

Abstract
Inosine 5′-monophosphate (IMP) cyclohydrolase catalyzes the cyclization of 5-formaminoimidazole-4-carboxamide ribonucleotide (FAICAR) to IMP in the final step of de novo purine biosynthesis. Two major types of this enzyme have been discovered to date: PurH in Bacteria and Eukarya, and PurO in Archaea. The structure of the MTH1020 gene product from Methanothermobacter thermoautotrophicus was previously solved without functional annotation but shows high amino acid sequence similarity to other PurOs. We determined the crystal structure of the MTH1020 gene product in complex with either IMP or 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) at 2.0 Å and 2.6 Å resolution, respectively. Based on the sequence analysis, ligand-bound structures, and biochemical data, MTH1020 is confirmed as an archaeal IMP cyclohydrolase, thus designated as MthPurO. MthPurO has a four-layered αββα core structure, showing an N-terminal nucleophile (NTN) hydrolase fold. The active site is located at the deep pocket between two central β-sheets and contains residues strictly conserved within PurOs. Comparisons of the two types of IMP cyclohydrolase, PurO and PurH, revealed that there are no similarities in sequence, structure, or the active site architecture, suggesting that they are evolutionarily not related to each other. The MjR31K mutant of PurO from Methanocaldococcus jannaschii showed 76% decreased activity and MjE102Q mutation completely abolished enzymatic activity, suggesting that these highly conserved residues play critical roles in catalysis. Interestingly, green fluorescent protein (GFP), which has no structural homology to either PurO or PurH but catalyzes a similar intramolecular cyclohydrolase reaction required for chromophore maturation, utilizes Arg96 and Glu222 in a mechanism analogous to that of PurO.
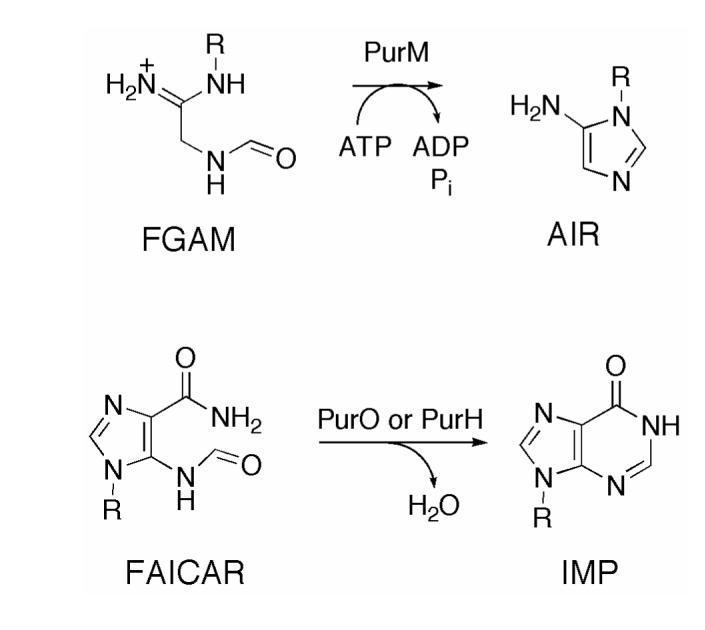
The final two steps of de novo purine biosynthesis are the conversion of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to 5-formaminoimidazole-4-carboxamide ribonucleotide (FAICAR) and the subsequent conversion of FAICAR to inosine 5′-monophosphate (IMP). These two reactions occur in different ways depending on the domains of life (Figure 1). In Bacteria and Eukarya, one bifunctional enzyme, AICAR transformylase/IMP cyclohydrolase (PurH), encoded by the purH gene, catalyzes both steps (1). The AICAR transformylase domain is located at the C-terminus of the enzyme and catalyzes formyl group transfer from N10-formyltetrahydrofolate (N10-formylTHF) to AICAR. The product FAICAR is then cyclized to IMP with elimination of water by IMP cyclohydrolase, which resides in the N-terminus of PurH. The locations of each domain and active site have been confirmed by the X-ray crystal structures of avian and human PurHs (2, 3).
Figure 1.

Two different pathways for the last two steps of de novo purine biosynthesis. In Bacteria and Eukarya, one bifunctional enzyme PurH performs these reactions, whereas some Archaea employ two separate enzymes for the same chemical reactions. PurP catalyzes the formylation of AICAR to FAICAR by using ATP and formate, and then PurO cyclizes FAICAR to IMP with the elimination of water.
Most Archaea lack folates and the reactions to convert AICAR to IMP occur in a completely different way compared to Bacteria and Eukarya (4). In Archaea FAICAR synthetase (PurP), encoded by the purP gene, catalyzes the ATP-dependent ligation of AICAR and formate to produce FAICAR. In some Archaea (e.g, Methanocaldococcus jannaschii, Methanothermobacter thermoautotrophicus, Methanococcus maripaludis, Halobacterium sp. NRC-1, Methanopyrus kandleri, Haloarcula marismortui, and Thermococcus kodakaraensis) IMP cyclohydrolase (PurO), encoded by the purO gene, catalyzes the conversion of FAICAR to IMP. PSI-BLAST searches (5) using PurO sequences show that PurO orthologs are found only in Archaea (6). The known PurO amino acid sequences show approximately 37-62% identity and 51-77% similarity. In Archaea there is no evidence for an IMP cyclohydrolase domain similar to PurH and there is no significant sequence similarity between PurH and either PurP or PurO. PurP is a signature gene in all known Archaeal genomes, whereas PurO is distributed in a limited range of Archaea (7). Therefore, in all Archaea PurP appears to catalyze the penultimate step of purine biosynthesis, while the final step is catalyzed by PurO in some Archaea, but in others the IMP cyclohydrolase activity derives from a yet to be identified gene product.
As a part of the M. thermoautotrophicus structural genomics project (8), the crystal structure of the MTH1020 gene product was determined and deposited without functional annotation in the Protein Data Bank (PDB ID 1KUU) (9). MTH1020 is composed of 202 amino acid residues with a molecular weight of 21,824 Da and has optimal activity at 65°C. MTH1020 adopts an N-terminal nucleophile (NTN) hydrolase fold, which is characterized by a four-layered αββα sandwich core structure (9). In general, members of the NTN hydrolase superfamily undergo autocatalytic self-processing to eliminate an N-terminal polypeptide. The resulting active enzyme is characterized by a nucleophilic residue (usually serine, threonine or cysteine) at the new N-terminus. The topological locations of the catalytic nucleophiles are roughly identical (10). However, MTH1020 does not have a nucleophilic residue at the corresponding active site position, suggesting a novel function for the NTN hydrolase fold.
Although the three-dimensional structure alone of MTH1020 did not provide obvious clues for the function of this protein, the high sequence similarity between MTH1020 and M. jannaschii PurO allowed us to predict that MTH1020 is an Archaeal IMP cyclohydrolase. Here, we report the crystal structure of M. thermoautotrophicus IMP cyclohydrolase (MTH1020, MthPurO) in complex with either IMP or AICAR at 2.0 Å and 2.6 Å resolution, respectively. Active site residues were identified using the structural information from MthPurO and mutational studies were carried out in the closely related PurO from M. jannaschii (MjPurO). Comparison of MthPurO and the IMP cyclohydrolase domain of PurH revealed that these enzymes have no resemblance in sequence, structure, or active site geometry in spite of having the same function. A mechanism for the MthPurO-catalyzed cyclohydrolase reaction is proposed and compared to that of PurH (3) and of green fluorescent protein (GFP), which catalyzes a similar cyclohydrolase reaction required for chromophore maturation (11).
MATERIALS AND METHODS
Materials
FAICAR was made according to the methods of Flaks et al. (12). All other chemicals were purchased from Sigma-Aldrich.
Cloning, Expression, and Purification of MthPurO and MjPurO
MthPurO was cloned into the expression vector pET15b, and transformed into Escherichia coli overexpression strain BL21. Cells were grown in LB medium containing ampicillin (0.1 mg/mL) at 37 °C with shaking. When an OD600 of 0.6 was reached, isopropyl β-D-thiogalactopyranoside to a final concentration of 0.2 mM was added for induction, and the cells were further incubated at 30 °C for 3-4 hours and harvested by centrifugation. Harvested cells were resuspended in the equilibration/wash buffer (50 mM sodium phosphate buffer pH 7.0 containing 300 mM NaCl) and lysed by sonication, followed by centrifugation at 11300 × g for 30 min. The supernatant was incubated in a water bath at 65 °C for 1 h to remove most of the contaminating E. coli proteins. The supernatant was collected and applied to a BD TALON cobalt metal affinity column (Qiagen, Valencia, CA) previously equilibrated with the equilibration/wash buffer. The bound MthPurO was eluted with an elution buffer containing 150 mM imidazole in the equilibration/wash buffer. The purified protein was buffer exchanged into 10 mM Tris-HCl pH 7.5 and concentrated to 30 mg/mL using Amicon Ultra 15 (MWCO 10 kDa) (Millipore) and stored at -80 °C for later use. The homogeneity of the purified protein was verified by SDS-PAGE analysis (data not shown). The enzyme concentration was determined by using the Bradford assay method (13) with bovine serum albumin as the standard.
The gene MJ0626 (Swiss-Prot accession number Q58043) was cloned in the BamHI and NdeI site of a pET-19b vector as previously reported (6). Site-directed mutagenesis of MjPurO was performed using a QuickChange site-directed mutagenesis kit (Stratagene). The mutagenic oligonucleotide primers used are listed in Supporting Information Table 1. The mutated plasmids were confirmed by 1% agarose gel electrophoresis and DNA sequencing by the Virginia Bioinformatics Institute at the Virginia Polytechnic Institute and State University. E. coli cells containing the mutated plasmids were inoculated into seed cultures (5 mL LB broth/0.1 mg/mL ampicillin) for overnight growth. LB broth (400 mL containing 0.1 mg/mL ampicillin) was inoculated with 4 mL overnight seed culture. Cultures were grown to an OD600 = 0.6 to 1.0 and then induced by the addition of lactose (10 mg/mL). After growing an additional 4 hours at 37 °C, the cells were harvested by centrifugation at 4400 × g for 10 min and stored at -20 °C.
Table 1.
Data collection statistics of MthPurO complexes
| PurO | PurO/IMP | PurO/AICAR | |
|---|---|---|---|
| Beamline | CuKα | APS 8-BM | APS 8-BM |
| Wavelength (Å) | 1.5418 | 0.97949 | 0.97779 |
| Space group | P32 | P32 | P32 |
| a, c (Å) | 84.3, 124.8 | 86.0, 124.4 | 85.9, 124.3 |
| Resolution limit (Å) | 50-2.6 (2.7-2.6) | 50-2.0 (2.10-2.0) | 50-2.6 (2.7-2.6) |
| Measured reflections | 119054 | 174603 | 89910 |
| Unique reflections | 29991 (2984) | 65280 (6678) | 31271 (3192) |
| Redundancy | 4.0 (4.2) | 2.7 (2.6) | 2.9 (2.8) |
| I/σ | 27.4 (4.9) | 27.0 (6.3) | 21.3 (3.8) |
| Completeness (%) | 99.1 (99.9) | 97.9 (99.4) | 98.5 (99.6) |
| Rsym (%)a | 6.0 (37.5) | 4.3 (21.5) | 7.0 (40.5) |
Values for the highest resolution shell are given in parentheses.
Rsym = ΣΣi | Ii-<I> | /Σ<I>, where <I> is the mean intensity of the N reflections with intensities Ii and common indices h,k,l.
E. coli cells expressing MjPurO were resuspended in assay buffer (50 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES)/K+ at pH 7, 10 mM Mg2+, 20 mM dithiothreitol and sonicated to lyse the cells. Following centrifugation at 13000 rpm for 15 min, the supernatant was separated from the pellet and heated at 70 °C for 15 min and the denatured E. coli proteins were removed by centrifugation at 20400 × g for 20 min. The supernatant was separated from the pellet and the MJ0626-derived protein was purified by anion exchange chromatography on a MonoQ HR column (1 × 8 cm, Amersham Bioscience) with a linear gradient from 0-1 M NaCl in 25 mM Tris pH 7.5 over 55 mL at 1 mL/min. SDS-PAGE analysis was used to confirm the expression of the desired protein, which appeared as a single band at approximately 22 kDa by Coomassie staining. The enzyme concentration was determined by using the Bradford assay method (13) with bovine serum albumin as the standard.
IMP Cyclohydrolase Activity Assay
The enzyme activity of mutant and wild-type MjPurO was tested using the following standard assay (6). Each 157 μL assay contained 18 mM TES/K+ pH 7.2, 9 mM Mg2+, 0.7 mM FAICAR, and 0.2-8.5 μg enzyme. The reaction was monitored by the increase in UV absorbance at 249 nm resulting from the production of IMP. Samples were incubated at 60 °C for 15 min. For the control assay, buffer was added in place of enzyme. The activities of the mutant and wild-type enzymes were measured by a difference spectrum using the control as the reference sample.
Crystallization of MthPurO
The initial trial for the cocrystallization of MthPurO with IMP using a previously known condition (10-15% 2-methyl-2,4-pentanediol (MPD), 0.2 M magnesium acetate, and 0.1 M HEPES pH 7.5) (9) was not successful. Under this condition, MthPurO crystallized as bipyramids belonging to space group I4122, but the structure revealed no ligand binding at the predicted active site. Efforts to soak the MthPurO crystals with IMP were also unsuccessful because the crystals severely cracked and dissolved after a few seconds. This might be caused by the crystal packing in the tetrahedral space group structure, where the active sites of the two adjacent molecules face each other and thus may not allow ligand binding to the active site without a distortion of the crystal packing.
To find a new crystallization form for MthPurO that would enable IMP binding, different crystallization conditions were screened using the hanging-drop vapor diffusion method. One condition allowing successful IMP binding was found from Crystal Screen I (Hampton Research) and optimized. The crystals were grown in 25-30% of MPD, 0.2 M ammonium acetate and 0.1 M sodium citrate buffer pH 5.6. Drops were prepared by mixing 1 μL of protein solution (30 mg/mL) with 1 μL of reservoir solution. Droplets were placed on siliconized cover slips and equilibrated against 0.5 mL of the reservoir solution at a temperature of 24 °C. Rod-shaped crystals appeared after 3-4 weeks with average dimensions of 0.6 mm × 0.2 mm × 0.1 mm.
Data Collection and Processing
MthPurO crystals were soaked with a solution containing either 5 mM IMP or 5 mM AICAR in the mother liquor for 3-4 min and immediately frozen with liquid nitrogen since the high concentration of MPD of the reservoir solution was an effective cryoprotectant. For the unliganded MthPurO, the crystal was directly mounted in a cold nitrogen gas stream at 100 K without soaking or further cryoprotection. The X-ray diffraction data of unliganded MthPurO were collected with CuKα radiation using a Rigaku RU-200 rotating anode generator and a RAXIS-II image plate detector. Data were collected by the oscillation method in 0.5° increments over a total range of 120° with an exposure time of 15 min per image and a crystal-to-detector distance of 200 mm. The X-ray diffraction data of MthPurO in complex with either IMP or AICAR were collected at the NE-CAT beamline 8-BM of the Advanced Photon Source (APS) at Argonne National Laboratory (Argonne, IL) using a Quantum 315 CCD detector (Area Detector Systems Corp.). The data for MthPurO/IMP were collected by the oscillation method in 0.5° increments over a total range of 90° with an exposure time of 30 s per image using a crystal-to-detector distance of 300 mm. For MthPurO/AICAR, the data were collected over a total range of 100° with a 0.5° oscillation angle. The exposure time per image and the crystal-to-detector distance were 20 s and 280 mm, respectively. Diffraction data were processed using the HKL2000 program package (14). The crystals of unliganded MthPurO, MthPurO/IMP and MthPurO/AICAR diffracted to resolutions of 2.6 Å, 2.0 Å and 2.6 Å, respectively. All crystals belonged to space group P32, with unit-cell parameters given in Table 1. The Matthews coefficient (VM) (15) was 2.9 Å3/Da, suggesting four protomers per asymmetric unit with a solvent content of 57.7%. The data collection statistics are summarized in Table 1.
Structure Determination, Model Building and Refinement
The structures of unliganded MthPurO and the MthPurO/IMP and MthPurO/AICAR complexes were determined by molecular replacement with the program Crystallography & NMR System (CNS) (16) using the previously deposited MTH1020 structure (PDB ID 1KUU) as a search model (9). The structures were refined by consecutive cycles of simulated annealing, energy minimization, and temperature factor refinements using CNS and manual rebuilding by the program O (17). 10% of the reflections were excluded for the calculation of Rfree. Non-crystallographic symmetry (NCS) restraints were heavily applied throughout the refinement. Fourfold NCS map averaging was performed using the program RAVE (18). Water molecules were picked automatically using CNS and were manually inspected using O. Atomic coordinates and structure factors for unliganded MthPurO, MthPurO/IMP and MthPurO/AICAR were deposited in the PDB as entry numbers 2NTM, 2NTK and 2NTL, respectively.
Figure preparation
Figures were created with Molscript (19) or Bobscript (20) and Raster3D (21). Surface models were prepared using PyMol (22). The multiple sequence alignment was prepared by ClustalX 1.8 (23) and ESPript (24).
RESULTS
Quality of the Final Models
The final models of unliganded MthPurO, MthPurO/IMP and MthPurO/AICAR contain four protomers in the asymmetric unit (labeled A, B, C and D) with IMP or AICAR bound in each protomer. One protomer of MthPurO is composed of 202 amino acid residues without any missing residues. Three N-terminal His-tag residues were seen in chain D of unliganded MthPurO, four N-terminal His-tag residues were seen in chain A of MthPurO/IMP, and one in chain C of MthPurO/AICAR. The R-factors for the final refined models were 22.5% (Rfree 27.4%) for the unliganded MthPurO, 21.8% (Rfree 24.5%) for the MthPurO/IMP structure, and 20.8% (Rfree 24.3%) for the MthPurO/AICAR structure. The final models were assessed with the program PROCHECK (25), and the Ramachandran plot (26) showed that 98.9% of the main-chain dihedral angles lie in the most favored and additionally allowed regions in both MthPurO/IMP and MthPurO/AICAR models. The Glu149 residue in each protomer falls into the disallowed region of the Ramachandran plot, as was seen in the unliganded structure (PDB ID 1KUU). All attempts to prepare an FAICAR complex were unsuccessful. The refinement statistics of the final models are summarized in Table 2.
Table 2.
Refinement statistics of MthPurO complexes
| PurO | PurO/IMP | PurO/AICAR | |
|---|---|---|---|
| Resolution range (Å) | 10-2.6 (2.7-2.6) | 10-2.0 (2.1-2.0) | 10-2.6 (2.7-2.6) |
| Unique reflections | 26707 (2193) | 59591 (5462) | 28765 (2556) |
| Completeness (%) | 87.6 (71.5) | 89.6 (81.7) | 91.2 (80.7) |
| Protein atoms | 6168 | 6179 | 6179 |
| Water molecules | 14 | 200 | 46 |
| R factor (%)a | 22.5 | 21.8 | 20.8 |
| Rfree (%)b | 27.4 | 24.5 | 24.3 |
| Average B-factor (Å2) rmsdc from ideal geometry | 70.7 | 44.6 | 61.4 |
| bond length rmsd (Å) | 0.011 | 0.006 | 0.007 |
| bond angles rmsd (°) | 1.6 | 1.3 | 1.4 |
| Ramachandran Plot | |||
| most favored regions (%) | 82.6 | 85.3 | 86.2 |
| additional allowed regions (%) | 16.2 | 13.5 | 12.7 |
| generously allowed regions (%) | 0.6 | 0.6 | 0.6 |
| disallowed regions (%) | 0.6 | 0.6 | 0.6 |
Values for the highest resolution shell are given in parentheses.
R factor = Σhkl | | Fobs | - k | Fcal | | / Σhkl | Fobs |, where Fobs and Fcal are observed and calculated structure factors, respectively.
For Rfree the sum is extended over a subset of reflections (10%) excluded from all stages of refinement.
rmsd = root mean square deviation
Structure of MthPurO
The MthPurO protomer adopts an NTN hydrolase fold showing a four-layered αββα core structure with two antiparallel β-sheets packed against each other as previously described (9). The overall protomer fold and topology numbering are illustrated in Figure 2. The IMP molecule is bound at a deep pocket between the two central β-sheet layers (Figure 2). The topological locations of the active sites within the NTN hydrolase superfamily are approximately equivalent, with the N-terminal catalytic nucleophiles (Ser, Thr, or Cys) coming from the β1 strand. MthPurO does not have a nucleophile at the corresponding position (Figure 2A) (10). The root mean square (rms) deviations for the 202 Cα coordinates of MthPurO/IMP and MthPurO/AICAR protomers with the deposited apo structure are 0.32 Å and 0.26 Å, respectively, indicating that the ligand binding did not induce a significant change in the overall structure.
Figure 2.

Protomer structure of MthPurO/IMP. (A) Protomer of MthPurO/IMP complex shown in ribbon diagram with α-helices in blue and β-strands in green. MthPurO has a typical NTN hydrolase fold with an αββα structure. The IMP molecule is bound between two antiparallel β-sheets. The IMP molecule is represented by a ball-and-stick model with carbon in black, oxygen in red, nitrogen in blue, and phosphate in magenta. (B) Topology diagram of MthPurO. The first and the last residue numbers are labeled for each secondary structural element. The location of the active site is marked with a red asterisk.
The overall structures of the unliganded PurO’s from the two different space groups P32 and I4122 are essentially the same. The rmsd between the two structures is 0.31 Å for 202 Cα atoms. The largest difference occurs for the side chain of Arg5, which has a χ1 of -77.8° for the P32 crystal form, and -13.8° for the I4122 crystal form. In the unliganded P32 PurO structure, there were some unclear residual electron densities near the phosphate binding site and the O6 position of the purine binding site. The maximum peak height in the difference Fourier map is 7σ at the phosphate binding site. Because of the ambiguity of the density at this resolution, we did not attempt to model these features. In the I4122 PurO structure, a water molecule fills the position corresponding to the O6 atom of IMP and forms hydrogen bonds with the Arg30 and the Try59. Another water molecule in the I4122 PurO structure is positioned just below the phosphate binding site.
Gel filtration chromatography from previous studies revealed that MthPurO exists as a tetramer in solution (9). MthPurO also exists as a tetramer in both the tetragonal crystal form (9) and the trigonal crystal form in this study. The MthPurO tetramer is box-shaped with overall dimensions of approximately 78 × 65 × 57 Å3 (Figure 3). The tetramer is organized by creating identical interfaces with α1, α2, α3, β9, and their connecting loop regions for which each protomer makes contacts with the other three protomers. There are roughly three types of interfaces: the interface between the protomers A and B (C and D), the protomers A and D (B and C), and the central core of the tetramer completed by all four protomers. The A-B (C-D) interface is mainly provided by the two β9 strands (residues 131-136) from each protomer that are packed antiparallel to each other, resulting in 14 consecutive antiparallel β-strands (seven from each protomer) in the β-sheet layer β II. The main-chain hydrogen bonds provided by the packing of antiparallel β-strands stabilize this interface, and the side chains of Val127, Ala129, Gly131, Leu132, Met133, and Val134 from each protomer provide hydrophobic interactions. In the A-D (B-C) interface, helix α1 from each protomer and their connecting loops participate in interactions. The reciprocal salt bridge between residues Glu48 and Arg49′, where the prime refers to the neighboring protomer, and hydrophobic residues Val51, Phe52, and Ala58 contribute to the interface formation. At the tetramer core region, four helix-loop-helix (α2-loop-α3) elements from each protomer are packed together creating the stable tetramer center. A total of 16 out of 26 residues composing this interface are hydrophobic (about 61.5%). The core formation makes additional interactions between protomers A and C (B and D) through the loop regions connecting α2 and α3 and burying 562 Å2 of surface area. An interprotomer ion-pair network formed by three protomers using Asp82, Lys83, Arg91, Asp92, Asp102 and Lys105 is located at the core and repeated four times. About 1584 Å2 of the surface area per protomer (18% of the protomer) is buried upon tetramer formation. Salt bridges, or ion-pairs, have long been thought to play a role in increasing the thermostability of some proteins (27-32). It is possible that the tetrameric structure and the resulting interprotomer ion-pair network of MthPurO is an adaptation to higher temperature conditions.
Figure 3.
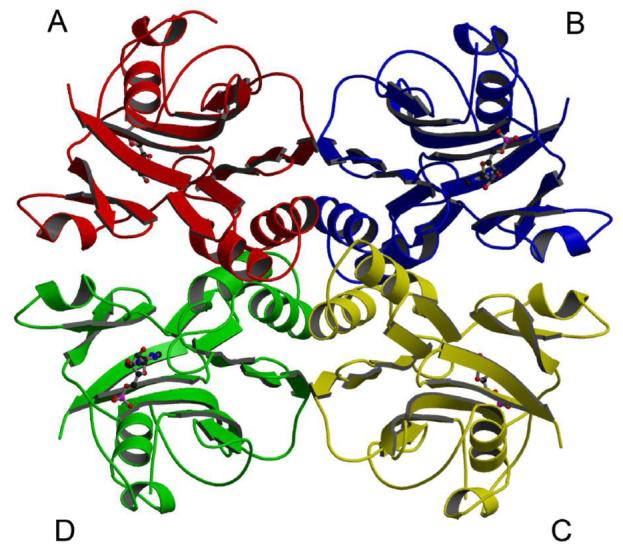
Quaternary structure of MthPurO/IMP. Tetramer of MthPurO/IMP with each protomer shown in different color. The IMP molecules shown in ball-and-stick representation are bound in each protomer.
Active Site of MthPurO
The active site of MthPurO was previously predicted by the superposition of MTH1020 with other NTN hydrolase superfamily members to be a deep pocket located between the two β-sheets in each protomer (9). Despite the good agreement of the active site location of MthPurO with that of other NTN hydrolase superfamily members, in MthPurO the corresponding NTN position is occupied by Arg5, which cannot act as a nucleophile, demonstrating a different function of MthPurO from the NTN hydrolases (9). After the initial round of refinement, strong and unambiguous difference density was found at the active site in each protomer for both the IMP and AICAR containing crystals (Figure 4). The model of the MthPurO/IMP complex revealed that the active site is located as predicted previously (Figure 5A). All amino acid residues in the active site making direct contact with the ligand are absolutely conserved within all PurO sequences (Figure 5C). The inosine moiety of the IMP molecule sits most deeply inside the binding pocket, well shielded from bulk solvent. There are four charged amino acid residues, Arg5, Arg30, Glu104, and Asp106 around the active site pocket, two of which (Arg30 and Glu104) are directly hydrogen bonded to the purine base ring (Figure 5B).
Figure 4.

Stereoview of Fobs-Fcalc electron density around the active site of MthPurO in complex with (A) IMP and (B) AICAR contoured at 3.0 σ levels.
Figure 5.

Surface representation of the MthPurO/IMP protomer. (A) Protomer surface in the same orientation as in Figure 2A. Strictly and strongly conserved residues within PurO sequences are mapped onto the molecular surface as red and yellow, respectively. The strictly conserved region coincides with a deep substrate binding pocket on the protein surface. (B) Close up view of the substrate binding pocket of MthPurO/IMP. The inosine moiety of the IMP sits deeply in the pocket shielded from the bulk solvent. Four charged residues, Arg5, Arg30, Glu104, and Asp106 are found in this region. (C) Multiple sequence alignment of PurOs. Strictly and strongly conserved residues within PurO sequences are shown in red and yellow boxes, respectively. The secondary structure of MthPurO is placed above the sequences. The residues that are involved in the purine base binding site (#), ribose binding (·), and phosphate binding (*) are marked below the sequences. All these active site residues are absolutely conserved in PurO sequences. METTH, Methanobacterium thermoautotrophicus; METJA, Methanocaldococcus jannaschii; METMP, Methanococcus maripaludis; HALN1, Halobacterium sp. NRC-1; METKA, Methanopyrus kandleri; HALMA, Haloarcula marismortui ATCC 43049; PYRKO, Thermococcus (Pyrococcus) kodakaraensis KOD1.
The active site of MthPurO is mainly composed of six loops; the N-terminal loop and the loops between β2 and β3, α1 and β5, β6 and α2, α3 and β7, and β10 and β11. The ligands are bound at the active site by an extensive hydrogen bonding network (Figure 6). The inosine moiety of the IMP is located between Arg30 and Glu104 on each side of the purine base ring. The guanidinium group of Arg30 is hydrogen bonded to the O6 and N7 atoms of the inosine moiety. The O6 atom of IMP also interacts with the hydroxyl group of Tyr59, which is connected to the hydroxyl group of Tyr20 via a hydrogen bond. The N1 atom of the inosine moiety has contacts with the main-chain carbonyl oxygen atoms of Tyr56 and Asn73, and the N3 atom hydrogen bonds with the carboxyl group of Glu104 (Figure 6).
Figure 6.

Active site of PurO with bound ligands. (A) Stereoview of the MthPurO/IMP active site. The IMP molecule and protein residues are represented by yellow and grey carbon atoms, respectively, with oxygen in red, nitrogen in blue, and phosphate in magenta. Hydrogen bonds are shown as broken lines. (B) Stereoview of the MthPurO/AICAR active site with the same atom colors as in (A).
In the MthPurO/IMP complex, Arg5 is involved in the interactions with all parts of the ligand; it stacks the purine base ring and also makes hydrogen bonds with the ribose and phosphate moieties of IMP. When compared to the deposited unliganded structure (PDB ID 1KUU), the side-chain of Arg5 undergoes a subtle conformational change upon substrate binding so that it is able to stack parallel to the purine base. This change results from hydrogen bonds between the guanidinium group of Arg5 and both the carbonyl oxygen atom of Ser23 and the hydroxyl group of Tyr148. In the ribose binding site, Phe27 and Leu108 provide a hydrophobic environment at each side of the ribose moiety. The O2′ atom of the ribose ring hydrogen bonds with the Oδ2 atom of Asp106 and the Nδ2 of Asn54, and the O4′ atom of the ribose interacts with the Nη2 of Arg5 and the Oη of Tyr148 (Figure 6A). In contrast, no contacts between the ribose ring and either Asn54 or Tyr148 were found in the MthPurO/AICAR complex because of a different sugar ring conformation (Figure 6B). The phosphate binding site is composed of the N-terminal loop and the loop connecting β2 and β3. Strictly conserved consecutive residues, Ser24, Arg25, and Ser26, and the N-terminal loop residues Tyr2 and Arg5 are involved in phosphate binding by providing extensive hydrogen bonding interactions. Arg25, located at the protein surface, showed different conformations among the four protomers and weak side-chain electron density consistent with high thermal motion or disorder (Figure 6).
Two water molecules are located near the N1 and C2 atoms of the purine base, the site of the cyclization reaction (Figure 6A). One water molecule, designated as “Wat A”, is 3.5 Å and 3.3 Å away from the N1 and C2 atoms, respectively, and directly interacts with the carboxyl group of Glu104 (2.8 Å) and the carbonyl oxygen atoms of Tyr56 and Asn73. On the opposite side of the purine base ring, the water molecule “Wat B” is positioned 3.6 Å and 3.2 Å away from the N1 and C2 atoms, respectively, and makes hydrogen bonds with Arg5 and the Nδ2 atom of Asn73. The positions of the water molecules suggest possible binding sites for the product water or a possible catalytic role for water in the cyclization reaction of FAICAR to IMP.
Site-Directed Mutagenesis of MjPurO
MthPurO (MTH1020) is 42% identical and 59% similar to MjPurO (MJ0626) with all active site residues strictly conserved (Figure 5C). Therefore site-directed mutagenesis studies were performed using MjPurO, which has been extensively studied biochemically (6). On the basis of the three-dimensional structure of MthPurO/ligand complexes, the amino acid residues of MjPurO corresponding to Arg30, Glu104, and Tyr59 of MthPurO were selected as targets for site-directed mutagenesis (Table 3). Substitution of Glu102 of MjPurO (corresponding to Glu104 of MthPurO) with glutamine resulted in a complete loss of enzyme activity, demonstrating that this glutamic acid is an essential residue for catalysis. The activity of MjR31K (corresponding to Arg30 of MthPurO) was 76% lower than that of the MjPurO wild-type enzyme. The mutation of Tyr59 to phenylalanine showed a relatively moderate decrease (34% reduction) in enzyme activity. The elimination of the hydroxyl group of the Tyr59 residue may disrupt the hydrogen bonding network around the O6 and Tyr20, resulting in a perturbation of accurate substrate binding (Table 3, Figure 6).
Table 3.
IMP cyclohydrolase activity of the wild-type and mutant MjPurOs
| MjPurO mutant | MthPurO number | Specific activity (μmol/min/mg) |
|---|---|---|
| wild-type | - | 1.8 |
| R31K | 30 | 0.44 |
| Y59F | 59 | 1.2 |
| C61A | 61 | 32 |
| E102Q | 104 | 0.0 |
In the early days of PurH research, it was suggested that His, Cys, and an unknown basic residue were directly involved in catalysis by human IMP cyclohydrolase (33). However, the crystal structures of human and avian PurH revealed that no such amino acid residues were located at the active site (2, 3). Likewise, MthPurO does not have His or Cys residues in the vicinity of the active site. The nearest Cys residue to the active site of MthPurO is Cys61, which is located about 8.6 Å away from the bound IMP molecule. Interestingly, the MjC61A mutant showed enhanced activity compared to the wild-type enzyme; however, the structural basis for this unexpected finding is not apparent.
DISCUSSION
Proposed Mechanisms for the PurO-Catalyzed Cyclization of FAICAR
We have considered two possible mechanisms for the cyclization of FAICAR 1 to IMP 6. In mechanism A (Figure 7), 4-carboxamide tautomerization and ionization gives 2. This tautomerization and ionization is facilitated by an unusual pairing of tyrosine residues (Tyr20 and Try59) in which the hydroxyl group of Tyr20 is buried in a completely hydrophobic environment except for a strong hydrogen bond to the hydroxyl group of Tyr59, thus reducing its pKa. The Tyr59 hydroxyl group is positioned to catalyze transfer of the amide proton from the nitrogen atom to the oxygen atom to form the iminol 3, which is in equilibrium with 2. This tautomerization is also assisted by the polarization of the amide carbonyl group by a hydrogen bond from Arg30. Ring closure of 2, activated by hydrogen bonding of the formamide carbonyl to Glu104, gives 4. Tautomerization of 4 to 5, assisted by Glu104 and Tyr59, followed by loss of water, also assisted by Glu104 and Tyr59, completes the reaction.
Figure 7.

Proposed mechanism A for the PurO-catalyzed cyclization of FAICAR. In mechanism A the reaction is initiated by ionization and tautomerization of the 4-carboxamide group of FAICAR 1 to the iminol 2/3. The tautomerization is assisted by an unusual pairing of tyrosine residues (Tyr20 and Tyr59) in which the environment of the Tyr20 hydroxyl group is entirely hydrophobic except for a strong hydrogen bond to the hydroxyl group of Tyr59. The tautomerization is further assisted by polarization of the carbonyl group by Arg30. Addition of the iminol nitrogen atom to the 5-formamide results in the tetrahedral intermediate 4. Tautomerization to 5 followed by loss of water results in IMP 6. R = ribose 5-phosphate.
Sequence comparisons show that Tyr20, Arg30, Tyr59 and Glu104 are absolutely conserved in PurO’s, consistent with this mechanism. The MjR31K and MjY59F mutants show reduction in activity of 76% and 34%, respectively. Replacement of arginine by lysine is a conservative mutation and retention of the positive charge is likely responsible for the modest reduction in activity. The replacement of tyrosine by phenylalanine would also be a conservative mutation if a water molecule were to replace the position previously occupied by the hydroxyl group. The critical involvement of MthGlu104 in catalysis is supported by the observation that the MjE102Q (MthE104) mutant has no activity.
Purine biosynthesis requires two cyclization reactions, both involving the addition of weakly nucleophilic nitrogen nucleophiles to a formamide functional group (Scheme 1). These two cyclization reactions are mechanistically distinct because the first cyclization to form the imidazole ring, catalyzed by PurM, requires ATP and most likely proceeds via an iminophosphate intermediate while the second cyclization to form the pyrimidine ring, catalyzed by either PurO or PurH, does not require ATP activation. One can argue that the conversion of FAICAR 1 to IMP 6 is more facile than the conversion of formylglycinamidine ribonucleotide (FGAM) to aminoimidazole ribonucleotide (AIR) because the imidazole double bond forces the amide nitrogen into close proximity to the formamide carbonyl group. However, the resulting reaction trajectory is not optimal for the addition of nucleophiles to carbonyl groups. We have therefore considered an alternative mechanism for the formation of the pyrimidine ring that is not possible for the formation of the imidazole ring.
SCHEME 1.
This alternate mechanism B is shown in Figure 8. Iminol 3 is generated as described for mechanism A. Glu104 catalyzed tautomerization of the 5-formamide would give 7, which is perfectly set up for a 6π electrocyclization reaction (34) to give 5. This is then converted to IMP 6 by the same dehydration chemistry used in mechanism A. The structural and mutagenesis studies described here are consistent with these two mechanistic proposals and do not yet differentiate between them. Such differentiation will require additional structural and kinetic studies using substrate and intermediate analogs.
Figure 8.

Alternate mechanism B for the PurO-catalyzed cyclization of FAICAR. In mechanism B tautomerization of the 4-carboxamide of FAICAR to iminol occurs as in mechanism A. Tautomerization of the 4-formamide, assisted by Glu104, results in 7. A 6π electrocyclization reaction gives 5 and the product IMP 6 is generated by the same chemistry as in mechanism A. R = ribose 5-phosphate.
Comparison of MthPurO with the IMP Cyclohydrolase Domain of PurH
Although MthPurO and the IMP cyclohydrolase domain of PurH carry out the same cyclization reaction of FAICAR to IMP, their overall structures are completely different. The IMP cyclohydrolase domain of PurH has a three-layered αβα structure (2) and the active site of the IMP cyclohydrolase domain of PurH is located at the dimer interface with only one bound ligand (3). The amino acid residues involved in catalysis and substrate binding are also very different (Figure 9). In contrast to the side-chain involvement of Arg30 and Tyr59 in interactions with the O6 atom of IMP in MthPurO, two main-chain nitrogen atoms from Lys66 and Thr67, and the hydroxyl group of Thr67 are involved in the corresponding interactions in PurH (Figure 9). The N1 position (4-carboxamide of FAICAR) interacts with a main-chain carbonyl oxygen in both enzymes, Arg64 in PurH, but Asn73 and Tyr56 in PurO. In PurH, it has been suggested that these backbone residues around the purine base ring are responsible for the correct binding of FAICAR from its stable solution state to its less favorable but more reactive form (3, 35).
Figure 9.

Active site comparison of MthPurO and PurH. The active sites are shown schematically in (A) MthPurO/IMP and (B) the IMP cyclohydrolase domain of human PurH/XMP. All key residues are different between PurO and PurH. In the PurH active site, most of the critical interactions between the protein and XMP are provided by backbone carbonyl atoms or nitrogen atoms. The Lys137′ from the other protomer completes the active site of PurH mediated by the water molecule. For the MthPurO/IMP active site, the hydrogen bonding distances are the average values for four protomers.
Although the involved amino acid residues are different in both enzymes, the hydrogen bonding network around O6 and N1 atoms could help precisely bind substrate and flip the nitrogen-oxygen positions to the more reactive geometry required for the cyclization. In MthPurO, it is also expected that the positively charged Arg30 would stabilize the partially negatively charged oxygen atom of FAICAR during the catalysis. The removal of its positive charge at this position using mutants such as R30M or R30Q can be suggested. The fact that the enzymatic activity of MjR31K is somewhat decreased despite the retention of a positive charge suggests that the existence of an arginine residue in a specific position is important for activity (Table 3). Possibly the flexibility of the lysine side chain compared to the guanidinium group of the arginine results in less precise hydrogen bonds to the O6 and N7 positions of the inosine moiety (Figure 6). Therefore, one of the roles of Arg30 might be the provision of the correct hydrogen bonds for substrate orientation to facilitate catalysis.
In both PurH and PurO, water molecules are positioned within 3.5 - 3.9 Å distances from the N1 atom of each ligand (4-carboxamide of FAICAR). The water molecule is hydrogen bonded to Glu104 in PurO and Lys137′ in PurH, where Lys137′ comes from an adjacent protomer. In both cases, the distances between this N1 position and the charged amino acid residues are too far to accomplish the direct attack. While it is possible that the water molecules are involved in nucleophilic attack at the 4-carboxamide position through activation by the charged residue, it is also possible that the water molecule marks the position of the product water molecule. The latter is consistent with the observation in PurH that the monomer also has IMP cyclohydrolase activity, although much lower than that of dimeric PurH. Therefore, Lys137′ in PurH is probably not a critical residue for catalysis but a contributor for more efficient catalysis when PurH is a dimer (36).
The PurH active site has two possible routes for nucleophilic attack on the N3 atom by either Tyr104 or Asp125 (3), while a similar result could be achieved by the absolutely conserved Glu104 directly attacking the N3 position in PurO. Although some active site mutants significantly decreased the cyclohydrolase activity of PurH, none of these mutants, or even double mutants, were completely inactive (35), whereas the MjE102Q mutant of PurO is inactive. Therefore, the active site requirements for maintaining the full enzymatic activity of PurO seem to be more restricted than for PurH. It has been suggested that the most important role of the IMP cyclohydrolase domain of PurH is the correct binding and reorientation of FAICAR for the cyclization rather than the catalytic power of the side-chain. This is also supported by the fact that the most important interactions are made by mainly the backbone residues and none of the active site residues are absolutely necessary for the catalysis (35).
PurO and PurH have some general similarities in chemical characteristics in the reaction such as a hydrogen bonding network for correct orientation of FAICAR binding, the possible involvement of the catalytic water molecule, which would be activated by a charged amino acid residue, and the involvement of a negatively charged residue near the 4-carboxamide and N3 atom of the substrate. Nevertheless, the active site amino acid residues and their contributions to catalysis seem completely different from each other, demonstrating the considerable evolutionary distance between PurH and PurO.
A comparison of the efficiencies of PurO and PurH shows that despite the differences in overall structure and active site residues, the wild type enzymes have comparable efficiencies. The recombinant purified human PurH has a specific activity of 2.8 μmol/min/mg protein at 25 °C (1). The specific activity of this enzyme was also recently reported to depend on the enzyme concentration and to change from 5.0 to 9.5 μmol/min/mg protein at 25 °C (35). The purified enzyme from chicken liver had a specific activity of 5.09 μmol/min/mg protein at 25 °C (37). These values are compared to our values of 1.8 μmol/min/mg protein for the wild type MjPurO and 32 μmol/min/mg protein for the C61A mutant, both at 60 °C.
Comparison of MthPurO with GFP
Green fluorescent protein (GFP) from jellyfish Aequorea victoria creates its chromophore spontaneously after protein folding without requiring cofactors or accessory proteins (38). The crystal structure of GFP (PDB ID 1EMA) is an 11-stranded antiparallel β-barrel protein with the chromophore located at the center of the β-barrel (39). The chromophore is created by the backbone cyclization of three amino acid residues, Ser65 (Thr65 in enhanced GFP), Tyr66, and Gly67, forming a five-membered imidazole ring (38). Interestingly, the backbone cyclization catalyzed by GFP and the IMP cyclization catalyzed by PurO both utilize arginine and glutamic acid residues for catalysis. In GFP, it has been suggested that Arg96 acts as an electrophile to modulate the pKa and enhance the cyclization reaction, and Glu222 acts as a general base to activate a water molecule (11, 40). Although the structural alignment of the active sites of GFP and MthPurO revealed that most elements of their catalytic sites were not equivalent, the relative positions of Arg30, Glu104, and IMP in MthPurO, and those of Arg96, Glu222, and the backbone chromophore in GFP superimposed surprisingly well (Figure 10), implying that a similar cyclization mechanism might be performed by these catalytic amino acid residues. Arg96 of GFP interacts with the chromophore at a distance of 2.7 Å, which is very similar to that found in the interaction between Arg30 and IMP in PurO. The Glu222 of GFP is not directly hydrogen bonded to the backbone ring, but there are a number of water molecules that are believed to act as a proton shuttle from Glu222 to the five-membered imidazole ring. As described earlier, a similar relationship between a glutamic acid residue and a water molecule was found in the PurO active site where the water molecule is positioned within a good hydrogen bond distance from Glu104 and also in the vicinity of the substrate where the cyclization occurs (Figure 10). In both proteins, glutamic acid residues appear to act as a general base to abstract the proton from a water molecule or the substrate, as supported by the structural and kinetic data. The similarities in the catalytic residues despite the clear differences in the overall fold and function are often found in enzyme systems because of a relatively limited number of amino acid residues that can be catalytically functional. All of these results indicate that PurO and GFP share common elements of catalytic strategy for cyclization that occurs with a similar acid-base catalytic mechanism using arginine and glutamic acid residues.
Figure 10.
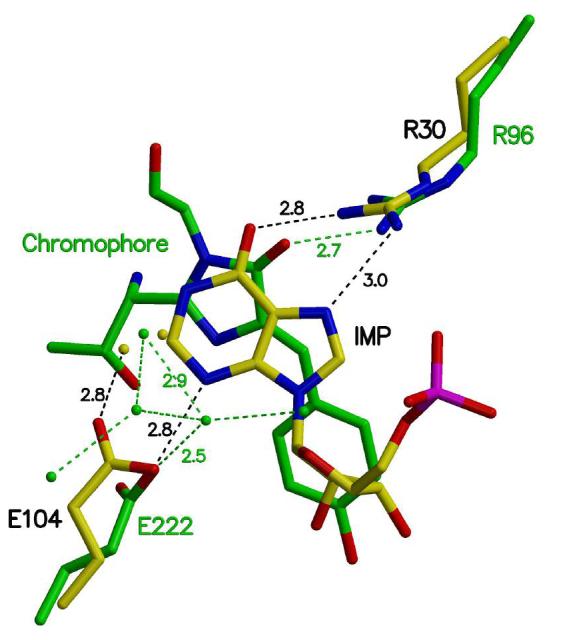
Active site comparison of MthPurO and GFP. The active sites are superimposed with the catalytically important residues and ligands shown: Arg30-Glu104-IMP in MthPurO (yellow carbon) and Arg96-Glu222-chromophore in GFP (green carbon). The hydrogen bonds are represented by broken lines. The relative positions of the arginine residues, glutamic acid residues, and ligands in the two proteins superimpose well despite their differences in sequence, structure, and function. Water molecules in GFP have been suggested as a proton shuttle.
PurO with NTN Hydrolase Fold - Structural Similarity with Functional Diversity
Many protein structures solved by the structural genomics projects have not been assigned biological functions, reflecting that there are still unknown factors directly determining the sequence-structure-function relationship. As described above, the previously solved crystal structure of MTH1020 did not provide any clue for its function. It is not unusual that the same structural fold is reused by different proteins because of the relatively limited number of structural folds compared to the vast number of proteins in nature. Nevertheless, from an evolutionary point of view, it is intriguing that the IMP cyclohydrolase function occurs with the NTN hydrolase structural fold in PurO. In both the first and last steps of the de novo purine biosynthetic pathway, enzymes adopting an NTN hydrolase fold are present, PurF and PurO. Futhermore, unlike all other NTN superfamily members, no autoprocessing is required to activate PurO. In contrast, PurF (glutamine phosphoribosylpyrophosphate amidotransferase) is an actual NTN hydrolase both in terms of function and structure whose NTN is a Cys (41). PurO only borrows the NTN hydrolase structural fold without relation to its actual function.
How has PurO evolved to catalyze the IMP cyclohydrolase activity being constrained by the NTN hydrolase structural fold? A systematic investigation of how enzymes evolve to catalyze totally different reactions within a similar structural fold has suggested that the evolutionary process is economical enough to substitute some residues for a different function within the existing active site and structure rather than shifting the whole active site position (42). Although the active site locations within the NTN hydrolase superfamily are almost identical even in PurO, there are no sequence similarities and no strictly conserved residues within the family, indicating that the proteins in this superfamily seem to be diversely evolved from a common ancestor with the same stable protein fold (43, 44). The relative plasticity of the amino acid sequences may induce a remarkable diversity of hydrolytic activity within the superfamily by subtle modification of the amino acid residues achieving a new function such as IMP cyclohydrolase activity. From this point of view, MthPurO is an interesting evolutionary offspring of the NTN hydrolase superfamily for which important mutations on the catalytic nucleophile position in MthPurO result in completely different biochemical properties. Triosephosphate isomerase-like (βα)8 barrel proteins are a classic example in which diverse functions occur in a similar structural fold (42, 45, 46). Because the NTN hydrolase fold has been identified very recently, more NTN hydrolase family members may be discovered. The increasing number of proteins belonging to this superfamily may provide promising clues for the evolutionary relationships within this superfamily.
Evolution of PurP and PurO in de novo Purine Biosynthesis
The reason for the evolution and continued existence of two separate groups of enzymes to catalyze the last two reactions in purine biosynthesis is unclear. PurP, the enzyme catalyzing the formation of FAICAR (7), is an Archaeal signature gene (47) and does not require folate. In Archaea, GAR transformylase T (PurT), which uses formate and ATP, replaces GAR transformylase N (PurN), which uses N10-formylTHF. Thus some Archaea can generate purines without folates and consequently we could view the Archaeal pathway as a more primitive pathway that predates the evolution of folates. Since the methanogens use methanopterin in place of folate and N10-formyltetrahydromethanopterin cannot function as a formate donor (48), these species are forced to continue to use the primitive PurP-containing pathway. This idea would also indicate that methanopterin evolved before folates. Since PurH always functions as a single bifunctional enzyme for both reactions, and its evolution/function was restricted by the tetrahydromethanopterin chemistry, then another enzyme had to evolve to catalyze the last, cyclization reaction. It should be noted that because PurO is not present in all Archaea, whereas PurP is always present, other enzymes catalyzing the cyclization of FAICAR remain to be identified.
Supplementary Material
ACKNOWLEDGMENTS
We thank Leslie Kinsland for assistance in the preparation of this manuscript. We thank the NE-CAT beamline 8-BM of the Advanced Photon Source, supported by the National Center for Research Resources of NIH for provision of beamtime.
ABBREVIATIONS
- IMP
inosine 5′-monophosphate
- FAICAR
5-formaminoimidazole-4-carboxamide ribonucleotide
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- NTN
N-terminal nucleophile
- GFP
green fluorescent protein
- PurH
AICAR transformylase/IMP cyclohydrolase
- PurP
FAICAR synthetase
- PurO
IMP cyclohydrolase
- THF
tetrahydrofolate
- PDB
Protein Data Bank
- MPD
2-methyl-2,4-pentanediol
- APS
Advanced Photon Source
- CNS
Crystallography & NMR System
- NCS
non-crystallographic symmetry
- XMP
xanthosine 5′-monophosphate
- AIR
aminoimidazole ribonucleotide
- FGAM
formylglycinamidine ribonucleotide
- GAR
glycinamide ribonucleotide.
Footnotes
This work was supported by a National Institutes of Health grant GM073220 to SEE and National Science Foundation grant MCB-0231319 to RHW. Beamline 8-BM is supported by NIH grant RR-015301. SEE is indebted to the Lucille P. Markey Charitable Trust and W.M. Keck Foundation.
The Brookhaven Protein Data Bank code for MthPurO is 2NTM, for the MthPurO/IMP complex is 2NTK and for the MthPurO/AICAR complex is 2NTL.
REFERENCES
- 1.Rayl EA, Moroson BA, Beardsley GP. The human purH gene product, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase. Cloning, sequencing, expression, purification, kinetic analysis, and domain mapping. J. Biol. Chem. 1996;271:2225–2233. doi: 10.1074/jbc.271.4.2225. [DOI] [PubMed] [Google Scholar]
- 2.Greasley SE, Horton P, Ramcharan J, Beardsley GP, Benkovic SJ, Wilson IA. Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nat. Struct. Biol. 2001;8:402–406. doi: 10.1038/87555. [DOI] [PubMed] [Google Scholar]
- 3.Wolan DW, Cheong CG, Greasley SE, Wilson IA. Structural insights into the human and avian IMP cyclohydrolase mechanism via crystal structures with the bound XMP inhibitor. Biochemistry. 2004;43:1171–1183. doi: 10.1021/bi030162i. [DOI] [PubMed] [Google Scholar]
- 4.White RH. Purine biosynthesis in the domain Archaea without folates or modified folates. J. Bacteriol. 1997;179:3374–3377. doi: 10.1128/jb.179.10.3374-3377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graupner M, Xu H, White RH. New class of IMP cyclohydrolases in Methanococcus jannaschii. J. Bacteriol. 2002;184:1471–1473. doi: 10.1128/JB.184.5.1471-1473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ownby K, Xu H, White RH. A Methanocaldococcus jannaschii archaeal signature gene encodes for a 5-formaminoimidazole-4-carboxamide-1-β-D-ribofuranosyl 5′-monophosphate synthetase. A new enzyme in purine biosynthesis. J. Biol. Chem. 2005;280:10881–10887. doi: 10.1074/jbc.M413937200. [DOI] [PubMed] [Google Scholar]
- 8.Christendat D, Yee A, Dharamsi A, Kluger Y, Savchenko A, Cort JR, Booth V, Mackereth CD, Saridakis V, Ekiel I, Kozlov G, Maxwell KL, Wu N, McIntosh LP, Gehring K, Kennedy MA, Davidson AR, Pai EF, Gerstein M, Edwards AM, Arrowsmith CH. Structural proteomics of an archaeon. Nat. Struct. Biol. 2000;7:903–909. doi: 10.1038/82823. [DOI] [PubMed] [Google Scholar]
- 9.Saridakis V, Christendat D, Thygesen A, Arrowsmith CH, Edwards AM, Pai EF. Crystal structure of Methanobacterium thermoautotrophicum conserved protein MTH1020 reveals an NTN-hydrolase fold. Proteins. 2002;48:141–143. doi: 10.1002/prot.10147. [DOI] [PubMed] [Google Scholar]
- 10.Oinonen C, Rouvinen J. Structural comparison of NTN-hydrolases. Protein Sci. 2000;9:2329–2337. doi: 10.1110/ps.9.12.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sniegowski JA, Lappe JW, Patel HN, Huffman HA, Wachter RM. Base catalysis of chromophore formation in Arg96 and Glu222 variants of green fluorescent protein. J. Biol. Chem. 2005;280:26248–26255. doi: 10.1074/jbc.M412327200. [DOI] [PubMed] [Google Scholar]
- 12.Flaks JG, Erwin MJ, Buchanan JM. Biosynthesis of the purines. XVIII. 5-Amino-1-ribosyl-4-imidazolecarboxamide 5′-phosphate transformylase and inosinicase. J. Biol. Chem. 1957;229:603–612. [PubMed] [Google Scholar]
- 13.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 15.Matthews BW. Solvent content of protein crystals. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 16.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 17.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 18.Kleywegt GJ, Jones TA. Halloween mask and bones. In: Bailey S, Hubbard R, Waller D, editors. From First Map to Final Model. SERC Daresbury Laboratory; Warrington, UK: 1994. pp. 59–66. [Google Scholar]
- 19.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 20.Esnouf R. An extensively modified version of Molscript which includes greatly enhanced colouring capabilities. J. Mol. Graphics. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 21.Merritt EA, Bacon DJ. Raster3D: Photorealistic Molecular Graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 22.DeLano WL. The PyMOL Molecular Graphics Systems. 2002. [Google Scholar]
- 23.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 26.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 27.Vogt G, Woell S, Argos P. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997;269:631–643. doi: 10.1006/jmbi.1997.1042. [DOI] [PubMed] [Google Scholar]
- 28.Szilágyi A, Závodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure Fold. Des. 2000;8:493–504. doi: 10.1016/s0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 29.Perutz MF, Raidt H. Stereochemical basis of heat stability in bacterial ferredoxins and in haemoglobin A2. Nature. 1975;255:256–259. doi: 10.1038/255256a0. [DOI] [PubMed] [Google Scholar]
- 30.Walker JE, Wonacott AJ, Harris JI. Heat stability of a tetrameric enzyme, D-glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 1980;108:581–6. doi: 10.1111/j.1432-1033.1980.tb04753.x. [DOI] [PubMed] [Google Scholar]
- 31.Yip KS, Stillman TJ, Britton KL, Artymiuk PJ, Baker PJ, Sedelnikova SE, Engel PC, Pasquo A, Chiaraluce R, Consalvi V. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure. 1995;3:1147–1158. doi: 10.1016/s0969-2126(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 32.Vetriani C, Maeder DL, Tolliday N, Yip KS, Stillman TJ, Britton KL, Rice DW, Klump HH, Robb FT. Protein thermostability above 100 °C: a key role for ionic interactions. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12300–12305. doi: 10.1073/pnas.95.21.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabados E, Hindmarsh EJ, Phillips L, Duggleby RG, Christopherson RI. 5-Aminoimidazole-4-carboxamide ribotide transformylase-IMP cyclohydrolase from human CCRF-CEM leukemia cells: purification, pH dependence, and inhibitors. Biochemistry. 1994;33:14237–14245. doi: 10.1021/bi00251a036. [DOI] [PubMed] [Google Scholar]
- 34.Boger DL. Diels-Alder reactions of azadienes. Tetrahedron. 1983;39:2869–2939. [Google Scholar]
- 35.Vergis JM, Beardsley GP. Catalytic mechanism of the cyclohydrolase activity of human aminoimidazole carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase. Biochemistry. 2004;43:1184–1192. doi: 10.1021/bi035139b. [DOI] [PubMed] [Google Scholar]
- 36.Vergis JM, Bulock KG, Fleming KG, Beardsley GP. Human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/inosine 5′-monophosphate cyclohydrolase. A bifunctional protein requiring dimerization for transformylase activity but not for cyclohydrolase activity. J. Biol. Chem. 2001;276:7727–7733. doi: 10.1074/jbc.M009940200. [DOI] [PubMed] [Google Scholar]
- 37.Mueller WT, Benkovic SJ. On the purification and mechanism of action of 5-aminoimidazole-4-carboxamide-ribonucleotide transformylase from chicken liver. Biochemistry. 1981;20:337–344. doi: 10.1021/bi00505a017. [DOI] [PubMed] [Google Scholar]
- 38.Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- 39.Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 40.Barondeau DP, Putnam CD, Kassmann CJ, Tainer JA, Getzoff ED. Mechanism and energetics of green fluorescent protein chromophore synthesis revealed by trapped intermediate structures. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12111–12116. doi: 10.1073/pnas.2133463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JH, Krahn JM, Tomchick DR, Smith JL, Zalkin H. Structure and function of the glutamine phosphoribosylpyrophosphate amidotransferase glutamine site and communication with the phosphoribosylpyrophosphate site. J. Biol. Chem. 1996;271:15549–15557. doi: 10.1074/jbc.271.26.15549. [DOI] [PubMed] [Google Scholar]
- 42.Bartlett GJ, Borkakoti N, Thornton JM. Catalysing new reactions during evolution: economy of residues and mechanism. J. Mol. Biol. 2003;331:829–860. doi: 10.1016/s0022-2836(03)00734-4. [DOI] [PubMed] [Google Scholar]
- 43.Artymiuk PJ. A sting in the (N-terminal) tail. Nat. Struct. Biol. 1995;2:1035–1037. doi: 10.1038/nsb1295-1035. [DOI] [PubMed] [Google Scholar]
- 44.Brannigan JA, Dodson G, Duggleby HJ, Moody PC, Smith JL, Tomchick DR, Murzin AG. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 45.Anantharaman V, Aravind L, Koonin EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr. Opin. Chem. Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 46.Nagano N, Orengo CA, Thornton JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 47.Graham DE, Overbeek R, Olsen GJ, Woese CR. An archaeal genomic signature. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3304–3308. doi: 10.1073/pnas.050564797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maden BE. Tetrahydrofolate and tetrahydromethanopterin compared: functionally distinct carriers in C1 metabolism. Biochem. J. 2000;350(Pt 3):609–629. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.