

Abstract
A series of β-benzylaspartate derivatives were prepared from N-trityl-L-aspartate dimethyl ester and evaluated as inhibitors of neuronal glutamate transporter EAAT3. The result of the structure-activity studies suggest that the position occupied by the aromatic ring of β-benzylaspartate within the binding site of EAAT3 may be different from that occupied by comparable groups in previously identified inhibitors, such as L-threo-benzyloxy aspartate (TBOA). Further, halogen substitutions at the 3-postition of the aromatic ring of β-benzylaspartate can increase the potency with which the analogues inhibit EAAT3.
Keywords: pharmacophore, bound conformation, diastereoselective, lipophilic pocket
1. Introduction
L-Glutamate is the most abundant excitatory neurotransmitter in the mammalian CNS and, as such, contributes to neuronal signaling and cognitive function through its activation of a wide variety of ionotropic and metabotropic excitatory amino acid (EAA) receptors 1. If extracellular glutamate concentrations in the CNS become excessive, it can lead to the over-activation of EAA receptors and the triggering of numerous neuropathological pathways 1–3. Termed excitotoxicity, glutamate-mediated neuronal injury is believed to contribute to CNS pathology in acute insults (ischemia, traumatic injury), as well as chronic neurological disorders (e.g., ALS, Alzheimer's disease, epilepsy, and Huntington's disease). Excitatory amino acid transporters (EAATs) present on neurons and glia play a critical role in regulating extracellular levels of glutamate and are thereby positioned to influence: i) the access of neurotransmitter to synaptic and extrasynaptic EAA receptors, ii) the recycling of the neurotransmitter and iii) the accumulation of excitotoxic levels of glutamate 4, 5. The expression of EAATs has also been found to be altered in neurological disorders such as epilepsy, ischemia, spinal cord injury, amyotrophic lateral sclerosis (ALS), Alzheimer’s and schizophrenia 6. Much of our understanding of the function of the EAATs has been dependent upon the development of substrates and inhibitors with which to probe transporter function and the physiological consequences of decreased activity.
Five distinct glutamate transporters (EAATs 1–5) have been identified by molecular cloning that, along with the sodium-dependent neutral amino acid transporters ASCT1-2, comprise a novel gene family (i.e., SLC1 in the Human Genome Organization HUGO nomenclature) 7. Uptake through the EAATs occurs via an alternate access mechanism that is electrogenic and driven by ionic gradients across the cell membrane. In this manner, the transport of one molecule of glutamate into a cell is stoichiometrically coupled to the import of three Na+ ions and one H+, and to the counter transport of one K+ 8. While the EAATs share a similar mechanisms and ionic dependence, each exhibits a distinct localization 4. EAAT1 and EAAT2 are primarily considered to be glial transporters that exhibit a preferential distribution in the cerebellum and forebrain, respectively. In contrast EAAT3 is present on neurons and is enriched in forebrain areas. EAAT4 is localized to purkinje neurons in the cerebellum and EAAT5 is restricted to the retina. To a large degree the EAATs can be pharmacologically distinguished from one another based upon the comparative actions of a number of substrates and inhibitors, although EAAT2 stands alone with respect to readily available, well-characterized subtype-selective inhibitors 5, 9. Not surprisingly, considerable effort has been focused on the development of inhibitors that can be used to selectively modulate the activity of the other individual transporters.
Toward this goal our laboratories prepared L-β-benzylaspartate (L-β-BA) and identified it as one of the few EAAT inhibitors that exhibits a marked selectivity for EAAT3 10. The more potent enantiomer, L-threo-β-benzylaspartate ((2S,3S) 3-benzylaspartate, L-t-β-BA), shows an approximate 10-fold preference for blocking EAAT3 compared to either EAAT1 or EAAT2 in C17.2 cells expressing the human isotypes. This contrasts with activity of the closely related and widely used inhibitor L-threo-benzyloxy aspartate (TBOA) which has been reported to more potently inhibit the activity of EAAT2 than EAAT3 11, 12, 14. In the present study a new series of L-β-benzylaspartate derivatives has been prepared from the easily accessible precursor N-trityl-L-aspartate dimethyl ester. The analogues were evaluated as inhibitors of 3H-D-aspartate uptake in C17.2 neuroprogenitor cells transiently transfected to express human EAAT3. We report that structure activity profile of these inhibitors is quite different from TBOA-based analogues suggesting that subtle variations in size, substituents and orientation of the aromatic ring appended to the β-position of aspartate can noticeably influence activity at the EAATs. In particular, substitutions at the 3-position of the aromatic ring of L-t-β-BA are most favorable for imparting potency at EAAT3.
2. Results and Discussion
2.1. Preparation of β-substituted Aspartates
The starting material N-trityl-L-aspartate dimethyl ester (2) was prepared following a general procedure. The aspartate dimethyl ester was formed using thionyl chloride in methanol. N-Tritylation is achieved with triphenylmethyl chloride and triethylamine 13. The resulting starting material was purified by two subsequent crystallizations from methanol. The triphenylmethyl protecting group was chosen due to ease of preparation, mild deprotection conditions, and the protection against alpha-carbon deprotonation it imparts to the molecule to avoid alpha-racemization (Scheme 1)
Scheme 1.

Synthesis of β-benzylaspartates. Tr = triphenyl methyl. See Table 1 and Experimental Section for base, E+, and conditions.
Initially, beta addition was achieved by the addition of lithium hexamethyldisilyl amide (LiHMDS, 2 equivalents in THF) to a 1M solution of 2 in anhydrous THF at −32°C under inert atmosphere. This was followed by quenching with the desired substituted-benzylic bromide (2–3 equivalents) and allowing the temperature to rise to 0°C 14. Substituted-benzylic bromides are shown in Table 2 as R, A–J. The reaction was terminated by the addition of 2N ammonium chloride. This produced a mixture of 3, SS:SR with SR as the major component in a 1:2 to 1:1 mixture of diastereomers. This was verified by detailed NMR analysis and compared to similar systems with similar reaction conditions reported that were accompanied by in-depth stereochemical determinations 15. HPLC separation of L-β-BA diastereomers 4A gave NMR spectra consistent with previous reports of similar aspartate syntheses 14–17. At this step, yields of the mixture ranged from 13.5% to 87% following chromatography. Yields generally improved with greater equivalents of substituted-benzylic bromide, Table 1: 5 vs. 6, as well as higher temperatures, 2 vs. 3, though diastereoselectivity decreased with increasing temperature. We found that the ratio of SS:SR increases to 1:11 if the temperature is decreased to −55°C and quenched at this cold temperature (entries 5 and 6, Table 1). If the reaction temperature is allowed to rise to 0°C after addition of substituted-benzylic bromide the ratio of SS:SR decreases to 1:2 to 1:1. Interestingly we found that adding DMPU to the starting material reverses the stereochemistry to give SS:SR in a 3:1 mixture of diastereomers 16.
Table 2.
Percent of control uptake in the presence of L-β-benzylaspartate derivatives (control uptake with no inhibitor present is 100%). C17.2 Cells assayed for their ability to block 25µM 3H-D aspartate uptake in the presence of 100µM inhibitor, n≥3. Inhibitors in an approximate 1:1 ration of (S,S) : (S,R) unless specified.
| Compound | R-group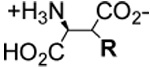
|
EAAT3 Uptake 3H-D-Asp (25 µM) (% of Control) | Compound | R-group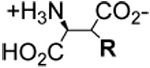
|
EAAT3 Uptake 3H-D-Asp (25 µM) (% of Control) |
|---|---|---|---|---|---|
| L-β-Benzyl-asp 4A | 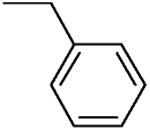 |
4 ± 2 | L-β-3-Br-benzyl-asp 4F | 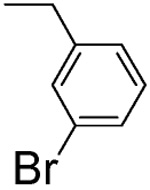 |
8 ± 3 |
| L-β-Benzyl-asp S,S-4i | 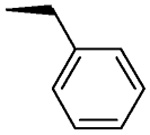 |
1 ± 1 | L-β-3-F-benzyl-asp 4G | 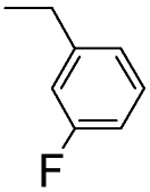 |
0 ± 0 |
| L-β-Methyl-2-napthyl-asp 4B | 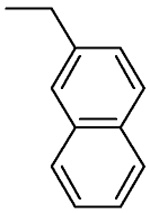 |
68 ± 6 | L-β-4-F-benzyl-asp 4H | 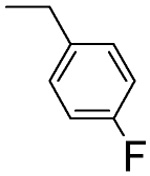 |
7 ± 3 |
| L-β-3,5 Dimethyl-benzyl-asp 4C | 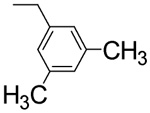 |
36 ± 7 | L-β-3-Nitro-benzyl-asp 4I | 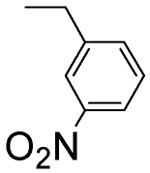 |
29 ± 5 |
| L-β-3-Methyl-benzyl-asp 4D | 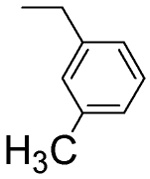 |
33 ± 3 | L-β-4-Nitro-benzyl-asp 4J | 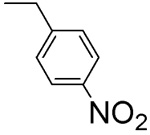 |
65 ± 8 |
| L-β-2,6- Dichloro-benzyl-asp 4E |  |
86 ± 5 |
Table 1.
Conditions used and results obtained in the benzylation reactions. S,S:S,R ratios as determined by 1H NMR.
| N-Protecting group + additives | Temp °C | Base: E+ | Time of rxn (hrs) | E+ | S,S : S,R (i:ii) | % yield |
|---|---|---|---|---|---|---|
| 1. Trityl | −35→0 | 2:2 LiHMDS |
4 | 3F | 1:2 | 57% |
| 2. Trityl | −55 | 2:2 LiHMDS |
6 | 3F | 1:11 | 55% |
| 3. Trityl | −55→0 | 2.5:3 LiHMDS |
6 | 3F | 1:2 | 65% |
| 4. Trityl DMPU | −55 | 2:3 LiHMDS |
21 | 3Cl | 3:1 | 74% |
| 5. Trityl | −65 | 2:2 LiHMDS |
21 | 3Cl | 1:11 | 68% |
| 6. Trityl | −65 | 2:3 LiHMDS |
21 | 3Cl | 1:11 | 76% |
| 7. Trityl | −55 | 2.1:2.3 KHMDS |
21 | 3Nitro | 99:1 | 51% |
| 8. Trityl | −55 | 2.1:2.3 KHMDS |
21 | 3F | 99:1 | 58% |
| 9. Trityl | −55→0 | 2.1:2.3 KHMDS |
21 | 3Br | 2.5:1 | 88% |
When KHMDS was used in place of LiHMDS we observed a switch in diastereoselectivity similar to that observed by Humphrey et al., with N-benzyl-N-9-phenylfluoren-9-yl dimethyl aspartate 14. We propose that Li complexes tightly to the enolate but may be forming a loose cyclic chelate between the enolate oxygen and the nitrogen (Figure 1), similar to that suggested by Fernàndez-Megía and Sardina 15. This is supported by the decrease in selectivity observed at higher temperatures, as well as the reversal of diastereoselectivity observed upon addition of DMPU.
Figure 1.

Low temperature chelation controlled diastereo-meric outcome of enolates using lithium and potassium bases.
This reversal of selectivity suggests that the metal is being complexed by DMPU, thereby disrupting the nitrogen oxygen chelate and allowing the molecule more rotational flexibility. This would increase the probability that the Si face is open for attack. We hypothesize that potassium forms a cyclic enolate - α-ester chelation as previously proposed 14, 15, giving the (S,S) product (Figure 1).
Deprotection was achieved by heating the alkylated product (3) in 6N HCl to afford the chloride salt of the product. The reaction was then neutralized to a pH of 7, loaded on an anion exchange resin (acetate form, 10g resin per gram product), and eluted using a gradient of 1–5N acetic acid. The zwitterion was obtained by concentrating the ninhydrin positive fractions followed by chasing the product with deionized water several times. Alternatively, the product can be precipitated out of ethyl acetate then water to give final product.
2.2. Inhibitory Activity at EAAT3
The compounds were evaluated as inhibitors of EAAT3 by quantifying their ability to reduce the uptake of 3H-D-aspartate in standard competition assays 10. In each instance the analogues (SS:SR ratio of approximately 1:1) were added simultaneously with the radiolabel to achieve a final concentration of 100 µM and 25 µM, respectively. The results are summarized in Table 2 as % of Control activity, i.e., uptake in the absence of any inhibitor. The replacement of the benzyl group on L-β-BA with a naphthyl moiety (4B) resulted in a marked reduction of inhibitory activity. Interestingly, when an analogous substitution is made with L-TBOA to produce L-threo-β-(1-naphthyl)methyloxaspartate (L-TNOA1) and L-threo-β-(2-naphthyl)methyloxaspartate (L-TNOA2), the inhibitory activity was retained (or increased) as reflected by a decrease in the IC50 with which the compounds blocked the uptake of 14C-glutamate into COS-1 cells expressing EAAT3 (i.e., L-TBOA, 7.0 µM; L-TNOA1, 4.8 µM; L-TNOA2, 6.5 µM) 11. This would suggest that the proposed lipophilic regions adjacent to the substrate binding domain on EAAT3 with which the benzyl groups of either L-TBOA or L-t-β-BA interact, can only accommodate a larger naphthyl group when its orientation is dictated by the methoxy linkage of TBOA and not the methylene linking group of L-t-β-BA. Recent structural insight into these substrate binding domains has emerged from crystallographic studies in which the archaeal aspartate transporter Gltph from Pyrococcus horikoshii has been crystallized in the presence of either L-aspartate or L-TBOA 18. It was concluded that the binding site is positioned between two hairpin loops that extend from opposite sides of the membrane and likely participate in the gating of substrate movement. While non-substrate inhibitors such as L-TBOA or L-t-β-BA can fit into this site normally occupied by L-glutamate, interaction with nearby lipophilic residues in the vicinity of the HP2 loop may preclude subsequent conformational movements that are necessary for substrate translocation, such as the closure of the external HP2 gate or providing access of external sodium to its requisite binding site. Interestingly, the ability of an inhibitor to interact with EAAT3 in this manner may increase its potency as an inhibitor, but decrease its ability to act as an alternative substrate. Comparisons between the naphthyl derivatives of L-TBOA or L-β-BA, suggest, however, that the exact placement of the aromatic rings within the binding site of EAAT3 is probably distinct and may additionally point to structural differences between the EAAT3 and EAAT2 subtypes.
The addition of a methyl (4D) or nitro (4I) group to the 3 position of L-β-BA, as well as a methyl group to both the 3 and 5 positions (4C), resulted in a retention of some inhibitory activity, although to a lesser degree than found in the parent molecule (e.g. about 30% of Control uptake vs. 4%). Again, this may be indicative of a more constrained binding arena surrounding the aromatic ring of L-β-BA when compared to L-TBOA. In contrast to these compounds, the di-substitution of chloro groups at the 2 and 6 positions (4E) produced an almost complete loss of inhibitory activity. In this instance the decreased ability to bind to EAAT3 may be attributable to steric clashes with the transporter itself or, given the locations on the aromatic ring, between the substituents and the carbon backbone of aspartate when it assumes the requisite conformation for binding. Comparisons among additions made at the 3 position revealed a rank order of inhibitory activity of: methyl (4D) ≈ nitro (4I) < bromo (4F) < fluoro (4G). Lastly, L-β-3-F-BA (4G) and L-β-4-F-BA (4H) inhibited EAAT3-mediated uptake to a similar or greater extent than observed with either L-β-3-Br-BA or the parent L-β-BA. This would be consistent with the steric argument that the lipophilic region interacting with the benzyl group of L-β-BA in the binding site of EAAT3 is spatially confined, as well as reflect the a ability of fluoro groups to favorably interact with both regional sidechain R groups and backbone peptide bonds 19.
To illustrate the diastereomeric preference of EAAT3 for ligand binding as well as highlight the diastereoselectivity of the benzylation reaction depending on base used, the individual diastereomers (S,S)4Gi and (S,R)4Gii of L-β-3-F-BA 4G were synthesized and evaluated as inhibitors of EAAT3. Competitive binding curves were obtained using 3H-D-asparate at 1µM and varying the concentration of inhibitor (Figure 2). Curve fitting using GraphPad Prism 4.0 software to the equation Y = 1/(1+10^(X-logIC50)), where Y is the fractional % of control and X is the log[Inhibitor] in M, IC50 values were obtained for the two diastereomers ((S,S)-3-F-βBA IC50 = 2.46+/−0.9 µM, (S,R)-3-F-βBA IC50 = 21.1+/−9.2 µM). In agreement with that reported previously, the (S,S) diastereomer is the more potent 10, 11, and in this case (S,S)4Gi exhibits a 10-fold higher affinity for EAAT3 than (S,R)4Gii.
Figure 2.

IC50 curves for (S,S)-3-F-βBA and (S,R)-3-F-βBA with [3H-D-aspartate] = 1 µM. (S,S)-3-F-βBA IC50 =2.46+/−0.9 µM (S,R)-3-F-βBA IC50 = 21.1+/−9.2 µM (IC50 +/− SEM).
2.3. Molecular Modeling
The recently published crystallographic study in which L-TBOA is bound to the archaeal aspartate transporter Gltph provides a strategy to conformationally compare these two inhibitors 18. The similarities are readily apparent when the bound conformation of L-TBOA is extracted from crystal structure and overlaid with a computationally minimized 10 conformation of L-threo-β-BA (Figure 3). While the aromatic rings are positioned in a similar direction it also appears that the benzyl group of L-threo-β-BA is considerably closer to the carbon backbone of the amino acid than that of L-TBOA. In this respect the aromatic rings of the two analogues are likely interacting with subtly different lipophilic regions within the binding domain on EAAT3. Our SAR data would suggest that in the instance of L-threo-β-BA, this region of the protein is less accommodating of steric bulk than the comparable region delineated by L-TBOA. Further, it is tempting to speculate that the ability of L-threo-β-BA to interact with these portions of the binding site may be a factor in its selectivity for EAAT3.
Figure 3.

Overlay of L-TBOA and L-threo-βBA using the bound conformation of L-TBOA and similar conformation of L-threo-βBA. Viewed by A) looking down perpendicular to the plane of the aromatic ring, and B) looking parallel to the plane of the aromatic ring.
3. Conclusions
A series of L-β-benzylaspartate derivatives have been prepared from N-trityl-L-aspartate dimethyl ester in which diastereomeric control was achieved using either lithium hexamethyldisilazide to yield predominantly the (2S,3R) adduct in one example or potassium hexamethyldisilazide to yield predominantly the (2S,3S) adduct in one example and evaluated as inhibitors of the neuronal glutamate transporter EAAT3. SAR data, including the inactivity of L-β-methyl-2-naphthyl-aspartate (4B) and the increased activity of L-β-3-F-benzylaspartate relative to the lead analogue L-β-benzylaspartate, suggest a confined interaction between lipophilic domains of EAAT3 and the aromatic moiety of L-β-benzylaspartate that is distinct from previously characterized inhibitors, such as L-TBOA. These finding will be further explored in pursuit of more potent and selective ligands for the EAAT subtypes.
4. Experimental Section
In order to verify the purity and composition of the compounds product analysis was performed by high resolution mass spectrometry. Intermediates were treated with trifluoroacetic acid prior to mass spectrometry to remove the trityl group so the signal of the molecular ions could be detected. Proton and carbon NMR were taken on a Varian 400 MHz NMR in CDCl3, D2O or DMSO with TMS as the internal standard. NMRs were reported as mixtures of SS:SR with each diastereomer being identified where possible. Diastereomers on carbon NMR are indicated in parentheses where carbon peaks of each molecule are resolved. Chirality was verified by optical rotation on a Perkin-Elmer 241 Polerimeter in 1N HCl unless otherwise specified in a 1.0dm tube. Reagents were purchased from Sigma (St. Louis, MO) and were used without further purification. Microanalyses were performed for C, H, and N (Midwest Microlab, Indianapolis, IN) and were within ± 0.4% of theoretical values. Compounds showing potency equal to or greater than L-β-Benzyl-Aspartate (approximate 1:1 S,S:S,R mixture) were resolved by semi-preparative reverse phase HPLC. The detection method was a Waters 486 tunable absorbance detector set at 254 nm.
4.1. N-trityl aspartate dimethyl ester (2)
L(+)-Aspartic acid (26 g, 100 mmol) was stirred in a round bottom flask in a 1M solution of methanol (200 ml). Thionyl chloride (22 ml, 140 mmol) was added dropwise. The solution was stirred at room temperature for 48 hours. The resulting dimethyl ester aspartate hydrochloride was washed 3 times with methanol, 3 times with methylene chloride, 2 times with toluene and once with water. The aspartate dimethyl ester was then suspended in methylene chloride (200ml) and dried with magnesium sulfate. Triphenylmethyl chloride (53 g, 95 mmol) was added followed by the dropwise addition of triethylamine (83.6 g, 300 mmol). The reaction was stirred overnight then diluted with ether and filtered through a silica plug with a 30/70 solution of ethyl acetate/hexanes. The filtrate was concentrated and rinsed 2 times with methylene chloride and recrystallized from methanol to yield N-trityl aspartate dimethyl ester 2 (70%). 1H NMR (400 MHz, CDCl3) δ: 7.54-7.48 (m, 6H), 7.32-7.15 (m, 9H), 3.77-3.71 (m, 1H), 3.69 (s, 3H), 3.28 (s, 3H), 2.99-2.91 (s, 1H), 2.73-2.62, (m, 1H), 2.58-2.48 (m, 1H). 13C NMR (100 MHz, CDCl3) δ: 173.86, 170.95, 145.61, 128.71, 127.83, 126.48, 71.13, 53.63, 51.91, 51.71, 40.16. [α]21D = 13.3° ( ethyl acetate).
4.2. General procedure for alkylation
Compound 2 was placed in a flame dried round bottom flask equipped with a stir bar. Anhydrous THF was added under argon and the solution was cooled. Once cooled 1M LiHMDS in THF (2–3 equivalents) was added via needle and syringe. After approximately 20 minutes the desired substituted-benzylic bromide (2–3 equivalents) was added. The temperature was then allowed to rise to 0°C (unless specified) and the reaction was stirred until quenched (4–24 hours) with 2N NH4Cl. Water and ethyl acetate (or ether) were added for separation. The water layer was subsequently washed three times and the organic layers were combined and concentrated. Flash chromatography, when needed, through silica gel using hexane:ethyl acetate in an approximate 85:15 ratio gave the desired protected substituted benzylaspartate in good to moderate yields.
4.3. N-tritylamino dimethyl ester β-benzylaspartate (3a)
To 2 (1.55 g, 3.84 mmol) in 1M THF under Argon at −23°C was added LiHMDS (9.6 ml, 11.53 mmol). After 15minutes benzyl bromide (1.15 ml, 9.6 mmol) was added and temperature allowed to rise to 0°C at which time it was stirred for a subsequent 2.5 hours. The reaction was quenched and ether was added for separation followed by a silica plug (85:15 Hexanes:ethyl acetate) (82.7%: 1.57 g). 1H NMR (400 MHz, CDCl3) δ: 7.57-7.48 (m, 6H), 7.32-7.20 (m, 14H), (3.94-3.90 (m, .5 H, S,S), 3.75-3.72 (m, .5 H, S,R)), (3.65 (s, 1.5H, S,S), 3.60 (s, 1.5H, S,R)), (3.28 (s, 1.5H, S,S), 3.23 (s, 1.5H, S,R)), 3.19-2.88 (m, 3H). 13C NMR (100 MHz, CDCl3) δ: 172.77, 172.51, 145.43, (139.15, 138.96), 129.12, 128.72, 127.71, 126.40, 126.22, (71.07, 70.93), (58.11, 57.67), 52.82, (51.65, 51.95), (51.53, 51.48), (34.05, 33.47). HRMS m/e calcd. for C13H17NO4+ 252.1236, found 252.1225.
4.4. N-tritylamino dimethyl ester β-2-naphthylmethyl-aspartate (3b)
To 2 (2.5 g, 6.2 mmol) in 1M THF under Argon at −35°C was added LiHMDS (18.6 mmol). 20 minutes later 2-naphthyl methyl bromide was added (3.4 g, 15.5 mmol) as a solid all at once. Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 4 hours before being quenched with 2N NH4Cl (9 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (1.68 g, 50%). 1H NMR (400 MHz, CDCl3) δ: 7.84-7.74 (m, 3 H), 7.66-7.59(m, 1 H), 7.47-7.43 (m, 7H), 7.37-7.29 (m, 1 H), 7.18-7.26 (m, 8H), 7.16-7.11(m, 1 H), 7.09-6.99 (m, 1 H) (3.91-3.87 (m, .4H, S,S), 3.73-3.70 (m, .6H, S,R)), (3.61 (s, 1.2 H, S,S), 3.56 (s, 1.8 H, S,R)), (3.27 (s, 1.2 H, S,S), 3.22 (s, 1.8 H, S,R)), 3.19-2.90 (m, 3H). 13C (100 MHz, CDCl3) δ: 173.20, 172.93, 145.76, 135.32, 133.38, 129.80, 129.07, 129.01, 128.93, 128.19, 128.08, 128.04, 127.96, 127.00, 126.81, 126.72, 126.64, (71.41, 71.31), 60.63, 52.98, 52.02, 49.66, 34.30. HRMS m/e calcd. for C17H20 NO4+ 302.1392, found 302.1388.
4.5. N-tritylamino dimethyl ester β-3,5 dimethyl-benzylaspartate (3c)
To 2 (3.2 g, 8.02 mmol) in 1M THF under Argon at −55°C was added LiHMDS (24 mmol). 20 minutes later 3,5-dimethylbenzyl bromide was added (6.4 g, 32.1 mmol) as a solid all at once. Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 1.5 hours before being quenched with 2N NH4Cl (12 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (61%, 3.9 g). 1H NMR (400 MHz, CDCl3) δ: 7.47 (m, 6H), 7.29-7.20 (m, 9H), 6.85-6.76 (m, 3H), (3.86-3.83 (m, .66 H, S,S), 3.72-3.68 (m, .33H, S,R)), (3.63 (s, 1H, S,S), 3.60 (s, 2H, S,R)), (3.25 (s, 1H, S,S), 3.20 (s, 2H, S,R)), 3.03-2.74 (m, 3H), (2.29 (5 H app. singlet, S,S), 2.17 (1H app. singlet S,R)). 13C (100 MHz, CDCl3) δ: (173.05, 172.96), (172.80, 172.69), 145.52, (139.05, 138.93), (137.78, 137.69), (128.81, 128.69), 127.93, 127.87, 127.78, 127.17, 126.72, 126.58, 126.48, 126.37, (71.16, 70.93), (58.19, 57.79), (52.86, 52.77), (51.78, 51.72), (51.65, 51.60), (33.93, 33.36), 21.21. HRMS m/e calcd. for C13H18NO4+ 280.1549, found 280.1540.
4.6. N-tritylamino dimethyl ester β-3 methyl-benzylaspartate (3d)
To 2 (1.86 g, 4.61 mmol) in 1M THF under Argon at −30°C was added LiHMDS (13.8 mmol) slowly. 20 minutes later 3 methyl benzyl bromide was slowly added (1.56 ml, 11.5 mmol). Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 4 hours before being quenched with 2N NH4Cl (7 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (1.05 g, 45%). 1HNMR (400 MHz, CDCl3) δ: 7.48-7.45 (m, 7H), 7.32-7.13 (m, 10H), 7.03-6.93 (m, 2H), (3.86-3.81 (m, .66H, S,R), 3.68-3.64 (m, p. obsc., .33H, S,S)), (3.63 (s, 1H, S,S), 3.59 (s, 2H, S,R)), (3.24 (s, 1H, S,S), 3.19 (s, 2H, S,R)), 3.08-2.77 (m, 3H), (2.33 (s, 2H, S,R), 2.30 (s, 1H, S,S)). 13CNMR (100 MHz, CDCl3) δ: 172.99, 172.74, 145.55, 139.15, 139.01, 129.71, 128.83, 127.81, 126.51, 125.82, (71.16, 70.98), (58.19, 57.79), (52.89, 52.86), (51.84, 51.77), (51.71, 51.65), (34.02, 33.46), 21.38. HRMS m/e calcd. for C14H19NO4+ 266.1392, found 252.1225.
4.7. N-tritylamino dimethyl ester β-2,6 dicloro-benzylaspartate (3e)
To 2 (4.35 g, 10.78 mmol) in 1M THF under Argon at −35°C was added LiHMDS (32.3 mmol). After stirring at −35°C for 15 minutes, 2,6-diclorobenzyl bromide was added by removing the septa and quickly adding the dry reactant (6.47 g, 26.95 mmol). Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 5 hours before being quenched with 2N NH4Cl (15 ml). Ethyl acetate was added for separation. No starting material was observed by TLC and NMR showed disappearance of starting material with no breakdown to aspartic acid. 1HNMR (400 MHz, CDCl3) δ: 7.53-7.49 (m, 6H), 7.29-7.15 (m, 11H), 7.10-7.06 (m, 1H), (3.94-3.90 (m, .5H, S,R), 3.78 (m, .5H, S,S)), (3.63 (s, 1.5H, S,S), 3.62, (s, 1.5H, S,R)), 3.55-3.38 (m, 2H), (3.20 (s, 1.5H, S,S), 2.19 (s, 1.5H, S,R)), 3.06-3.02 (m, 1H). 13CNMR (100 MHz, CDCl3) δ: (172.72, 172.51), (172.31, 172.17), 145.46, 135.75, 135.29, 134.90, 129.40, 128.84, 127.74, 127.22, 127.02, 126.42, 126.34, (71.25, 70.90), (58.08, 57.70), (51.86, 51.74), (51.68, 51.60), (50.11, 49.25), (30.28, 29.62). HRMS m/e calcd. for C13H15Cl2NO4+ 320.0456, found, 320.0441.
4.8. N-tritylamino dimethyl ester β-3 bromo-benzylaspartate (3f)
To 2 (2.27 g, 5.63 mmol) in 1M THF under Argon at −35°C was added LiHMDS (16.9 mmol) slowly. 20 minutes later 3-bromo-benzyl bromide was added (3.5 g, 14.1 mmol) as a solid all at once. Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 2.5 hours before being quenched with 2N NH4Cl (5 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (56%, 1.8 g). Spectra S,R, separated via column chromatography. S,R comes off first followed by S,S. Spectra reported for S,R 1HNMR (400 MHz, CDCl3) δ: 7.46-4.44 (m, 5H), 7.34-7.29 (m, 5H), 7.26-7.08 (m, 9H), 3.86-3.82 (m, 1H), 3.58 (s, 3H), 3.21 (s, 3H), 2.99-2.78 (m, 3H). 13CNMR (100MHz, CDCl3) δ: 173.04, 172.55, 145.68, 141.96, 132.15, 130.16, 129.72, 129.05, 128.11, 127.85, 126.82, 122.63, 71.34, 58.32, 52.77, 51.96, 33.21. HRMS m/e calcd. for C13H16BrNO4+ 330.0341, found 330.0336.
4.9. N-tritylamino dimethyl ester β-3 fluoro-benzylaspartate (3g)
To 2 (.5 g, 1.24 mmol) in 1M THF under Argon at −35°C was added LiHMDS (2.5 ml) slowly. 20 minutes later 3-fluoro-benzyl bromide was slowly added (.304 ml, 2.40 mmol). Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 4 hours before being quenched with 2N NH4Cl (3 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (57%, .363 g). 1HNMR (400 MHz, CDCl3) δ: 7.47-7.43 (m, 6H), 7.32-7.18 (m, 10H), 6.98-6.83 (m, 3H), (3.86-3.82 (m, .66H, S,R), 3.64-3.60 (m, .33H, S,S)); (3.64 (s, 1H, S,S), 3.59 (s, 2H, S,R)); (3.25 (s, 1H, S,S), 3.20 (s, 2H, S,R)); 3.05-2.87 (m, 3H). 13CNMR (100 MHz, CDCl3) δ: 173.04, 172.64, 161.81, 145.86, 142.09, 130.02, 129.05, 128.11, 126.82, 124.82, (116.13, 115.91), (113.62, 113.41), 71.32, 58.30, 52.86, 52.08, 51.99, 33.33. HRMS m/e calcd. for C13H16FNO4+ 270.1142, found 270.1141.
4.10. N-tritylamino dimethyl ester β-3 fluoro-benzylaspartate (3g, S,S)
Trityl aspartate (.555 g, 1.38 mmol) was placed in a flame dried round bottom flask equipped with stir bar. Anhydrous THF (5.5 ml) was added under argon and reaction was cooled to −35°C. Once cooled 20% KHMDS in THF (3.2 ml) was slowly added, 20 minutes later 3-Floro benzyl bromide was slowly added (.39 ml, 3.16 mmol). The reaction was stirred for an additional 21 hours before being quickly quenched with 2N NH4Cl (4 ml). Separate by adding water and ethyl acetate. Wash water layer two more times with ethyl acetate, combine organic layers and concentrate. Purification by chromatography on silica gel using ethyl acetate and hexanes 15:85 gave product in 58% yield (.407 g). 1HNMR (400MHz, CDCL3) δ: 7.51-7.46 (m, 7H), 7.27-7.20 (m, 11H), 7.00-6.86 (m, 1H), 3.69 (s, 3H), 3.64-3.60 (p.obsc. m, 1H-α), 3.28 (s, 3H), 2.95 (m, 1H-β); (2.69-2.65 (m, 1H), 2.54-2.50 (m, 1H,). 13CNMR (100MHz, CDCl3) δ: 173.04, 172.64, 161.81, 145.86, 142.09, 130.02, 129.05, 128.11, 126.82, 124.82, (116.13, 115.91), (113.62, 113.41), 71.32, 58.30, 52.86, 52.08, 51.99, 33.33.
4.11. N-tritylamino dimethyl ester β-3 fluoro-benzylaspartate (3g. S,R)
Trityl aspartate (.5 g, 1.24 mmol) was placed in a flame dried round bottom flask equipped with stir bar. Anhydrous THF (5 ml) was added under argon and reaction was cooled to −55°C. Once cooled LiHMDS in THF (2.5 ml) was slowly added, 20 minutes later 3-Floro benzyl bromide was slowly added (.3 ml, 2.48 mmol). The reaction was stirred for an additional 21 hours before being quickly quenched with 2N NH4Cl (4 ml). Separate by adding water and ethyl acetate. Wash water layer two more times with ethyl acetate, combine organic layers and concentrate. Purification by chromatography on silica gel using ethyl acetate and hexanes 15:85 gave product in 55% yield (.35 g). 1HNMR (400MHz, CDCL3) δ: 7.51-7.46 (m, 6H), 7.31-7.27 (m, 9H), 7.22 (d, 1H, J=7.33), 7.00 (d, 1H, J=7.33), 6.94-6.92 (m, 2H), 3.95 (m, .1H, S,S), 3.68 (m, .9H, S,R), 3.65 (s, .3H, S,S), 3.60 (s, 2.7H, S,R,), 3.27 (s, .3H, S,S), 3.22 (s, 2.7H, S,R), 3.08-2.85 (m, 3H).
4.12 N-tritylamino dimethyl ester β-4-fluoro-benzylaspartate (3h)
To 2 (.536 g, 1.33 mmol) in 1M THF under Argon at −30°C was added LiHMDS (2.66 mmol) slowly. 20 minutes later 4-fluoro-benzyl bromide was added (.33 ml, 2.66 mmol) dropwise. Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 2 hours before being quenched with 2N NH4Cl (6 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (.517 g, 76%). (Found: C, 75.06; H, 6.00; N, 2.74 C32H30FNO4 requires C, 75.13; H, 5.91; N, 2.74). 1H NMR (400 MHz, CDCl3) δ: 7.48-7.44 (m, 6H), 7.29-7.15 (m, 9H), 7.13-7.05 (m, 2H), 6.99-6.89 (m, 2H), (3.86-3.82 (m, .5H, S,R), 3.69-3.65 (m, .5H, S,S)), (3.63 (s, 1.5H, S,S), 3.58 (s, 1.5H S,R)), (3.24 (s, 1.5H, S,S), 3.19 (s, 1.5H, S,R)), 3.09-2.78 (m, 3H). 13C NMR (100 MHz, CDCl3) δ: (172.83, 172.78), 172.51, (172.71, 160.28), 145.44, (134.85, 134.82), (134.65, 134.62), (130.45, 130.38), (130.35, 130.27), 128.77, 128.65, 127.78, 126.52, 126.42, (115.26, 115.20), (115.05, 115.01), (71.13, 70.99), (58.05, 57.52), 53.00, (51.89, 51.75), (51.72, 51.65), (33.26, 32.67).
4.13. N-tritylamino dimethyl ester β-3-nitro-benzylaspartate (3i)
To 2 (3.546 g, 8.8 mmol) in 1M THF under Argon at −23°C was added LiHMDS (26.4 mmol) slowly. 20 minutes later 3-nitro-benzyl bromide was added (4.7 g, 21.97 mmol) as a solid all at once. Temperature was allowed to rise to 0°C, at which time it was stirred for an additional 4 hours before being quenched with 2N NH4Cl (13 ml). Ethyl acetate was added for separation, followed by silica column 85:15 hexane:ethyl acetate (13.5%, .64g). 1H NMR (400 MHz, CDCl3) δ: 8.20-8.10 (m, 3H), 8.07-8.04 (m, 1H), 7.60-7.17 (m, H), (3.95-3.90 (m, .75H, S,R), 3.76-3.70 (m, .15H, S,S)), (3.66 (s, .45H, S,S), 3.63, (s, 2.55H, S,R)), (3.28 (s, .45H, S,S), 3.25 (s, 2.55H, S,R)), 3.08-2.98 (m, 3H), 2.91 (s, .75H), 2.88 (s, .25H). 13C (100 MHz, CDCl3) δ: 172.68, 182.36, 145.32, 135.47, 135.40, 129.81, 129.25, 128.77, 128.63, 128.33, 128.18, 127.93, 127.87, 126.99, 126.69, 126.55, 123.88, 123.75, 121.64, 121.56, 71.23, (57.99, 57.59), 52.25, 52.12, 51.92, (33.71, 32.70). HRMS m/e calcd for C13H16N2O6+, 297.087, found 297.1076.
4.14. N-tritylamino dimethyl ester β-3-nitro-benzylaspartate (3i, S,S)
Trityl aspartate (.5 g, 1.34 mmol) was placed in a flame dried round bottom flask equipped with stir bar. Anhydrous THF (5 ml) was added under argon and the solution was cooled to −55°C. Once cooled KHMDS (20% in THF, 2.6 mmol) was slowly added, 20 minutes later 3-nitro-benzyl bromide was added (.634 g, 2.9 mmol) as a solid all at once. The reaction was stirred for an additional 21 hours before being quenched with 2N NH4Cl (6 ml). Ethyl acetate was added for separation. The water layer was washed two more times and the organic layers were concentrated down for separation on silica in 15% ethyl acetate, 85% hexanes (51%, .34 g). 1H NMR (400 MHz, CDCl3) δ: 8.26-8.00 (m, 1H), 7.52-7.37 (m, 4H), 7.52-7.16 (m, 15H), 3.63 (s, 3H), 3.58 (m, 1H β), 3.30 (s, 3H), 3.27-3.25 (m, 1H α), 3.22-3.15 (m, 2H), 3.03-2.94 (m, 2H). 13C (100 MHz, CDCl3) δ: 172.36, 145.34, 141.06, 135.47, 129.01, 128.77, 128.63, 127.87, 127.66, 126.55, 123.75, 121.64, 71.23, 57.59, 52.24, 52.10, 51.91, 33.71.
4.15. N-tritylamino dimethyl ester β-P-nitro benzylaspartate (3j)
Compound 2 (3.123 g, 7.74 mmol) in 1 M anhydrous THF under argon was cooled to −40°C. Once cooled 1M LHMDS (23.2 mmol) was added. After 20 minutes P-nitro benzyl bromide, dissolved in anhydrous THF, was added (4.18 g, 19.35 mmol). The temperature was then allowed to rise to 0°C and the reaction was stirred for 4 hrs at which time the reaction was quenched with 2N NH4Cl (10ml). Water and ether were added for separation. The water layer was subsequently washed three times and the ether layers were combined and concentrated down. Chromatography through silica gel (90% hexanes 10% ethyl acetate and .5% triethyl amine), methylene chloride was for easier loading to give (3.63g, 87.0%) as a yellow solid. 1H-NMR (400 MHz, CDCl3) δ: (8.14(d, 1.55 H, S,R, J=8.06), 8.08 (d, .44H, S,S, J=8.06)), 7.43 (dd, 6H, J=7.33, 8.79), 7.34 (d, 2H, J=8.79), 7.28 (dd, 2H, J=7.33), 7.26 (m, 2H), 7.26 (d, 1.55H, S,R, J=8.06), 7.22 (d, .44H, S,S, J=8.06), 7.21 (d, 2H, J=7.33), 7.19, (d, 1H, J=7.33). 3.90-3.87 (m, .8H, S,R), (3.63 (s, .65H, S,S), 3.57 (s, 2.33H, S,R)), (3.25 (s, .65H, S,S), 3.24 (d, 2.33H, S,R)), 3.03-2.94 (m, 3H). 13C (100 MHz, CDCl3) δ: 172.65, 171.98, 145.34, 129.83, 128.77, 127.92, 126.69, 123.62, 58.03, (52.25; 51.92), (32.94, 30.88). HRMS m/e Calcd for C13H16N2O6+, 297.087, found 297.1076.
4.16. N-tritylamino dimethyl ester β-3-chloro benzylaspartate
Trityl aspartate (.50 g, 1.2 mmol) was placed in a flame dried round bottom flask equipped with stir bar. Anhydrous THF (5 ml) was added under argon and the solution was cooled to −65°C. Once cooled 1M LHMDS in THF (2.5 ml) was slowly added, 20 minutes later 3-cloro-benzyl bromide was added (.47 ml, 3.6 mmol) at which time the reaction was stirred for an additional 21 hours before being quenched with 2N NH4Cl (6 ml). Ethyl acetate was added for separation. The water layer was washed two more times and the organic layers were concentrated down for separation on silica in 15% ethyl acetate, 85% hexanes (63%, .654 g). 1H NMR (400 MHz, CDCl3) δ: 7.46-7.42 (m, 6H), 7.25-7.22 (m, 7H), 7.19-7.16 (m, 5H), 7.06-7.05 (m, 1H), (3.85-3.83 (m, 1H), (3.61 (s, .3H, S,S), 3.56 (s, 2.7, S,R)), (3.23 (s, .3H, S,S), 3.19 (s, 2.7, S,R)), 3.02-2.78 (m, 3H). 13C (100 MHz, CDCl3) δ: 173.10, 172.66, 146.81, 145.37, 141.31, 134.05, 129.59, 128.93, 128.74, 127.81, 127.13, 126.54, 71.02, 58.00, 52.48, 51.77, 51.71, 32.94. HRMS m/e calcd. for C13H16ClNO4+ 286.0846, found 286.0840.
4.17. General Procedure for Deprotection
The protected β-substituted aspartate was taken up in 6N HCl and refluxed for 3–12 hours. The reaction was then diluted with water and washed three times with ether. The water layer was concentrated and neutralized with NH4OH and loaded onto an Ag 1X 8 acetate form ion exchange resin in the ratio of 1g product to 10g resin. The product was eluted with varying concentrations of acetic acid from .1 to 5N. Product came off around 2N. Water was lyophilized 2–3 times to remove the acetic acid.
4.18. β-Benzylaspartate (4A)
The protected benzyl aspartate (1.496 g) was taken up in 6N HCl and refluxed for 3 hours. Product came off at 2N as indicated by NMR (8.6 %, .058 g). 1H NMR (400 MHz, D2O) δ: 7.32-7.21 (m, 2H), 7.25-7.21 (m, 3H), (4.04 (d, .25H, S,S, J=3.88), 3.96 (d, .75H, S,R, J=3.88)), (3.40 (ddd .75H, S,R, J=3.88, 6.47, 9.06), 3.24 (ddd, .25H, S,S, J=3.88, 6.47, 9.06)) 3.09-3.01 (m, 1H), 2.95-2.89 (m, 1H). 13C (100 MHz, CDCl3) δ: 172.69, 169.64, (139.69, 138.69), 129.07, 128.42, 126.54, (54.85, 52,91), (49.66, 47.87), (33.46, 32.99). HRMS m/e calcd. for C11H14NO4+ 224.0923, found 224.0923. [α]21D = +29.8°.
4.19. S,S L-β-Benzylaspartate (4A)
The solvent system comprised of a buffered solution of ammonium acetate and water (0.1 M at pH 6.4). Reversed phase C18 3μ analytical and 10μ semi-prep columns were used with a retention time for S,S being 2.59 minutes and 16.00 minutes, respectively and for S,R 3.19 minutes and 21.00 minutes, respectively. 1H NMR (400MHz, D2O) δ: 7.29-7.25 (m, 2H), 7.21-7.17 (m, 3H), 3.89 (d, 1H, J=3.24), 2.99 (ddd, 1H, J=3.24, 4.53, 11.00), 2.79-2.66 (m, 2H). 13C NMR (400MHz, D2O) δ: 22.20, 33.46, 49.41, 55.96, 126.96, 128.96, 139.02, 176.693. S,S: [α]25D = +14.11.
4.20. β-2 Naphthylmethylaspartate (4B)
The protected β-alkylated aspartate (1.68 g, 3.09 mmol) was taken up in 6N HCl and refluxed for 4 hours. Ethyl ether was added for separation; the water layer was washed 3 times and concentrated. Product crashes out of solution upon neutralization with NaOH. The pH was adjusted to about 12 and precipitated out of methanol (.5g, 51%). 1HNMR (400MHz, D2O) δ: 7.83-7.78 (m, 3H), 7.66 (app.singlet, 1H), 7.47-7.35 (m, 3H), 3.60 (m, 1H, S,S), 3.27-3.25 (m, 1H, S,R), 3.03-2.94 (m, 1H), 2.86-2.83 (m, 1H), 2.69-2.60 (m, 1H). 13CNMR (100MHz, DMSO) δ: (172.93. 172.82), (170.44, 170.12), 136.99, 133.65, 132.50, 129.93, 129.29, 128.29, 128.13, 127.70, 127.50, 126.65, 126.11, (55.27, 53.18) (50.54, 48.62), (33.53, 31.39). 18.27 = acetic acid. HRMS m/e Calcd for C15H16NO4+ 349.9889, found 349.9902. [α]21D = +9.8.
4.21. β-3,5 Dimethyl-benzylaspartate (4C)
The protected β-alkylated aspartate (1.29 g, 2.47 mmol) was taken up in 6N HCl and heated for 24 hours at 70°C. Ethyl ether was added for separation; the water layer was washed and concentrated. NH4OH was added to adjust the pH to 7 and loaded onto an anion exchange resin. Product came off at 2N (.032 g, 8%). 1H NMR (400 MHz, D2O) δ: (6.92 (app. Singlet, .67H, S,R.); 6.91 (app. Singlet, .33H, S,R)), (6.88 (1.33H, app. Singlet, S,R,); 6.87 (.66H, app singlet, S,S)), (4.08-4.07 (m, .33H, S,S), 4.00-3.99 (m, .66H, S,R)), (3.45-3.40 (m, .66H, S,R), 3.26-3.21 (m, .33H, S,S)), 3.05-2.98 (m, 1H), 2.89-2.81 (m, 1H), (2.18 (br singlet, 5H), 2.12 (br singlet, 1H S,S)). 13C (100MHz, D2O) δ: (172.54,172.07), (169.43,169.37), (138.52,138.20), (137.34,137.14), (128.05,127.87; 126.78, (52.68,52.36), (47.90,47.46), (34.23, 32.03), 21.00. HRMS m/e calcd. for C13H18NO4+ 252.1236 found 252.1230. [α]21D = +34.8°.
4.22. β-3 Methyl-benzylaspartate (4D)
The protected β-alkylated aspartate (.9 g, 1.77 mmol) was taken up in 6N HCl and heated for 24 hours at 70°C. Ethyl ether was added for separation; the water layer was washed and concentrated. NH4OH was added until the pH reached 6. THF was added for solubility and the product was loaded onto an Ag 1X 8 acetate form ion exchange resin in the ratio of 1g product to 10g resin. Product was eluted with varying concentrations of acetic acid from .1 to 5N in a 4:1 ratio of water:THF. Product comes off around 2N. (.0765 g, 18.3%). 1HNMR (400MHz, D2O) δ: 7.24-7.19 (m, 1H), 7.11-7.05 (m, 3H), (4.14-4.13 (m, .36H, S,S), 4.09-4.08 (m, .64H, S,R)), (3.52-3.47 (m, .64H, S,R), 3.31-3.28 (m, .36H, S,S)), 3.12-3.07 (m, 1H), 2.98-2.88 (m, 1H), 2.25 (s, 3H). 13CNMR (100MHz, CDCl3) δ: 174.89, 170.46, 139.13, 137.49, 129.78, 128.96, 127.93, 126.06, 52.86, (47.55, 46.88), (33.76, 33.65), 20.47. HRMS m/e calcd. for C12H16NO4+ 238.1079, found 238.1079. [α]21D = +7.8° (in DI H2O, adjusted to 7.5 with NaOH).
4.23. β-2,6 Dicloro-benzylaspartate (4E)
The protected β-alkylated aspartate was dissolved in THF and 6N HCl and refluxed for 5 hours. The reaction was washed with ethyl acetate and the water layers were combined and concentrated. The reaction was neutralized to pH 5 using NaOH, loaded onto an anion exchange resin and eluted with 4:1 H2O:THF mixture followed by a gradient of acetic acid prepared from concentrated acetic acid in the same 4:1 H2O:THF ratio to give a gradient from .1N to 5N. Product was isolated from 2N and 5N acetic acid (1g, 37.2%). 1HNMR (400MHz, D2O) δ: 7.37-7.32 (m, 2H), 7.21-7.14 (m, 1H), (4.05 (m, .4H, S,S), 3.96 (m, .6H, S,R)), 3.47-3.33 (m, 2H), 3.24-3.21 (m, .6H), 3.12-3.09 (m, .4H)‥ 13CNMR (100MHz, CDCl3) δ: 171.75, 169.40, 135.23, 134.03, 129.18, 128.37, 52.80, 46.16, 28.07. HRMS m/e calcd. for C11H12Cl2NO4+ 320.0456, found 320.0441. [α]21 D = +31.7°.
4.24. β-3 Bromo-benzylaspartate (4F)
The protected β-alkylated aspartate(.5 g, .873mmol) was taken up in 6N HCl and heated for 24 hours. Ethyl ether was added for separation; the water layer was washed 3 times and concentrated. NH4OH is added until the pH is 7. THF is added for solubility and is loaded onto an Ag 1X 8 acetate form ion exchange resin in the ratio of 1g product to 10g resin. Product is eluted with varying concentrations of acetic acid from .1 to 5N in a mixture of water:THF in a ratio of 4:1. Product comes off around 2N. Lyophilize off water 2–3 times to remove the acetic acid (.1 g, 38.5%). 1HNMR (400 MHz, DMSO) δ: 7.47 (m, 1H), 7.41-7.38 (m, 1H), 7.28-7.22 (m, 2H), 4.01 (br s, .85H, α S.S) 3.82, (br s, .15H, α S,R), 3.25-3.21 (m, .15H, β S,R), 3.12-3.10 (m, .85H, β S,S), 3.04-3.00 (m 1H), 3.28-2.74 (m, 1H). 13CNMR (100MHz, DMSO) δ: 171.80, 169.23, 141.69, 131.65, 130.42, 129.27, 128.22, 121.52, 52.68, (48.16, 47.35, β C), (31.62, 30.76, CH2), acetic acid 21.15, 172.33. HRMS m/e calcd for C11H13BrNO4+ 302.0028, found 302.0014. [α]21D = +26.9°.
4.25. β-3 Fluoro-Benzylaspartate (4G)
The protected β-alkylated aspartate (.678 g, 1.33 mmol) was taken up in 6N HCl and heated for 24 hours at 70°C. Ethyl ether was added for separation; the water layer was washed 3 times and concentrated. NH4OH was added until the pH reached 7 and loaded onto an ion exchange resin where product comes off around 2N (.115 g, 36.2%). 1H NMR (400 MHz, DMSO) δ: 7.34-7.29 (m, 1H), 7.08-6.99 (m, 3H), (3.99 (d, .33H, S,S, J=4.40), 3.88 (d, .66H, S,R, J=4.40)), (3.33 (ddd, .66H, S,R, J=4.40, 6.59, 9.52), 3.20 (ddd, .33H, S,S, J=4.40, 5.86, 10,26)), 3.08-2.86 (m, 2H). 13C (100 MHz, DMSO) δ: (172.54, 171.89), 169.17, (163.18, 160.77), (141.60, 141.53), (130.15, 130.03, 129.94), 125.05, (115.63, 115.42), (113.31, 113.16, 112.94), (52.97, 52.48), (48.89, 47.25), (33.70, 32.03, 30.65). HRMS m/e for C11H13FNO4+ calcd. 242.0829, found 242.0834. [α]21D = +40°.
The mixture of diasteromers was resolved by HPLC as well as synthetically prepared in ratios of 99:1, S,S:S,R and 11:1 S,R:S,S. For HPLC resolution the solvent system comprised of a buffered solution of ammonium acetate and water (0.1 M at pH 6.4). Reversed phase C18 3u analytical and 10u semi-prep columns were used with a retention time for S,S being 2.3 minutes and 18.48 minutes, respectively and for S,R 2.8 minutes and 27.22 minutes, respectively.
4.26. β-3 Fluoro-Benzylaspartate (4G, S,S)
1H NMR (400 MHz, D2O) δ: 7.30-7.26 (m, 1H), 7.06-6.94 (m, 3H), 3.94 (m, 1H), 3.08-3.05 (m, 1H), 2.88-2.81 (m, 1H), 2.77-2.73 (m, 1H). 13C (100 MHz, DMSO) δ: 178.20, 172.02, 164.62, 162.21, 143.35, 131.50, 126.19, 116.81, 114.49, 56.53, 49.31, 33.91. [α]21D = +18.3°
4.27. β-3 Fluoro-Benzylaspartate (4G, S,R)
1H NMR (400 MHz, D2O) δ: 7.32-7.25 (m, 1H), 7.05-6.95 (m, 3H), 3.64-3.59 (m, 1H), 3.07-3.02 (m, 1H), 2.96-2.88 (m, 2H). 13C (100 MHz, DMSO) δ: 179.81, 172.63, 163.22, 160.81, 140.12, 129.93, 125.11, 115.43, 112.90, 52.68, 49.74, 28.22. [α]21D = +38.23°
4.28. β-4 Fluoro-benzylaspartate (4H)
The protected β-alkylated aspartate (.457 g, .893 mmol) was taken up in 6N HCl and heated for 24 hours at 75°C. Ethyl ether was added for separation; the water layer is washed and concentrated. NH4OH is added until the pH reached 7. THF 1:7 ratio was added for solubility and loaded onto an Ag 1X 8 acetate form ion exchange resin. Product is eluted with varying concentrations of acetic acid in a mixture of water:THF in a ratio of 4:1. Product comes off at 2N and 5N acetic acid. Lyophilize off water 2–3 times to remove the acetic acid (.156, 73%). Found C, 53.48; H, 4.83; N, 5.60; C11H11FNO4 1/3 H2O requires C, 53.66; H, 4.78; N, 5.69. 1HNMR (400 MHz, D2O) δ: 7.26-7.20 (m, 2H), 7.06-7.00 (m, 2H), (3.98 (d, .5H, S,S, J=3.24), 3.88 (d, .5H, S,R, J=3.24)), (3.31 (ddd, .5H, S,R, J=3.24, 7.12, 9.71), 3.18 (ddd, .5H, S,S, J=3.24, 5.18, 9.06)), 3.05-2.84 (m, 2H). 13CNMR (100 MHz, D2O) δ: (176.21, 176.00), (172.29, 171.81), (163.01, 162.93), (160.60, 160.53), (133.80, 133.39), (130.72, 130.68), (130.65, 130.59), (115.69, 115.58), (115.47, 115.37), 54.74, (48.19, 48.03), (33.82, 32.82). [α]21D = +116.6° in DI H2O adjusted to 7.5 with NaOH.
4.29. β-3 Nitro-benzylaspartate (4I)
The protected β-alkylated aspartate (.24 g, .446 mol) was taken up in 6N HCl and heated for 24 hours at 65°C. Ethyl ether was added for separation; the water layer was washed and concentrated. NH4OH was added until the pH reached 7. THF was added for solubility and loaded onto an ion exchange resin. Product was eluted with a mixture of water:THF in a ratio of 4:1. Product came off around 2N. Lyophilized off water 2–3 times to remove the acetic acid (.022 g, 18.4%). Product was sparingly insoluble in H2O. 1H NMR (400 MHz, D2O) δ: 8.13-8.06 (m, 2H), 7.67-7.61 (m, 1H), 7.54-7.48 (m, 1H), (3.99 (d, .6H, S,S, J=3.88), 3.87 (d, .4H, S,R, J=3.88)), (3.30 (ddd, .4H, S,R, J=3.88, 5.18, 9.17), 3.21 (ddd, .6H, S,S, J=3.88, 5.18, 9.06)), 3.11-3.05 (m, 1.4H), 2.97 (d,, .45 H, J=5.18) 2.94 (d, .25H, J=5.18). 13C (100 MHz, DMSO) δ: (173.92, 172.48), (169.50, 168.75), (146.79, 146.56), 139.37, 136.19, (131.76, 130.60), (123.72, 123.55), (122.23, 122.02), 52.92, 48.83, 29.69. HRMS m/e calcd for C11H13N2O6 269.0774, found 269.0779. [α]21D = + 2.7° in DI H2O adjusted to 7.5 with NaOH.
4.30. β-3 Nitro-benzylaspartate (4I, S,S)
The protected β-alkylated aspartate(.325 g, .603 mmol) is taken up in 6N HCl with a little acetone and heated for 24 hours at 65°C. The reaction is then concentrated down, washed with water 2 times and placed under vacuum. Ethyl acetate is added and product precipitates out, white powder. For ease this was centrifuged down for 10 minutes at 4000 rpm and the supernant poured off. The precipitate was dissolved in a small amount of water from which it almost immediately crashed out as white crystals. This is washed with water and concentrated down 2 more times (.045 g, 28%). 1H NMR (400 MHz, D2O) δ: 8.10 (s, 1H), 8.06 (d, 1H, J=8.06), 7.64 (d, 1H, J=7.33), 7.50 (t, 1H, J=8.06,7.33), 4.13 (1H, α), 3.40-3.36 (m, 1H, β), 3.28-3.23 (m, 1H), 3.11-3.06 (m, 1H). 13C (100MHz, D2O) δ: 172.28, 169.29, 147.75, 141.07, 136.05, 129.69, 123.67, 121.64, 52.79, 47.28, 33.85. [α]21D = −20°.
4.31. β-para nitro benzylaspartate (4J)
The protected β-alkylated aspartate (1.35 g, 2.51 mmol) was placed in a round bottom flask with 6N HCl and THF, to dissolve. Reaction was refluxed for three hours then washed with ethyl acetate. The ethyl acetate layer was washed 2 more times and the aqueous layer was concentrated down. 7.4g AgX1-8 resin 200–400mesh size was loaded into a column and washed with two columns of water. Product was loaded and the column was washed with a gradient of acetic acid from .1N to 5N. Faction’s 1–5 are lyophilized down (.378 g, 56%). 1H-NMR (400 MHz, D2O) δ: 8.04-8.00 (m, 2H), 7.39-7.34 (m, 2H), (3.99-3.88 (d, .65H, S,S), 3.92-3.91 (d, .35H, S,R)), (3.35-3.33 (m, .35H, S,R), 3.23-3.22 (m, .65H, S,S)), 3.12-2.91 (m, 2H). 13CNMR (100MHz, D2O) δ: (178.86,178.41), (173.37,172.64), (147.30,146.96), (146.42,146.35), (129.92,129.83), (123.86,123.80), (55.73,55.67), (49.40,48.99), (36.12,33.29). HRMS m/e Calcd for C11H14N2O6+ calcd. 252.1236, found 252.1225. [α]21D = +18.52°.
4.32. β-3 Chloro-benzylaspartate S,R
1H-NMR (400 MHz, D2O) δ: 7.30-7.24 (m, 3H), 7.18-7.17 (m, 1H), (3.96 (app. d, .07H, S,S), 3.81 (app. d, .93H, S,R)), (3.27-3.22 (m, .91H, S,R), 3.15-3.10 (m, .09H, S,S)), (3.02-2.89 (m, 1.9H, S,R), 2.83-2.79 (m, .1H, S,S). 13CNMR (100MHz, D2O) δ: 172.01, 169.34, 141.26, 132.73, 130.06, 128.65, 127.75, 126.34, 52.44, 49.61, 32.14. [α]21 D = +25.7°.
4.33 EAAT3-Mediated transport in C17.2 cells
EAAT3 was transiently expressed in C17.2 cells (obtained from Dr. Evan Snyder, Burnham Inst., La Jolla, CA) using an AAV-based vector (kindly provided by Dr. Mathew During, University of Auckland, NZ) pAM-CAG-EAAT3-WPRE as previously described 10. Cells between passages 10–20 were seeded at 5 × 104 cells/well in 12 well plates and grown in complete DMEM supplemented with 10% fetal bovine serum, 1mM sodium pyruvate, 0.1 mM nonessential amino acids solution. At 24 hours after plating, cells were transfected using FuGENE6 Transfection Reagent (Roche, Indianapolis, IN) in a ratio of 4µl of FuGENE6 to 3µg of purified plasmid DNA in accordance with manufacturers instructions. The cells were used in transport assays 24 hours following transfection as described by Esslinger et al. 10. Briefly, transfected C17.2 cells were grown in DMEM containing 10% FCS in a humid atmosphere of 5% CO2. Near-confluent cells (plated at 5×104 cells/well) were rinsed with a physiological buffer (138 mM NaCl, 11 mM D-glucose, 5.3 mM KCl, 0.4 mM KH2PO4, 0.3 mM Na2HPO4, 1.1 mM CaCl2, 0.7 mM MgSO4, 10 mM HEPES, pH 7.4) and allowed to preincubate at 37°C for 5 min. Uptake was initiated by replacing the pre-incubation buffer with buffer containing 3H-D-aspartate (25µM) and inhibitors at the concentrations indicated. Following a 5 min incubation, the media was removed by rapid suction and the cells rinsed 3 times with ice-cold buffer. The cells were dissolved in 0.4N NaOH for 24 hours and analyzed for radioactivity by LSC and protein by the BCA (Pierce) method. Transport rates were corrected for background: i.e., radiolabel accumulation at 4°C. Initial studies confirmed that uptake quantified in this manner was linear with time and protein levels and that uptake in untransfected C17.2 cells was indistinguishable from background.
Supplementary Material
Acknowledgements
The authors wish to thank J. Gerdes, M. Kavanaugh, S. Patel, C.M. Thompson, and T. Denton for their insightful advice regarding the studies presented in this manuscript. Molecular modeling studies were conducted in the Molecular Computational Core Facility of the Center for Structural and Functional Neuroscience and HRMS were obtained in the Proteomics and Mass Spec. Core Facility of the Center for Structural and Functional Neuroscience. This work was supported in part by NINDS NS30570 (RJB), NINDS NS045704 (CSE) and NCRR COBRE RR15583 (RJB, CSE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Balazs R, Bridges RJ, Cotman CW. Excitatory amino acid transmission in health and disease. ed. New York: Oxford University Press; 2006. [Google Scholar]
- 2.Hynd MR, Scott HL, Dodd PR. Neurochem. Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Foster A, Kemp J. Curr Opin Pharmacol. 2006;6:7–17. doi: 10.1016/j.coph.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Danbolt NC. Prog. Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 5.Bridges RJ, Esslinger CS. Pharmacol. Therap. 2005;107(3):271–285. doi: 10.1016/j.pharmthera.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Beart PM, O'Shea RD. Br. J. Pharmacol. 2007;150(1):5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. Pflugers Arch. -Eur. J. Physiol. 2004;447:465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 8.Zerangue N, Kavanaugh MP. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 9.Kanner BI, Kavanaugh MP, Bendahan A. Biochem. Soc. Trans. 2001;29:707–710. doi: 10.1042/0300-5127:0290707. [DOI] [PubMed] [Google Scholar]
- 10.Esslinger CS, Agarwal S, Gerdes JM, Wilson PA, Davies ES, Awes AN, O'Brien E, Mavencamp T, Koch HP, Poulsen DJ, Chamberlin AR, Kavanaugh MP, Bridges RJ. Neuropharmacol. 2005;49(6):850–861. doi: 10.1016/j.neuropharm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Shimamoto K, Shigeri Y, Yasuda-Kamatani Y, Lebrun B, Yumoto N, Nakajima T. Bioorg. Med. Chem. Lett. 2000;10:2407–2410. doi: 10.1016/s0960-894x(00)00487-x. [DOI] [PubMed] [Google Scholar]
- 12.Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Mol. Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- 13.Zervas L, Theodorpoulos D. J. Am. Chem. Soc. 1956;78:1359. [Google Scholar]
- 14.Humphrey JM, Bridges RJ, Chamberlin AR. J. Org. Chem. 1994;59:2467–2472. [Google Scholar]
- 15.Fernandez-Megia P, Sardina J. Org. Chem. 1994;59:7643–7652. [Google Scholar]
- 16.Hanessian S, Margarita R, Hall A, Luo X. Tet. Lett. 1998;39:5883. [Google Scholar]
- 17.Sakaguchi K, Yamamoto M, Kawamoto T, Yamada T, Shinada T, Shimamoto K, Ohfune Y. Tet. Lett. 2004;45:5869–5872. [Google Scholar]
- 18.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 19.Muller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.