Ek/LIC vector using ligation-independent cloning procedure as described by Novagen (Madison, WI) to give pCDF-2Ek/LIC-SgcC2 (pBS1040). The PCR products of sgcC3, sgcE6 and fre were similarly cloned into the pET-30Xa/LIC vector (Novagen, Madison, WI) to yield pET-30Xa/LIC-SgcC3 (pBS1041), pET-30Xa/LIC-SgcE6 (pBS1042), and pET-30XaLIC-Fre (pBS1043), respectively. All cloned PCR products were confirmed by DNA sequencing. The sgcC1 expression construct pBS1033 has been described previously.13
Overproduction and Purification of SgcC1, SgcC2, SgcC3, SgcE6 and Fre
E. coli BL21 (DE3) introduced with pBS1033 (for SgcC1), pBS1040 (for SgcC2), pBS1041 (for SgcC3), pBS1042 (SgcE6), or pBS1043 (for Fre) was cultured in LB medium33 supplemented with streptomycin (50 μg mL−1 for pBS1040) or kanamycin (50 μg mL−1 for pBS1033, pBS1041, pBS1042, pBS1043), respectively. All cells were grown at 18 °C and induced with IPTG (final concentration of 0.1 mM) when OD600 reached ~ 0.5. They were further cultured at 18 °C for additional 15 hrs. Cells were harvested by centrifugation (4 °C, 8000 rpm for 15 min) and resuspended in Buffer A (100 mM sodium phosphate, pH 7.5, containing 300 mM NaCl) supplemented with a complete protease inhibitor tablet, EDTA-free (Roche Applied Science, Indianapolis, IN). The cells were lysed by sonication (4 × 30 sec pulsed cycle), and the debris was removed by centrifugation (4 °C, 15000 rpm for 50 min). The clarified supernatant was loaded onto a pre-equilibrated Ni-NTA agarose column (Qiagen, Valencia, CA) with Buffer B (Buffer A plus 10% glycerol). The column was washed with 5-column volumes of Buffer B followed by 5-column volumes of Buffer B containing 20 mM imidazole. The His6-tagged proteins were eluted with 6-column volumes of Buffer B containing 250 mM imidazole. SgcC2 was dialyzed in 50 mM Tris-HCl, pH 7.5, 50 mM NaCl and 1mM dithiothreitol. SgcC1, SgcC3, SgcE6, and Fre were desalted using a PD-10 column (GE Healthcare, Piskataway, NJ). After concentration with an Amicon Ultra-4 (5K or 10K), the purified proteins were stored at −25 °C in 40% glycerol. Protein purity was assessed as > 90% by 12–15% SDS-PAGE. Protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA)
Determination of the SgcC3 Flavin Cofactor
SgcC3 was denatured by heat or with 50% methanol (final concentration) to release any noncovlently-bound cofactor. After centrifugation the supernatent was analyzed on C18 reverse phase column (250 × 4.6 mm, Alltech Associates Inc. Deerfield, IL) using a linear gradient from 0 to 60% acetonitrile (in water) at a flow rate of 1 mL min−1 with detection at 266 nm.
Enzymatic Synthesis of β-Aminoacyl-S-SgcC2
Post-translational modification of apo-SgcC2 into holo-SgcC2 was achieved in a reaction mixture containing 100 mM Tris-HCl, pH 7.5, 160 μM apo-SgcC2, 0.8 mM coenzyme A (CoA), 12.5 mM MgCl2, 2.0 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 5 μM Svp.34 After incubation at room temperature for 45 min, an equal volume of loading solution consisting of 2 mM 4 [4 mM for (R)- β-tyrosine (11) or 3-hydroxy-β-tyrosine], 4 mM adenosine triphosphate, 2.0 mM TCEP and 12.5 mM MgCl2 (final concentration in the loading reaction mixture) was added. Amino acid loading was initiated by the addition of SgcC1 to a final concentration of 2 μM (6 μM for 11 or 3-hydroxy-β-tyrosine), and the resulting solution was incubated at room temperature for an additional 60 min as described previously.13 The resulting β-aminoacyl-S-SgcC2 substrates [i.e., 6, (R)- β-tyrosyl-S-SgcC2 (12), or 3-hydroxy-β-tyrosyinl-S-SgcC2] were purified from loading mixture with a 5-mL HiTrap Q anion-exchange column (GE Healthcare, Piskataway, NJ). The column was pre-equilibrated with 20 mM sodium phosphate buffer, pH 7.0, and the crude SgcC2-tethered substrate preparations were loaded and eluted using a linear gradient of 0 to 100 % of 1 M NaCl for 25-column volumes and flow rate of 3 mL min−1. The purified β-aminoacyl-S-SgcC2 substrates, which were eluted between 0.35 M and 0.4 M NaCl, were desalted using size-exclusion chromatography (Superose® 12, GE Healthcare, Piskataway, NJ) and concentrated prior to use in SgcC3 assays.
In vitro Activity Assay for SgcC3
The SgcC3 assay solution contained 50 μM 6 (or 12 or 250 μM 3-hydroxy-β-tyrosyl-S-SgcC2), 5 mM β-nicotinamide adenine dinuceotide, reduced (NADH), 0.10 mM FAD, 100 mM NaCl, 1 mM TCEP, 20 μM SgcC3, and 5 μM SgcE6 in 50 mM sodium phosphate buffer, pH 6.0. Reactions were incubated at 37 °C for 1 hr. The reaction was terminated by the addition of 70 % trichloroacetic acid (TCA) to a final concentration of 10 % to precipitate all proteins. After incubation on ice for 15 min, the precipitate was separated by centrifugation (4°C, 14,000 rpm for 15 min). The resulting pellet was washed twice with 200 μL of 5% TCA and once with 200 μL of ethanol. After drying by speed-vac, the protein pellet was re-dissolved in 150 μL of 0.1 M KOH and incubated at 70 °C for 15 min to hydrolyze all SgcC2-tethered β-amino acids. After removal of the proteins by centrifugation, the solution was concentrated by speed-vac and analyzed for 4 (or 11 or 3-hydroxy-β-tyrosine) and the expected product (S)-3-chloro-β-tyrosine (13) [or (R)-3-chloro-β-tyrosine (14) or 3-chloro-5-hydroxy-β-tyrosine] by a Varian ProStar 210 HPLC System equipped with a C18 reverse-phase column (250 × 4.6 mm, Alltech Associates Inc., Deerfield, IL), using a 24-min linear gradient from 0 to 40% (25% for 3-hydroxy-β-tyrosine and 3-chloro-5-hydroxy-β-tyrosine) acetonitrile (0.1 % TFA) at a flow rate of 1 mL min−1 and detection at 280 nm and authentic 4, 11, 13, 14, 3-hydroxy-β-tyrosine, or 3-chloro-5-hydroxy-β-tyrosine as references. Control experiments were carried out under identical conditions but with boiled SgcC3.
Formation of 3-Bromo-β-Tyrosyl-S-SgcC2
Both SgcC3 and SgcE6 were prepared by purification using Ni-NTA columns in buffers without chloride. After 4 was loaded onto holo-SgcC2 by SgcC1, the resultant product 6 was purified from the loading reaction mixture through a 5-mL HiTrap Q column. Chloride was then completely removed by passing through a size-exclusion column twice (Superose® 12). The SgcC3-catalyzed bromination of 6 was performed in a solution identical to above except that 0.1 M NaBr replaced NaCl in the assay mixture and TCEP was excluded. The reaction was quenched with 10% TCA to precipitate proteins at 10, 30 and 60 min as described above. The precipitate was treated with 150 μL of 0.1 M KOH to release all SgcC2-tethered β-amino acids. The resultant free β-amino acids [i.e., 4 and the expected product 3-bromo-β-tyrosine (15)] were subjected to HPLC and MS analysis. The standard curve was calibrated with synthetic 15 to correlate peak area with product amount formed in each reaction. The product formation was fitted into a linear equation to obtain the initial velocity.
Initial Velocity of SgcC3 with (S)- β-Tyrosyl-S-SgcC2 and (R)- β-Tyrosyl-S-SgcC2
Conversion of apo-SgcC2 to holo-SgcC2 was carried out in a 1.8 mL reaction containing 400 μM SgcC2, 1.6 mM CoA, and 10 μM Svp at room temperature for 60 min. For preparation of 6 from 4, the loading condition was identical to the standard assay condition described above. For preparation of 12 from 11, the reaction proceeded with 4 mM 11 and 6 μM SgcC1 and was incubated at room temperature for 90 min. Chlorination assays were performed in 200 μL of reaction mixture containing 200 μM 6 or 12, 0.1 M NaCl, 5 mM NADH, 0.1 mM FAD, 25 μM SgcC3, and 3 μM SgcE6. The reaction was quenched by the addition of 30 μl of 70% TCA at 10, 20, 40 and 60 min and analyzed as described above to determine the rate for (S)-3-chloro-β-tyrosyl-S-SgcC2 (16) or (R)-3-chloro-β-tyrosyl-S-SgcC2 (17) formation. A standard calibration curve was created with synthetic 13, and product formation was fitted to a linear equation to obtain the initial velocity. The specific activity was calculated from the initial velocity divided by the concentration of SgcC3 as determined using the Bradford dye-binding procedure.
Results
Overproduction and Purification of SgcC1, SgcC2, SgcC3, SgcE6, and Fre
The sgcC2 gene was subcloned from pBS103413 into pCDF-2Ek/LIC vector to eliminate most of the extra N-terminal residues engineered into the recombinant protein used in previous studies.13,14 After overproduction in E. coli, SgcC2 was purified to homogeneity as an N-terminal His6-tagged fusion protein containing an additional 13 amino acids. Purification of SgcC3 to homogeneity afforded a yellow solution whose UV-Vis spectrum showed the characteristic absorption maxima of 375 nm and 450 nm, indicative of the presence of a flavin prosthetic group. Since it has been previously reported that FADH2-dependent halogenases require a separate flavin reductase to supply flavin cofactor in FADH2 form,22–31 the putative flavin reductase SgcE6 within the C-1027 biosynthetic gene cluster (Figure 2A) and E. coli Fre were individually overproduced and purified to homogeneity. Similar to SgcC3, the purified SgcE6 and Fre also had UV-Vis profiles characteristic of flavoproteins. SgcC1 was overproduced, purified, and the activity accessed as previously described.13,14 The homogeneity of the purified proteins was confirmed by SDS-PAGE analysis (Figure 3).
Figure 3.
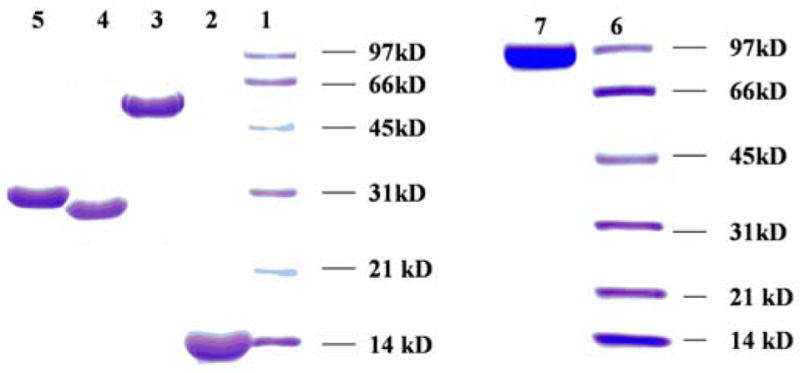
Purification and characterization of proteins used in this study as judged by SDS-PAGE: lane 1, SgcC2; lane 3, SgcC3; lane 4, SgcE6; lane 5, Fre; lane 7, SgcC1; lanes 1 and 6, low range molecular weight standards.
Identity of the SgcC3 Flavin Cofactor
As predicted from bioinformatics analysis, the purified SgcC3 had a UV-vis spectrum reminiscent of a flavin cofactor. Heat denaturation of SgcC3 completely released the flavin group, indicating that the cofactor is noncovalently bound to SgcC3. HPLC analysis of the released flavin prosthetic group with authentic FMN and FAD as standards established its identity as FAD with a 1:0.18 molar ratio of SgcC3: FAD (see Figure S1 in Supporting Information).
Substrate Preparation and Activity Assay of SgcC3 with (S)- β-Tyrosyl-S-SgcC2
Holo-SgcC2 was generated enzymatically using the Svp phosphopantetheinyl transferase (PPTase).34 Subsequently, the 4-specific adenylation enzyme SgcC1 was used to load holo-SgcC2 with 4 to generate 6. After purification using anion exchange, 6 was incubated with NaCl in the presence of SgcC3, SgcE6, FAD, and NADH under aerobic condition at 37°C for 1 hr, and the reaction was monitored by subjecting aminoacyl-S-SgcC2 substrate 6 and product 10 to alkaline hydrolysis followed by HPLC analysis. A new peak appeared eluting after 4 (obtained from hydrolysis of substrate 6), and this new peak had an identical retention time to authentic 13 (Figure 4). The identity of the hydrolyzed product as 13 was confirmed by ESI-MS, yielding a pair of [M+H]+ ions at m/z = 216.2 and 218.1 with 3:1 ratio, characteristic for the mono-chlorinated 13 (molecular formula C9H10O3NCl, calcd 215.0 and 217.0) (see Figure S2 in Supporting Information). Time course analysis showed that SgcC3-catalyzed the conversion of 6 to 10 in a time-dependent reaction with a specific activity of 0.93 ± 0.14 hr−1 (Figure 4).
Figure 4.
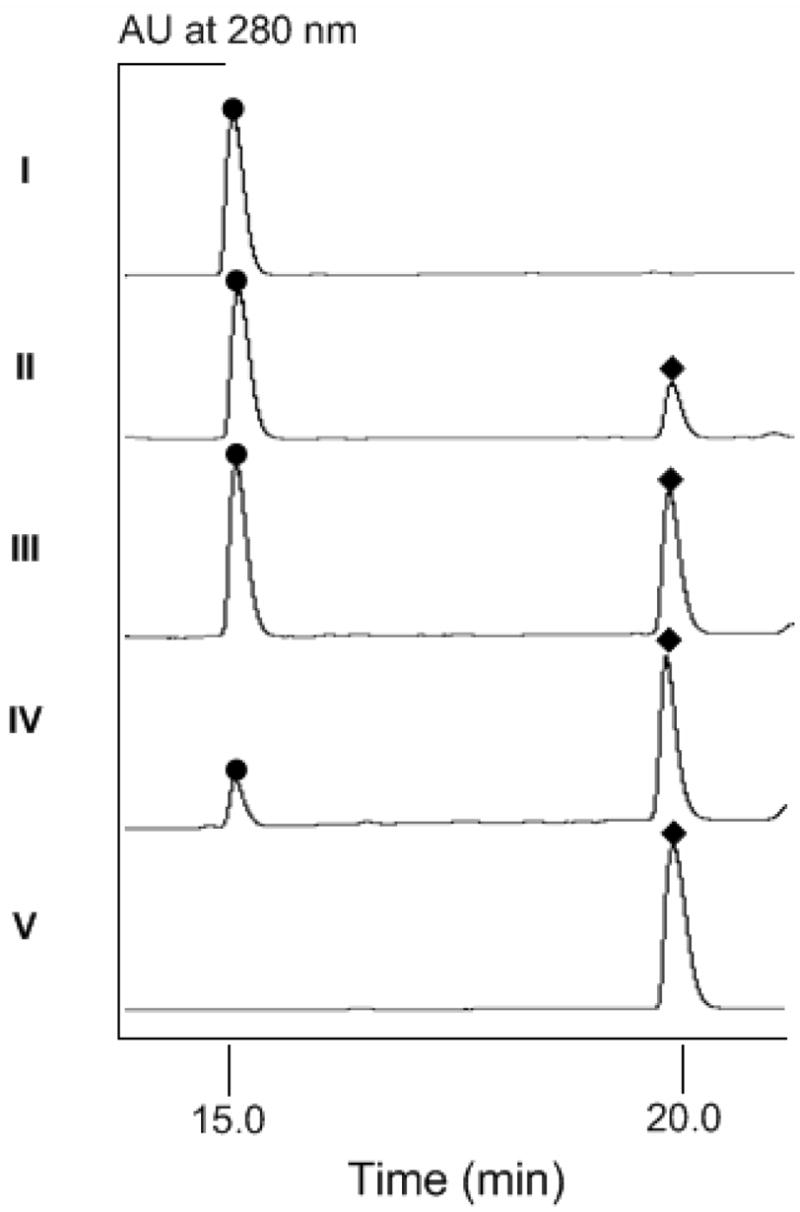
HPLC chromatograms of SgcC3 assays showing authentic (S)- β-tyrosine (4) standard (I), incubation of (S)- β-tyrosyl-S-SgcC2 (6) with SgcC3 at 37°C for 15 min (II), 30 min (III), 60 min (IV), and synthetic 3-choro-β-tyrosine (V). (●), 4 and (◆), 13 or 3-choro-β-tyrosine.
Cofactor and Co-substrate Requirements for SgcC3-Catalyzed Chlorination
The substrate and cofactor requirement for the SgcC3-catalyzed chlorination of 6 to 10 as depicted in Figure 5A was next examined (results summarized in Table 1). Both O2 and NADH are required for SgcC3 catalysis – the removal of either completely abolished 10 formation (Figure 5B, VI and VII). When FAD was excluded from the assay solution, a minute amount of 10 was detected by HPLC, consistent with the observation that SgcC3 is co-purified with sub-stoichiometric amounts of FAD that can be utilized for SgcC3 catalysis (Figure 5B, V). In the absense of a flavin reductase, a small amount of 10 was produced, and this activity is likely due to co-purification of SgcC3 with E. coli Fre as has been reported for other halogenases27 and flavin-dependent oxygenase.35,36 However, optimal SgcC3 activity requires the exogenous supply of a flavin reductase (Figure 5B, IV vs II and III). Finally, there appears to be no specific interaction between SgcC3 and the flavin reductase since SgcE6 and Fre can equally support the halogenase activity of SgcC3 (Figure 5B, II and III). This finding is consistent with the observation that SgcE6 is the only flavin reductase within the C-1027 cluster,8 which likely serves all of the flavin-dependent enzymes involved in C-1027 biosynthesis.
Figure 5.

SgcC3-catalyzed halogenation of (S)- β-tyrosyl-S-SgcC2 (6) or (R)- β-tyrosyl-S-SgcC2 (11). (A) The reaction scheme for SgcC3 depicting a catalytic cycle involving the FAD reductase SgcE6. (B) HPLC chromatograms examining substrate and cofactor requirement for SgcC3-catalyzed chlorination of 6. (C) HPLC chromatograms examining alternative substrates for SgcC3. (●), 4 or 11; (◆), 13, 14 or 3-choro-β-tyrosine; (◇), 15 or 3-bromo-β-tyrosine.
Table 1.
Cofactor and co-substrate requirement for SgcC3-catalyzed chlorination of (S)-β-tyrosyl-S-SgcC2 (6) in vitro.
| Entrya | Condition | Conversion | Relative rate |
|---|---|---|---|
| II | Complete assay | 38% | 100 |
| III | Complete assay – SgcE6 + E. Coli Fre | 39% | 100 |
| IV | Complete assay – SgcE6 | 6% | 16 |
| V | Complete assay – FAD | 11% | 30 |
| VI | Complete assay – NADH | 0 | 0 |
| VII | Complete assay – O2 | 0 | 0 |
Entry number corresponds to the chromatogram shown in Figure 5B.
pH Optimum for SgcC3 Activity
To estimate the pH optimum for SgcC, chlorination of 6 was carried out in 50 mM sodium acetate, sodium phosphate, and Tris-HCl buffer, ranging from pH 5.0 to pH 9.0, under the standard assay conditions. SgcC3 showed the highest activity at pH 6.0 (see Figure S3 in Supporting Information), and therefore all subsequent assays were performed in 50 mM phosphate buffer at pH 6.0.
Substrate Specificity of SgcC3
The substrate specificity of SgcC3 was initially examined with free amino acids as substrates, including 4, 11, and 3-hydroxy-β-tyrosine. HPLC analysis showed no activity under all conditions tested, consistent with the previous proposal that chlorination occurs after 4 is tethered to SgcC2 as 6 (Figure 2B).8,10–14
The substrate specificity of SgcC3 was next investigated by using a series of β-aminoacyl-S-SgcC2 as substrates (Figure 5A and 5C). Taking advantage of SgcC1’s ability to activate and load other β-amino acids to SgcC2,13,14 12 and 3-hydroxy-β-tyrosyl-SgcC2 were prepared and tested, in comparison with 6, for activity with SgcC3. Unexpectedly, SgcC3 catalyzes the chlorination of 12 to generate 16 with a specific activity of 1.1 ± 0.20 hr−1, nearly identical to that found with 6 (Figure 5C II and III). In contrast, no chlorination was detected with 3-hydroxy-β-tyrosyl-SgcC2 in spite of the inclusion of a 5-fold amount of SgcC3 and prolonged incubation time. The latter finding is consistent with 6 as the bona fide substrate of SgcC3 and supports the timing of the individual steps proposed for 3 biosynthesis (Figure 2B).8,10–14
Finally, halogen specificity of SgcC3 was examined. After purification of all proteins in the absence of chloride, SgcC3 efficiently catalyzed the bromination of 6 to afford the corresponding brominated product 3-bromo-β-tyrosyl-SgcC2 (17) with a specific activity of 0.36 ± 0.10 hr−1, which is approximately 2-fold less than chlorination under similar reaction conditions (Figure 2C, V and VI). However, neither fluorination nor iodination was observed under the identical condition, a finding that is consistent with all flavin-dependent halogenases known to date.16,22 The identity of 17 was confirmed by HPLC analysis of the free acid 15 using synthetic 15 as a standard. ESI-MS analysis of 15 yielded a pair of [M+H]+ ions at m/z = 260.0 and 262.0 with 1:1 ratio, in agreement with the mono-bromonated 15 (molecular formula C9H10O3NBr, calcd 259.0 and 261.0 around 1: 1 ratio).
Discussion
Like all enediyne natural products, C-1027 shows a potent antitumor activity. Despite sharing a common enediyne core, however, C-1027 is unique within the family in that it is estimated to 1000-times more potent than other 9-membered enediynes such as neocarzinostatin or 10-membered enediynes such as calicheamicin.37 This difference in cytotoxicity can be attributed in part to the moieties decorating the enediyne core, for which C-1027 has three – a deoxy aminosugar, a benzoxazolinate moiety, and a β-amino acid moiety (Figure 1). Indeed, as we have recently reported, small structural permutations to the benzoxazolinate and β-amino acid moieties have profound effects on the biological activity of C-1027, including the ability to induce DSB, the in vivo cytotoxicity, and the mechanism of the cellular response upon treatment with C-1027.9
The 3-chloro-4,5-dihydroxy-β-phenylalanine moiety of 1 contributes critical interactions with the apo-protein CagA and, as suggested from the studies regarding the bioactivity of 7 and 9,9 modulates the stability and reactivity of the enediyne core via π-π stacking interaction.38 This moiety is activated and incorporated into C-1027 by first generating a (S)- β-tyrosyl-S-SgcC2 intermediate 6, which subsequently serves as a substrate for chlorination and likely hydroxylation prior to attachment to the enediyne core.8,13 The process of activation and incorporation is achieved by enzymes with homology to protein domains found in nonribosmal peptide synthetases (NRPS), and these enzymes – SgcC1, SgcC2, and SgcC5 – are located in a sub-clustered region within the ~ 80 kb C-1027 biosynthetic gene cluster (Figure 2A). In contrast, a single candidate for halogenation, SgcC3, was located upstream and distant from the NRPS locus. It only became apparent that SgcC3 was involved in chlorination of the β-amino acid moiety upon inactivation of sgcC3 and isolation of the expected deschloro analog 7.13 To unravel the molecular details of this transformation, we cloned sgcC3 and overproduced and purified the recombinant protein for in vitro characterization.
Initial activity tests revealed that SgcC3 does not catalyze the chlorination of free amino acid substrates, including 4. As a result, 6 was enzymatically prepared from 4 in vitro using the Svp PPTase,34 the SgcC1 adenylation enzyme, and the SgcC2 PCP, all of which have been previously characterized.13,14 After purification of the resulting aminoacyl-S-SgcC2 and prior to activity tests, hydrolysis of β-amino acids from SgcC2 was optimized to afford a stoichiometric release for detection by HPLC. Activity of SgcC3 was examined with NaCl as a halide donor, and a single, new peak was observed only when (i) exogenous FAD was supplied to the reaction mixture, (ii) an NADH-dependent flavin reductase was included to provide diffusible FADH2, and (iii) the reaction was performed in an aerobic environment. This new peak was confirmed to be 13 based on comparisons to synthetic standard and ESI-MS analysis. These results provided unambiguous evidence that SgcC3 is a FADH2, O2-dependent halogenase that requires a carrier protein-tethered substrate for activity. The cofactor and co-substrate requirements are similar to those observed for the halogenase PltA involved in pyoluteorin biosynthesis,27 and these two enzymes now represent a growing family of FADH2, O2, and carrier protein- dependent halogenases for which several have been predicted from bioinformatics analysis.16
We previously reported that SgcC1 specifically activates 4, but also activates other β-tyrosine analogues including 11, 3-chloro-β-tyrosine, 3-hydroxy-β-tyrosine and 3-chloro-5-hydroxy-β-tyrosine, albeit with minimally 25-fold less efficiency.13,14 This indicates that C-3 chlorination and C-5 hydroxylation of 4 occurs most likely after 4 is tethered to SgcC2, a prediction that is now confirmed by the finding that 4 is not a substrate for SgcC3. However, these results fell short of revealing any insight into the timing of halogenation after 4 is tethered to SgcC2. Therefore, both 12 and 3-hydroxy-β-tyrosyl-S-SgcC2 were enzymatically prepared by taking advantage of the promiscuous nature of SgcC1. Remarkably, SgcC3 catalyzes the chlorination of 12 into 16 with a specificity activity that is almost identical to that for 6. While this may appear to be unexpected, it in fact is consistent with the early finding that SgcC1 has a 25-fold selectivity for 4 over 11, serving as the “gate-keeper” that controls theβ-amino acid to be incorporated into 1 in vivo.13,14 In contrast, no chlorination was detected with 3-hydroxy-β-tyrosyl-S-SgcC2, the alternative substrate for SgcC3 if C-5 hydroxylation occurred prior to C-3 halogenation. This is consistent with 6 as the preferred substrate of SgcC3 and further supports the timing of the individual steps proposed for 3 biosynthesis (Figure 2B). Finally, similar to other halogenases, chloride can be substituted with bromide but not fluoride or iodide, and structural analysis of SgcC3 should provide insights into this selectivity.
In conclusion, SgcC3 has been shown to catalyze the regioselective chlorination of 6, and this activity is dependent on O2 and FADH2. The requirements for SgcC3 catalysis are consistent with the catalytic cycle proposed for FADH2, O2-dependent halogenases as depicted in Figure 5A. The fact that recombinant strains of S. globisporus can produce C-1027 analogs 7, 8, and 9 suggests enzymes downstream of SgcC3 have relaxed substrate specificity. Along with the results for SgcC3 provided here, application of precursor-directed biosynthesis and combinatorial biosynthesis methods to the C-1027 biosynthetic machinery presents a unique opportunity to generate novel C-1027 analogs some of which could have improved biological activity.
Supplementary Material
Full experimental details for the synthesis of β-tyrosine analogs as substrates and authentic standards of products, PCR primers used for amplification of the sgcC2, sgcC3, sgcE6, and E. coli fre genes for heterologous expression (Table S1), HPLC chromatogram for determination of the SgcC3 cofactor (Figure S1), ESI-MS spectra of (S)-3-chloro-β-tyrosine (Figure S2), and pH profile for SgcC3 (Figure S3). This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. Y. Li, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Beijing, China for the wild-type S. globisporus strain and the Analytical Instrumentation Center of the School of Pharmacy, University of Wisconsin-Madison for support in obtaining MS and NMR data. This work is supported in part by NIH grants CA78747 and CA113297. SVL is the recipient of an NIH postdoctoral fellowship (CA1059845).
Abbreviations used
- CoA
coenzyme A
- DSB
double-stranded breaks
- ESI-MS
electrospray ionization-mass spectrometry
- FAD
flavin adenine dinucleotide
- FMN
flavin adenine mononucleotide
- FADH2
reduced flavin adenine dinucleotide
- NADH
β-nicotinamide adenine dinuceotide, reduced
- NRPS
nonribosomal peptide synthetase
- PCP
peptidyl carrier protein
- PPTase
phosphopantetheinyl transferase
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
References
- 1.Hu J, Xue YC, Xie MY, Zhang R, Otani T, Minami Y, Yamada Y, Marunaka T. J Antibiot. 1988;41:1575–1579. doi: 10.7164/antibiotics.41.1575. [DOI] [PubMed] [Google Scholar]
- 2.Maeda H, Edo K, Ishida N, editors. Drug. Springer; Tokyo: 1997. Neocarzinostatin: the Past, Present, and Future of an Anticancer. [Google Scholar]
- 3.Schroeder DR, Closon KL, Klohr SE, Zein N, Langley DR, Lee MS, Matson JA, Doyle TW. J Am Chem Soc. 1994;116:9351–9352. [Google Scholar]
- 4.Otani T, Yasuhara T, Minami Y, Shimazu T, Zhang R, Xie MY. Agric Biol Chem. 1991;55:407–417. [PubMed] [Google Scholar]
- 5.Sugimoto Y, Otani T, Oie S, Wierzba K, Yamada Y. J Antibiot. 1990;43:417–421. doi: 10.7164/antibiotics.43.417. [DOI] [PubMed] [Google Scholar]
- 6.Sugiura Y, Matsumoto T. Biochemistry. 1993;32:5548–5553. doi: 10.1021/bi00072a008. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Zhen Y, Zhen Y, Goldberg IH. Biochemistry. 1995;34:12451–12460. doi: 10.1021/bi00038a044. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, Christenson SD, Standage S, Shen B. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy DR, Gawron LS, Ju J, Liu W, Shen B, Beerman TA. Cancer Res. 2007;67:773–781. doi: 10.1158/0008-5472.CAN-06-2893. [DOI] [PubMed] [Google Scholar]
- 10.Christenson SD, Liu W, Toney MD, Shen B. J Am Chem Soc. 2003;125:6062–6063. doi: 10.1021/ja034609m. [DOI] [PubMed] [Google Scholar]
- 11.Christenson SD, Wu W, Spies MA, Shen B, Toney MD. Biochemistry. 2003;42:12708–12718. doi: 10.1021/bi035223r. [DOI] [PubMed] [Google Scholar]
- 12.Christianson CW, Montavon TJ, Van Lanen SG, Shen B, Bruner SD. Biochemistry. 2007;46:7205–7214. doi: 10.1021/bi7003685. [DOI] [PubMed] [Google Scholar]
- 13.Van Lanen SG, Dorrestein PC, Christenson SD, Liu W, Ju J, Kelleher NL, Shen B. J Am Chem, Soc. 2005;127:11594–11595. doi: 10.1021/ja052871k. [DOI] [PubMed] [Google Scholar]
- 14.Van Lanen SG, Lin S, Dorrestein PC, Kelleher NL, Shen B. J Biol Chem. 2006;281:29633–29640. doi: 10.1074/jbc.M605887200. [DOI] [PubMed] [Google Scholar]
- 15.Van Pee KH. Arch Microbiol. 2001;175:250–258. doi: 10.1007/s002030100263. [DOI] [PubMed] [Google Scholar]
- 16.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 17.Vaillancourt FH, Yeh E, Vosberg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 18.Kelly WL, Boyne MT, II, Yeh E, Vosburg DA, Galonic DP, Kelleher NL, Walsh CT. Biochemistry. 2007;46:359–368. doi: 10.1021/bi061930j. [DOI] [PubMed] [Google Scholar]
- 19.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci US A. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 21.Galonic DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 22.Van Pee KH, Patallo EP. Appl Microbiol Biotech. 2006;70:631–641. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 23.Zehner S, Kotzsch A, Bister B, Sussmuth RD, Mendez C, Salas JA, Van Pee KH. Chem Biol. 2005;12:445–452. doi: 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Keller S, Wage T, Hohaus K, Holzer M, Eichhorn E, Van Pee KH. Angew Chem Int Ed. 2000;39:2300–2302. doi: 10.1002/1521-3773(20000703)39:13<2300::aid-anie2300>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 25.Dong C, Flecks S, Unversucht S, Haupt C, Van Pee KH, Naismith JH. Nature. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeh E, Garneau S, Walsh CT. Proc Natl Acad Sci USA. 2005;102:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc Natl Acad Sci USA. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeh E, Cole LJ, Barr EW, Bollinger JM, Ballou DP, Walsh CT. Biochemistry. 2006;45:7904–7912. doi: 10.1021/bi060607d. [DOI] [PubMed] [Google Scholar]
- 29.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 30.Holzer M, Burd W, Reissig HU, van Pee KH. Adv Synth Catal. 2001;343:591–595. [Google Scholar]
- 31.Wynands I, van Pee KH. FEMS Microbiol Lett. 2004;237:363–367. doi: 10.1016/j.femsle.2004.06.053. [DOI] [PubMed] [Google Scholar]
- 32.Tan CYK, Weaver DF. Tetrahedron. 2002;58:7449–7461. [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A Laboratory Mannual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. [Google Scholar]
- 34.Sanchez C, Du LC, Edwards DJ, Toney MD, Shen B. Chem Biol. 2001;8:725–738. doi: 10.1016/s1074-5521(01)00047-3. [DOI] [PubMed] [Google Scholar]
- 35.Eichhorn E, van de Ploeg JR, Leisinger T. J Biol Chem. 1999;274:26639–26646. doi: 10.1074/jbc.274.38.26639. [DOI] [PubMed] [Google Scholar]
- 36.Fieschi F, Niviere V, Frier C, Decout JL, Fontecave M. J Biol Chem. 1995;270:30392–30400. doi: 10.1074/jbc.270.51.30392. [DOI] [PubMed] [Google Scholar]
- 37.Wang XW, Hong X. Drugs of the Future. 1999;24:847–852. [Google Scholar]
- 38.Okuno Y, Otsuka M, Sugiura Y. J Med Chem. 1994;37:2266–2273. doi: 10.1021/jm00041a004. [DOI] [PubMed] [Google Scholar]
- 36.van Berkel WJH, Kamerbeek NM, Fraaije MW. J Biotech. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 37.Orser CS, Lange CC, Xun L, Zahrt TC, Schneider BJ. J Bacteriol. 1993;175:411–416. doi: 10.1128/jb.175.2.411-416.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details for the synthesis of β-tyrosine analogs as substrates and authentic standards of products, PCR primers used for amplification of the sgcC2, sgcC3, sgcE6, and E. coli fre genes for heterologous expression (Table S1), HPLC chromatogram for determination of the SgcC3 cofactor (Figure S1), ESI-MS spectra of (S)-3-chloro-β-tyrosine (Figure S2), and pH profile for SgcC3 (Figure S3). This material is available free of charge via the internet at http://pubs.acs.org.