

Abstract
The Mre11-Rad50-Nbs1 complex and autophosphorylated Ser1981-ATM are involved in recognizing and repairing DNA damage, such as double-strand breaks (DSBs). However, the role of these factors in response to stalled replication forks is not clear. Nucleoside analogues are agents that are incorporated into DNA during replication, which cause stalling of replication forks. The molecular mechanisms that sense these events may signal for DNA repair and contribute to survival, but are poorly understood. Cellular responses to both DSBs and stalled replication forks are marked by H2AX phosphorylation on Ser139 (γ-H2AX), which forms nuclear foci at sites of DNA damage. Here, concentrations of the nucleoside analogues, ara-C, gemcitabine, and troxacitabine, that inhibited DNA synthesis by 90% within 2 h were determined for each agent. Using γ-H2AX as a marker for changes in chromatin structure, we demonstrate that Mre11, Rad50, Nbs1 and phosphorylated ATM respond to nucleoside analogue-induced stalled replication forks by forming nuclear foci that co-localize with γ-H2AX within 2 h. Since neither DSBs nor single-strand breaks were detectable after nucleoside analogue exposure, we conclude that this molecular response is not due to the presence of DNA breaks. Deficiencies in ATM, Mre11, or Rad50 led to a two- to five-fold increase in clonogenic sensitization to gemcitabine, while Nbs1 and H2AX deficiency did not effect reproductive growth. Taken together, these results suggest that ATM, Mre11, and Rad50 are required for survival after replication fork stalling, whereas Nbs1 and H2AX are in-consequential.
Keywords: Stalled replication forks, DNA damage response, gemcitabine, ara-C, troxacitabine
INTRODUCTION
Endogenous and exogenous events that hinder replication fork progression threaten both cellular survival and genome integrity. Therefore, molecular mechanisms that monitor and regulate fork progression are necessary to safeguard DNA replication under these circumstances (1). However, this capability likely contributes to resistance to pharmacological agents that target DNA replication, such as those that cause steric hindrance (i.e. aphidicolin), decrease the deoxynucleotide pool (i.e. hydroxyurea), or block DNA synthesis of the nascent strand (i.e. nucleoside analogues). Upon cellular entry and phosphorylation to their active metabolites, many nucleoside analogues are incorporated into replicating DNA and cause replication forks to stall, as they are poor substrates for chain extension (2). Cells respond to these events by activating the ATR-Chk1-Cdk2 S-phase checkpoint pathway (3, 4), which blocks firing of new replication origins and enforces cell cycle arrest. This likely enables DNA repair mechanisms to react to sites of DNA damage and contribute to survival.
The molecules that recognize nucleoside analogue-induced stalled replication forks and signal for repair processes are not well characterized. Our prior studies demonstrated that cells respond rapidly to nucleoside analogue exposure by causing phosphorylation of the DNA damage responsive histone, H2AX, and the phosphoinositol kinase-like kinase, ATM (4). The function of H2AX is not fully understood, but it is likely involved in anchoring damage-response proteins to sites of DNA damage, while ATM is a regulator of DNA repair and cell cycle checkpoints (5). Because phosphorylation of H2AX and ATM are most closely associated with recognition of DNA double-strand breaks (DSBs, ref. 6), it was not expected that such pronounced responses would occur upon nucleoside analogue exposure. It is now well-established that H2AX is rapidly phosphorylated in response to additional forms of DNA damage, such as single-stand breaks (SSBs, 7), stalled replication forks (4, 8), and hyperthermia-induced stresses (9). The molecules that are associated with the recognition and repair of ionizing radiation-induced DNA damage have been well characterized and may offer insight into cellular responses to blocked DNA replication.
The Mre11-Rad50-Nbs1 (MRN) complex is involved in the cellular responses to DSBs, including non-homologous end joining and telomere maintenance, and may also function during normal DNA replication (10, 11). It is among the first responders to bind to DNA ends (12, 13) and has been demonstrated to tether DNA fragments through the self-association of Rad50 coiled-coil domains (14). Mre11 possesses 3’→5’ exonuclease and single strand endonuclease activities (15, 16), and therefore potentially has the capability to remove nucleoside analogues from DNA. The DNA damage response is a highly regulated concert of many molecules in which the MRN complex functions both upstream and downstream of ATM. Here, we examined molecules that are known regulators of the DSB-response to determine if they recognize nucleoside analogue-induced stalled replication forks and contribute to survival.
MATERIALS AND METHODS
Cell culture
The following cell lines were used: OCI-AML3 adult myelogenous leukemia cell line (gift from M. Andreeff, The University of Texas M. D. Anderson Cancer Center, Houston, TX) was maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA). Ataxia-telangiectasia (AT) fibroblasts, AT22IJET+vector, and those that have been repleted with full length ATM (AT22IJE-T+ATM, gifts from Y. Shiloh, Sackler School of Medicine, Tel Aviv, Israel, ref. 17) were maintained in DMEM plus 20% FBS. Nijmegen breakage syndrome (NBS) fibroblasts, GM7166VA7, and those repleted with full length Nbs1 (GM7166VA7+Nbs1, gifts from K. Komatsu, Kyoto University, Kyoto, Japan, ref. 18), and H2AX−/− and H2AX+/+ mouse embryonic fibroblasts (gifts from A. Nussenzweig, NCI, Bethesda, MD, ref. 19) were maintained in DMEM plus 15% FBS.
Chemicals and antibodies
The nucleoside analogues, gemcitabine and troxacitabine, were kindly provided by Dr. L. W. Hertel (Lilly Research Laboratories, Indianapolis, IN) and Dr. Henriette Gourdeau (Biochem Pharma, Montreal, Canada), respectively. The ara-C used in these investigations was purchased from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal antibodies used were purchased from Upstate Biotechnology (Charlottesville, VA; pSer139-H2AX, pSer1981-ATM), Abcam (Cambridge, MA; ATM), Novus Biologicals (Littleton, CO; Mre11), GeneTex (San Antonio, TX; Rad50), Lab Vision (Fremont, CA; Ku70, Ku80), and Sigma-Aldrich (St. Louis, MO; β-actin). Rabbit polyclonal antibodies were purchased from Bethyl Laboratories (Montgomery, TX; pSer139-H2AX) and Novus Biologicals (Littleton, CO;.Nbs1). Alexa Fluor 488, 594, and 680 fluorescent secondary antibodies were purchased from Molecular Probes (Eugene, OR).
Immunoblotting analysis
Cell lysate preparations and detection of specific proteins were done according to previously described procedures (4). The Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) was used according to the manufacturer's instructions for protein visualization.
DNA synthesis assay
Measurement of DNA synthesis was done as previously described (4). Briefly, [3H]thymidine (5 μCi/ml, 60.9 Ci/mmol; Moravek Biochemicals, Brea, CA) was added 30 min. prior to harvesting. Labeled cells were collected on glass fiber filters (Schleicher & Scheull, Riviera Beach, FL) and radioactivity retained on the filter was determined by a liquid scintillation counter (Packard, Ramsey, MN).
Confocal microscopy
OCI-AML3 cultures were centrifuged onto slides, fixed, and immunostained according to previously described procedures (4). Images were obtained using an Olympus FluoView IX71 confocal microscope system (Olympus, Melville, NY) and processed using FluoView FV500 ver. 5 software. The objective lens was a 40×/1.30 NA oil lens. A profile of overlapping red and green fluorescence was measured by the image processing and analysis software, ImageJ (National Institute of Health, Bethesda, MD). Pearson's correlation coefficient (r) was calculated for each image using the Olympus FluoView FV500 ver. 5 software (Olympus, NY).
Cell cycle analysis
Cells were washed with ice-cold PBS, fixed, and stained with propidium iodide as previously described (4). At least 10,000 cells were evaluated for fluorescence using a Becton Dickinson FACSCalibur flow cytometer (San Jose, CA).
Comet assay
OCI-AML3 cells were mixed with agarose, placed onto a microscope slide, and assessed for DNA damage, as previously described (20). Briefly, the slides were left in lysis solution for 1 h at 4°C in the dark, washed twice with 1× TBE buffer (neutral conditions) or incubated in alkaline solution (pH>13) for 1 h, and subjected to electrophoresis (300 mA) for 15 min at 4°C. Comet images analyzed by a computer-based image analysis system (Kinetic Imaging Komet system, Version 5.5, Nottingham, United Kingdom) for tail moment, DNA content, and percentage of DNA in the tail.
Pulsed-field gel electrophoresis (PFGE)
Exponentially growing cultures were pre-labeled with [14C]thymidine for 48 h (0.02 μCi/ml every 12 h × 4), exposed to drugs or ionizing radiation (3.17 Gy/min of 137Cs), and harvested immediately. Cells were washed, embedded in agarose plugs, lysed, digested with proteinase K, and the DNA was separated by PFGE using a CHEF-DR III system (Bio-Rad Laboratories), as previously described (21). The gel was stained with ethidium bromide and imaged using the ImageQuant software program (Molecular Dynamics, Sunnyvale, CA). Radioactivity was quantitated using the InstantImager system (PerkinElmer, Shelton, CT) after the gel had been dried at 60°C under vacuum.
siRNA transfections
AT22IJE-T+ATM cells were plated in 6-well plates and incubated overnight. Medium was replaced with fresh medium containing 100 nM siControl non-targeting siRNA (D-001810−10−20, Dharmacon), Mre11 siRNA (L-009271−00−0005, Dharmacon), Rad50 siRNA (M-005232−01, Dharmacon), or Nbs1 siRNA (L-009641−00−0005, Dharmacon) and DharmaFECT 1 transfection reagent (Dharmacon). Seventy-two hours after transfection, cells were harvested, and plated for clonogenic survival assays. The remaining cells were lysed and subsequently examined for protein knockdown by immunoblotting.
Clonogenic assays
Exponentially growing cultures were placed in a 6-well plate for 8−24 h before being exposed to the indicated drugs for one cell cycle period. Cells were then washed twice with PBS (37°C), fresh medium was added, and allowed to grow undisturbed for 7−10 days in normal growing conditions. Cells were fixed (50% methanol, 5% acetic acid, 45% H2O) for 1 h, stained with Accustain Giemsa (1:20 dilution; Sigma-Aldrich, St. Louis, MO) for 1 h, and washed twice with water. Colonies of 50 cells were counted using a dissecting microscope.
RESULTS
Inhibition of DNA synthesis after exposure to nucleoside analogues
The deoxycytidine nucleoside analogues, cytarabine (ara-C), gemcitabine, and troxacitabine (Figure 1A), were utilized to cause stalled replication forks in the acute myelogenous leukemia cell line, OCI-AML3. A range of nucleoside analogue concentrations was first investigated to determine what concentrations of each drug caused a similar inhibition of DNA replication (data not shown). Exposure of 0.5 μM ara-C, 0.1 μM gemcitabine, or 2 μM troxacitabine caused inhibition of DNA synthesis by approximately 70% within 1 h and 90% within 2 h, as measured by a decrease in [3H]thymidine incorporation (Figure 1B). The decrease in DNA replication caused by drug incorporation into DNA was associated with an accumulation of cells in S-phase (>60%) within 24 h, as determined by propidium iodide staining of DNA content (Figure 1C). These investigations demonstrate that the deoxycytidine nucleoside analogues, ara-C, gemcitabine, and troxacitabine, are incorporated into DNA, causing an early inhibition of DNA synthesis and eventual arrest in S-phase.
Figure 1. The effect of nucleoside analogues on DNA synthesis.

(A) Chemical structures of (i) 2’-deoxycytidine, (ii) ara-C, (iii) gemcitabine, and (iv) troxacitabine. (B) Effect of 0.5 μM ara-C (•), 0.1 μM gemcitabine (○), or 2 μM troxacitabine (▲) on DNA synthesis in OCI-AML3 cultures, as measured by [3H]thymidine incorporation after a 30-minute pulse. Error bars represent the standard deviation of the mean. (C) Accumulation of OCI-AML3 cells in early S-phase after exposure to 1 μM ara-C, 10 nM gemcitabine, or 250 nM troxacitabine for 24 h, as measured by DNA content (propidium iodide). Cell populations representing G1, S, and G2/M phases are appropriately marked.
Co-localization of phosphorylated ATM and the Mre11-Rad50-Nbs1 complex at sites of nucleoside analogue-induced DNA damage
The histone variant, H2AX, is rapidly phosphorylated and forms distinct nuclear foci in response to many forms of DNA damage, such as DSBs (22) and stalled replication forks (4, 8). We investigated if other molecules that are important for the sensing and repairing of DNA DSBs also accumulate at sites of nucleoside analogue-induced stalled replication forks. Short drug incubations were chosen to minimize the effect of DSBs possibly caused by the collapse of replication forks. OCI-AML3 cultures exposed to ara-C, gemcitabine, and troxacitabine, which caused an inhibition of DNA synthesis by 90% within 2 h (Figure 1B), demonstrated an increase in phosphorylation of Ser1981 on ATM (Figure 2A). Distinct pSer1981-ATM foci, which co-localized with γ-H2AX, were evident within 2 h of exposure to all three nucleoside analogues, as analyzed by confocal microscopy. Red-green profiles illustrate overlapping nuclear foci, as depicted by overlapping red and green fluorescence peaks, for a region in which a line was randomly drawn through the center of a nucleus (Figure 2A, bottom panels). Pearson's correlation coefficient (r) was calculated for each image to determine the relationship between phosphorylated ATM and γ-H2AX, where one represents 100% correlation and zero for no correlation (Table 1, ref. 23). The correlation of pSer1981-ATM and γ-H2AX significantly (p<0.001) increased from 30% to 61−71% after nucleoside analogue exposure, which was comparable to that found in cultures treated with 10 Gy of ionizing radiation (74%, Table 1).
Figure 2. Co-localization of phosphorylated Ser1981-ATM, Rad50, Nbs1, and Mre11 at sites of stalled replication forks.

Exponentially growing OCI-AML3 cultures were incubated with 0.5 μM ara-C, 0.1 μM gemcitabine, or 2 μM troxacitabine for 2 h or exposed to 10 Gy ionizing radiation, harvested, and subjected to fluorescent staining. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI) and are shown in the upper panels in blue. A single line was randomly drawn through the center of nuclei to generate a red and green fluorescence profile, as shown in the bottom panels. Overlapping peaks illustrate overlapping red/green nuclear foci. Representative images of two independent experiments taken by confocal microscopy of (A) pSer1981-ATM (green)/γ-H2AX (red), (B) Rad50 (green)/γ-H2AX (red), (C) Nbs1 (red)/γ-H2AX (green), and (D) Mre11 (green)/γ-H2AX (red), are shown.
Table 1. Pearson's correlation coefficients of DNA damage response proteins with γ-H2AX.
Exponentially growing OCI-AML3 cultures were incubated with 0.5 μM ara-C, 0.1 μM gemcitabine, or 2 μM troxacitabine for 2 h or exposed to 10 Gy ionizing radiation, harvested, and subjected to two-color fluorescent staining. The mean Pearson's correlation coefficients and standard deviations were calculated from at least six separate fields of view obtained from two independent experiments to evaluate the co-localization of proteins with γ-H2AX. Zero represents no co-localization and one represents 100% co-localization. P-values were determined for each condition, compared to untreated controls.
| pATM | Rad50 | Nbs1 | Mre11 | |
|---|---|---|---|---|
| control | 0.30 ± 0.14 | 0.55 ± 0.08 | 0.51 ± 0.16 | 0.66 ± 0.10 |
| ara-C | 0.61 ± 0.05*** | 0.71 ± 0.03*** | 0.72 ± 0.04** | 0.66 ± 0.06 |
| gemcitabine | 0.71 ± 0.09*** | 0.74 ± 0.06*** | 0.64 ± 0.08 | 0.74 ± 0.07 |
| troxacitabine | 0.68 ± 0.13*** | 0.69 ± 0.09** | 0.69 ± 0.05* | 0.75 ± 0.04* |
| 10 Gy IR | 0.74 ± 0.06*** | 0.80 ± 0.03** | 0.72 ± 0.13* | 0.71 ± 0.15 |
, p<0.05
, p<0.01
, p<0.001
Further investigations demonstrated that Mre11, Rad50, and Nbs1 form nuclear foci in OCI-AML3 nuclei that also co-localize at sites of stalled replication forks within 2 h of exposure to nucleoside analogues (Figure 2B-D). An increased association of Rad50 and Nbs1 with γ-H2AX was observed after exposure to these agents or ionizing radiation, as determined by increased co-localization of foci and Pearson's coefficient (Figure 2B-C, Table 1). DNA damage-associated foci containing Mre11 and γ-H2AX also increased after exposure nucleoside analogues or ionizing radiation, suggesting an Mre11 response (Figure 2D). High levels of Mre11-associated fluorescence was evident throughout the nuclei of untreated cells (Figure 2D, green) and likely explains a lack of an increase in Pearson's correlation coefficient between Mre11 and γ-H2AX under all conditions (Table 1). DNA damage-induced nuclear foci were found in only a fraction of cells exposed to nucleoside analogues (Figure 2, data not shown), which is likely because these agents cause stalled replication forks in cells actively undergoing DNA replication (∼35% of total cell population). Conversely, an increase in DNA damage foci was evident in a majority of cells after ionizing radiation treatment, which is not cell cycle specific (Figure 2A-D, right panels). A similar response was previously reported for γ-H2AX (4). Although ara-C, gemcitabine, and troxacitabine have slightly different overall mechanisms of action (2), differences in co-localization was not dependent on the agent used. These experiments demonstrate that molecules that are closely associated with the DSB response also co-localize at sites of nucleoside analogue-induced stalled replication forks.
Undetectable levels of DNA breaks caused by nucleoside analogues
We investigated if the molecular response to nucleoside analogue-induced DNA damage was associated with the presence of DSBs because the functions of ATM, H2AX, and the Mre11-Rad50-Nbs1 complex are most closely associated with the sensing, signaling, and repair of these lesions. OCI-AML3 cultures were exposed to nucleoside analogues or ionizing radiation and examined for the presence of DSBs by pulsed field gel electrophoresis (PFGE). Cultures exposed to nucleoside analogues for 2 h demonstrated similar levels of DSB induction compared to untreated controls, as determined by both fluorescence (Figure 3A, upper) and [14C]-labeling of DNA (Figure 3A, lower). Conversely, 10−40 Gy of ionizing radiation caused a 1.5- to 4-fold increase in DSB detection. Since, the number of DSBs caused by 10 Gy of ionizing radiation approached the lower-limit of detection using PFGE, cells were further examined by the comet assay under both neutral and alkaline conditions, as a potentially more sensitive method to measure DSBs and SSBs, respectively. Similar to PFGE experiments, OCI-AML3 cultures exposed to nucleoside analogues for 2 h did not demonstrate increased levels of either DSBs (Figure 3B) or SSBs (Figure 3C), compared to untreated controls. Conversely, 10 Gy of ionizing radiation caused an increase in mean tail moment of 2−3 fold (neutral conditions) and 13-fold (alkaline conditions), which likely represents the measurement of DNA damage caused by approximately 400 DSBs per cell (24). The limit of sensitivity for the comet assay has been estimated at approximately 50 strand breaks per diploid mammalian cell (25). As there are opportunities for nucleoside analogues to incorporate into an estimated 100,000 replication forks throughout S-phase, in which 10% are engaged in DNA synthesis at any one time (26, 27), it does not seem likely that the molecular response seen under these conditions is due to the presence of DNA breaks.
Figure 3. Undetectable levels of DNA breaks after nucleoside analogue exposure.
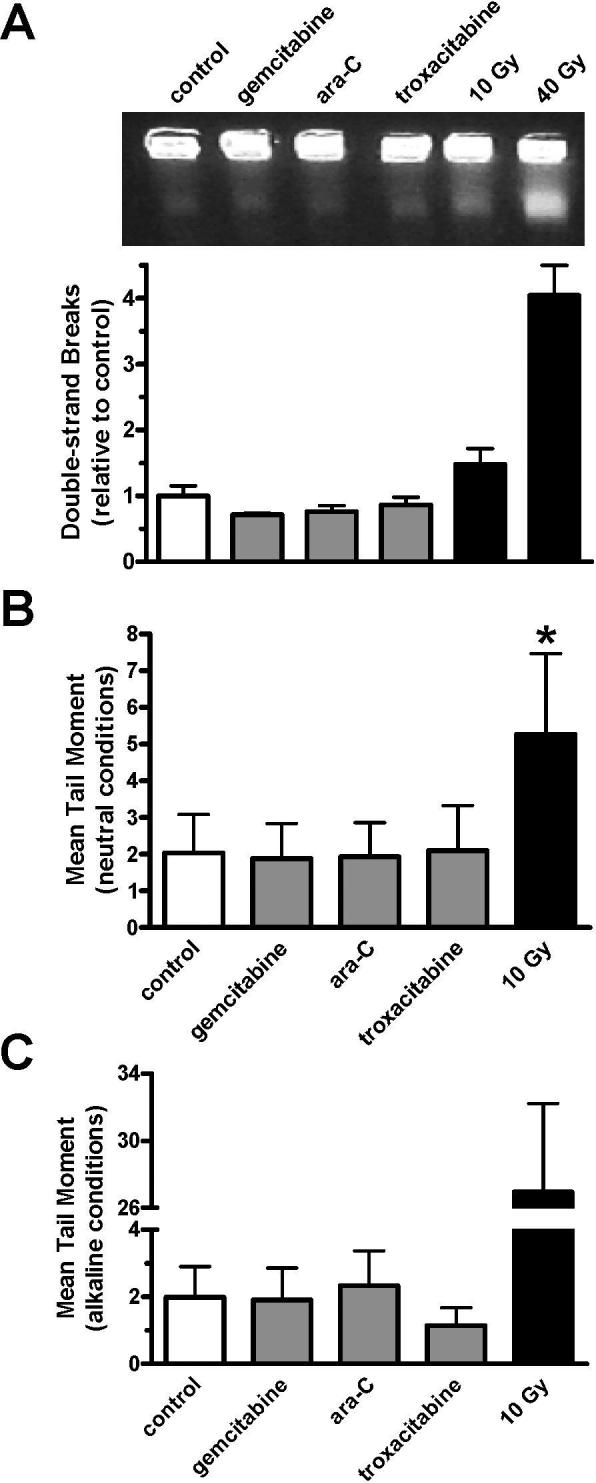
OCIAML3 cultures were incubated with 0.1 μM gemcitabine, 0.5 μM ara-C, or 2 μM troxacitabine for 2 h or exposed to 10−40 Gy ionizing radiation, harvested, and examined for DNA break induction. (A) Representative gel image of DNA stained with ethidium bromide in a typical experiment after PFGE (upper). The lower band represents the fraction of DNA with DSBs. The DNA in the lower band divided by the total DNA in each lane determined by [14C]-labeling, as compared to untreated controls is graphed. Error bars represent the standard error of the mean for two independent experiments performed in duplicate. (B-C) Calculated mean tail moment after exposure to nucleoside analogues or ionizing radiation, as determined by comet assay under neutral (B) or alkaline (C) conditions. Error bars represent the standard deviation of the mean for two independent experiments in which 100 cells per experiment were analyzed. *, P<0.001
The contribution of DNA damage sensor and repair molecules to survival
ATM, H2AX, Mre11, Rad50, and Nbs1 play important roles in sensing and repairing DSBs (10). Deficiencies in these DNA damage response molecules cause increased sensitivity to ionizing radiation (19, 28-31), implying that their functions relate to survival in regards to DSBs. To determine if these proteins contribute to survival after the formation of stalled replication forks, we analyzed cell lines that are deficient or have decreased levels of ATM, Mre11, Rad50, Nbs1, or H2AX for their sensitivity to a nucleoside analogue. When immortalized ataxia-telangiectasia (AT) fibroblasts deficient for ATM (Figure 4A) were exposed to gemcitabine, a significant difference in reproductive viability (P<0.001) was observed, as compared to the repleted line. Exposure of 10 nM gemcitabine for 24 h prior to drug removal killed 70% of AT cells whereas <10% of ATM repleted cells lost repopulation capacity (Figure 4A, middle). A 4.8-fold difference in gemcitabine sensitivity was observed between AT and AT+ATM cultures after exposure to a range of gemcitabine concentrations, as determined by IC50 values (Figure 4A, lower). These results are consistent with a previous report that demonstrated a 2-fold increase in sensitivity to gemcitabine after ATM knockdown using siRNA in A549 and HeLa cells (32).
Figure 4. Effect of ATM, Mre11, and Rad50 on colony growth after exposure to gemcitabine.

(A) Exponentially growing AT and AT+ATM fibroblasts were analyzed for ATM protein level by immunoblotting of whole cell lysates (upper), exposed to 10 nM gemcitabine for 24 h (middle) or a range of gemcitabine concentrations (lower) prior to analysis of clonogenic survival after 10 days. (B-D) Exponentially growing AT+ATM cultures were untreated, transfected with control siRNA, Mre11 siRNA, or Rad50 siRNA. Seventy-two hours after transfection, cells were harvested and split into 2 samples. One sample was lysed and saved for immunoblotting (B). Cells from the other sample were replated and allowed to grow undisturbed for 8 h before being exposed to 10 nM gemcitabine (B, lower) or 1−100 nM gemcitabine (C, D) for 24 h. Then, gemcitabine was washed out of the medium and colonies of untreated (◆), control siRNA (•), and Mre11/Rad50 siRNA (○) were counted after 10 days of undisturbed growth. The mean of two independent experiments performed in triplicate (n=6) ± S.E. is graphed. *, P<0.001. Gem.: 10 nM gemcitabine
In experiments in which Mre11 (85−90% knockdown, Figure 4B upper, Supplementary Figure 1) or Rad50 (75−78% knockdown, Figure 4B, middle) was depleted in the AT+ATM cell line using targeted siRNA, a 35−50% decrease in clonogenic survival was observed when cells were exposed to 10 nM gemcitabine, as compared to siRNA controls (P<0.001, Figure 4B, lower). When exposed to a range of gemcitabine concentrations, an approximate 2-fold increase in gemcitabine sensitivity was calculated in Mre11 and Rad50 depleted cells, as determined by calculated IC50 values (Figure 4C-D). Consistent with other reports (33, 34), a decrease in Mre11 protein caused by siRNA also led to a reduction in Rad50 (85%) and Nbs1 (40%), suggesting that Mre11 contributes to Rad50 and Nbs1 protein stability (Figure 4B, Supplementary Figure 1). Decreases in Mre11 did not affect protein levels of other DNA damage sensors, Ku70 or Ku80 (Figure 4B, upper). Neither the Mre11 nor the Nbs1 protein was affected following knockdown of Rad50 (data not shown).
Immortalized Nijmegen breakage syndrome (NBS) fibroblasts, which lack Nbs1 but were proficient for Mre11 and Rad50, did not demonstrate a difference in gemcitabine sensitivity, as compared to Nbs1-repleted cultures (Figure 5A). The lack of increased sensitivity was confirmed by using Nbs1-targeted siRNA in AT+ATM cultures (Figure 5B). Although Nbs1 has been reported to be required for the S-phase checkpoint in response to UV irradiation, ionizing radiation, and hydroxyurea (35, 36), Nbs1-deficient cells exposed to nucleoside analogues arrested in S-phase at similar concentrations as the paired Nbs1-proficient cell line (Supplementary Figure 2). Similarly, immortalized H2AX−/− mouse embryonic fibroblasts were not sensitized to gemcitabine (Figure 5C). This result supports previous data that the DNA damage response after nucleoside analogue exposure was not due to the presence of DSBs (Figure 3), as an increase in sensitivity is expected under DSB conditions (19). Figure 5D summarizes the IC50 values, 95% confidence intervals, and fold-differences in gemcitabine sensitivity for all cell lines. This investigation demonstrates that ATM, Mre11, and Rad50, but not Nbs1 and H2AX, contribute to cell survival and recovery from gemcitabine-induced stalled replication forks.
Figure 5. Effect of DNA damage response molecules on colony growth after exposure to gemcitabine.

Exponentially growing (A) NBS (○) and NBS+Nbs1 (•) human fibroblasts, (B) AT+ATM human fibroblasts that were untreated (◆), transfected with control siRNA (•), or exposed to Nbs1 siRNA (○) prior to drug exposure, and (C) H2AX−/− (○) and H2AX+/+ (•) mouse embryonic fibroblasts were exposed to a range of gemcitabine concentrations for one cell cycle (15−24 h) prior to fresh medium replacement. Cells were allowed to grow undisturbed for 5−10 days in normal growing conditions before colonies of ≥50 cells were counted. Mean of two independent experiments performed in triplicate are shown. Error bars represent the standard error of the mean. IR: 10 Gy ionizing radiation. (D) IC50 values, 95% confidence intervals (CI), and fold differences between compared conditions is summarized.
DISCUSSION
ATM and the Mre11-Rad50-Nbs1 complex are critical to sensing DSBs and initiating repair of damaged DNA. They may also function during DNA replication (11). Here, we report that these molecules respond to stalled replication forks, and that ATM, Mre11, and Rad50 are required for survival after such agitation. A previous study, demonstrated a similar increase in sensitivity after depletion of ATR, which is a known regulator of the DNA damage response to stalled replication forks (32). However, the relationship between ATR and the DNA damage response molecules investigated here is not well understood. Nbs1 and H2AX are not likely critically involved in the response to stalled replication forks since their absence did not affect reproductive viability (Figure 5). It has been reported that Nbs1 is required for the S and G2/M cell cycle checkpoints in response to UV irradiation, hydroxyurea, or ionizing radiation (35, 36). However, we did not find that Nbs1 was required for a nucleoside analogue-induced S-phase arrest (Supplementary Figure 2). Therefore, the molecular sensors investigated here likely have damage-specific functions and contribute to cell survival after inhibition of DNA synthesis by a different mechanism than signaling for cell cycle arrest. Differences may be due to the mechanisms that cause stalled replication forks. Nucleoside analogues physically block DNA synthesis of extending DNA strands, whereas UV and hydroxyurea cause fork stalling by other means. Because Nbs1 was not required for survival under these conditions, it is not clear if Mre11, Rad50, and Nbs1 function as a complex, as they do after DSBs, or individually in response to stalled replication forks.
Dysfunction of ATM or the MRN complex subunits results in embryonic lethality in eukaryotes and hypomorphic mutations are associated with a variety of human disorders, including ataxia-telangiectasia, ataxia-telangiectasia-like disorder and Nijmegen breakage syndrome (5, 34, 37, 38). Patients with these disorders display similar characteristics of cancer susceptibility, extreme sensitivity to ionizing radiation, and neurological defects. These phenotypes indicate that ATM, Mre11, and Nbs1 may play a role in normal DNA replication (11). In support of this hypothesis, it has been demonstrated Mre11 is required to prevent DSBs during normal chromosomal replication in Xenopus eggs extract (39). Further, Mre11 associates with chromatin in an S-phase specific manner and localizes to sites of ongoing DNA replication in human fibroblasts, as visualized by proliferating cell nuclear antigen (PCNA) and incorporated bromodeoxyuridine (40). The results reported here extend the functions of ATM, Mre11, and Rad50 to promoting cell survival in response to blocked DNA extension of nascent strands.
Although evidence supports that nucleoside analogues incorporate into DNA and exert a cytotoxic effect by blocking DNA synthesis (Figure 1, ref. 2), we sought to confirm that the molecular response observed here was not due to the presence of DNA breaks. While the assays were sensitive enough to detect DSBs caused by ionizing radiation, these lesions were not measurable after nucleoside analogue exposure (Figure 3). It is estimated that 1 Gy of ionizing radiation causes 40 DSBs per cell, resulting in a similar number of γ-H2AX nuclear foci (24). The same 1:1 ratio is not likely true in response to agents that cause stalled replication forks. An estimated 50,000 replication origins containing two forks each are brought together to form many replication factories at the start of replication in human cells (41, 42). Therefore, it is likely that the number of DNA damage nuclear foci observed here represent replication factories containing stalled replication forks. These factories are stably anchored in the nucleus but changes in the patterns occur during coordinated assembly and disassembly throughout S-phase (43). The pattern which forms after nucleoside analogue exposure resembles similar patterns reported previously during early S-phase DNA replication (Figure 2, ref. 44, 45), supporting our earlier finding that H2AX phosphorylation first occurs after nucleoside analogue treatment in cells initiating DNA synthesis (4).
The DNA repair mechanisms involved in removing nucleoside analogues from DNA are not well understood. Proof-reading 3’→5’ exonuclease activities associated with replicative DNA polymerases (46) and base excision repair processes (47) are capable of removing fraudulent nucleotides from DNA. However, a slow rate of drug removal (48) and sustained cell cycle arrest after exposure to nucleoside analogues (49) suggests that these mechanisms do not significantly promote survival. It is unknown what role ATM, Mre11, and Rad50 play at replication forks when DNA synthesis is inhibited. These molecules may have replication fork surveillance or DNA repair mechanisms that respond to chemotherapeutic agent-induced replication blocking, and thus contribute to drug resistance. Because Mre11 possesses 3’→5’ exonuclease and single strand endonuclease activities (15, 16), it may have the capacity to remove nucleoside analogues from DNA. Excision kinetics are enhanced when Mre11 is coupled with Rad50 (15), which may offer an explanation for the increase in gemcitabine sensitivity observed in Rad50-depleted cultures (Figure 4). Simplified models utilizing purified enzymes/enzyme complexes, oligonucleotides, and primer extension assays may be useful to answer these questions and may uncover a novel mechanism for the removal of nucleoside analogues from DNA, which is currently poorly understood (50).
Here, we present mechanistic evidence for the importance of a DNA repair pathway not previously known to be required for survival after the induction of stalled replication forks. These results provide a rationale for further studies to determine the mechanisms by which these molecules contribute to survival.
ACKNOWLEDGEMENTS
We thank Dr. Raymond Meyn and Colin Brooks for their assistance with PFGE investigations and Dr. V. Ashutosh Rao for his assistance with confocal microscopy data analysis.
Supported in part by grants CA32839, CA55164, and Cancer Center support grant CA16672 from the National Cancer Institute, Dept. of Health and Human Services
REFERENCES
- 1.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007;6:953–66. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene. 2003;22:9063–74. doi: 10.1038/sj.onc.1207229. [DOI] [PubMed] [Google Scholar]
- 3.Sampath D, Shi Z, Plunkett W. Inhibition of cyclin-dependent kinase 2 by the Chk1-Cdc25A pathway during the S-phase checkpoint activated by fludarabine: dysregulation by 7-hydroxystaurosporine. Mol Pharmacol. 2002;62:680–8. doi: 10.1124/mol.62.3.680. [DOI] [PubMed] [Google Scholar]
- 4.Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–48. doi: 10.1158/1535-7163.MCT-06-0633. [DOI] [PubMed] [Google Scholar]
- 5.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–10. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 7.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 9.Hunt CR, Pandita RK, Laszlo A, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res. 2007;67:3010–7. doi: 10.1158/0008-5472.CAN-06-4328. [DOI] [PubMed] [Google Scholar]
- 10.Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 2004;3:845–54. doi: 10.1016/j.dnarep.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 11.D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–27. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 12.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5:255–60. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 13.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Moreno-Herrero F, de Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature. 2005;437:440–3. doi: 10.1038/nature03927. [DOI] [PubMed] [Google Scholar]
- 15.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–79. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 16.Trujillo KM, Yuan SS, Lee EY, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem. 1998;273:21447–50. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- 17.Ziv Y, Bar-Shira A, Pecker I, et al. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–67. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 18.Tauchi H, Kobayashi J, Morishima K, et al. The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50-hMRE11-NBS1 complex DNA repair activity. J Biol Chem. 2001;276:12–5. doi: 10.1074/jbc.C000578200. [DOI] [PubMed] [Google Scholar]
- 19.Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res. 2001;7:3580–9. [PubMed] [Google Scholar]
- 21.Nishikawa T, Munshi A, Story MD, et al. Adenoviral-mediated mda-7 expression suppresses DNA repair capacity and radiosensitizes non-small-cell lung cancer cells. Oncogene. 2004;23:7125–31. doi: 10.1038/sj.onc.1207917. [DOI] [PubMed] [Google Scholar]
- 22.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103(Pt 3):857–62. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 24.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 2003;100:5057–62. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–9. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 26.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–95. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berezney R, Dubey DD, Huberman JA. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma. 2000;108:471–84. doi: 10.1007/s004120050399. [DOI] [PubMed] [Google Scholar]
- 28.Barlow C, Hirotsune S, Paylor R, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–71. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 29.Theunissen JW, Kaplan MI, Hunt PA, et al. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell. 2003;12:1511–23. doi: 10.1016/s1097-2765(03)00455-6. [DOI] [PubMed] [Google Scholar]
- 30.Luo G, Yao MS, Bender CF, et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci U S A. 1999;96:7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taalman RD, Jaspers NG, Scheres JM, de Wit J, Hustinx TW. Hypersensitivity to ionizing radiation, in vitro, in a new chromosomal breakage disorder, the Nijmegen Breakage Syndrome. Mutat Res. 1983;112:23–32. doi: 10.1016/0167-8817(83)90021-4. [DOI] [PubMed] [Google Scholar]
- 32.Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636–44. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 33.Olson E, Nievera CJ, Lee AY, Chen L, Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J Biol Chem. 2007;282:22939–52. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- 34.Stewart GS, Maser RS, Stankovic T, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–87. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 35.Stiff T, Reis C, Alderton GK, Woodbine L, O'Driscoll M, Jeggo PA. Nbs1 is required for ATR-dependent phosphorylation events. Embo J. 2005;24:199–208. doi: 10.1038/sj.emboj.7600504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim DS, Kim ST, Xu B, et al. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–7. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 37.Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–86. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 38.Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–76. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 39.Costanzo V, Robertson K, Bibikova M, et al. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell. 2001;8:137–47. doi: 10.1016/s1097-2765(01)00294-5. [DOI] [PubMed] [Google Scholar]
- 40.Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol. 2001;21:281–8. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frouin I, Montecucco A, Spadari S, Maga G. DNA replication: a complex matter. EMBO Rep. 2003;4:666–70. doi: 10.1038/sj.embor.embor886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meister P, Taddei A, Gasser SM. In and out of the replication factory. Cell. 2006;125:1233–5. doi: 10.1016/j.cell.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 43.Leonhardt H, Rahn HP, Weinzierl P, et al. Dynamics of DNA replication factories in living cells. J Cell Biol. 2000;149:271–80. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maser RS, Mirzoeva OK, Wells J, et al. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol. 2001;21:6006–16. doi: 10.1128/MCB.21.17.6006-6016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. Embo J. 2007;26:2719–31. doi: 10.1038/sj.emboj.7601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–7. [PubMed] [Google Scholar]
- 47.Chou KM, Kukhanova M, Cheng YC. A novel action of human apurinic/apyrimidinic endonuclease: excision of L-configuration deoxyribonucleoside analogs from the 3′ termini of DNA. J Biol Chem. 2000;275:31009–15. doi: 10.1074/jbc.M004082200. [DOI] [PubMed] [Google Scholar]
- 48.Kufe DW, Munroe D, Herrick D, Egan E, Spriggs D. Effects of 1-beta-D-arabinofuranosylcytosine incorporation on eukaryotic DNA template function. Mol Pharmacol. 1984;26:128–34. [PubMed] [Google Scholar]
- 49.Shi Z, Azuma A, Sampath D, Li YX, Huang P, Plunkett W. S-Phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 2001;61:1065–72. [PubMed] [Google Scholar]
- 50.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]