

Abstract
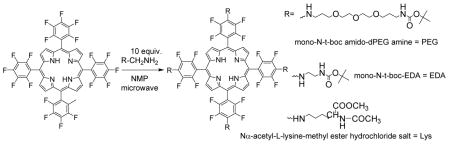
We report an efficient and rapid means for the synthesis of tetrapentafluorophenyl porphyrin (TPPF20) derivatives by microwave irradiation in an environmentally acceptable solvent. The selective displacement of the para-fluorine groups in TPPF20 by primary amines occurs in yields between 70–95%. This method demonstrates that TPPF20 is an ideal platform for the rapid formation of porphyrin conjugates for therapeutic, catalytic and other applications.
Applications of porphyrin derivatives range from catalysts,1 materials, devices2 to photodynamic therapeutic agents (PDT)3 because of their rich photochemistry and redox chemistry. However, most applications require modification of the porphyrin macrocycle to allow attachment of additional substituents with various other functionalities.
Microwave-assisted reactions have become increasingly important in chemical synthesis in the last 20 years due to the advantages it provides over conventional heating methods.4 Significant reduction in reaction times, side reactions, increased yields, ease of purification and minimization of the amount of solvent used are only a few of these desirable qualities.5 Shorter reaction times (usually <15 min) allow rapid investigation into new methodologies and reaction optimization. Microwave-assisted reactions are believed to facilitate polarization of the substrates thereby promoting the reactions.6
We previously demonstrated a solvent-free7 porphyrin synthesis, and solid-supported reactions minimizes the amount of subsequent purification.8 In view of the significant need for rapid synthesis of porphyrin derivatives – especially for therapeutic applications – we present methods for the facile preparation of porphyrin derivatives around a core platform of 5,10,15,20-tetrakis(2,3,4,5,6-pentafluorophenyl)porphyrin (TPPF20). As a demonstration of the method, a variety of amphipathic moieties are grafted onto the core for evaluation as photodynamic therapeutic (PDT) agents.
Previously reported TPPF20 derivatives were typically synthesized in refluxing DMF with a large excess of the amine or thiol reagents,9 but the yields were relatively poor and the substitution was sometimes incomplete. Better yields, ~90%, are obtained by using thiol reagents, and stirring for ~12 h at room temperature in DMF with a stoichiometric amount of dialkylamine.3a,c To decrease the reaction time, improve yields, and employ a greener solvent, we developed a synthesis using microwave irradiation (MW) in N-methylpyrrolidone.
A diverse array of primary amines were chosen to demonstrate the method. Polyethylene glycols (PEG) are relevant to drug design and delivery as they enable the facile uptake of drugs into cells10; polylysine derivatives because these moieties are known to impart selectivity towards cancer cells; and a polyamine because these impart both cancer cell selectivity and cell permeability. The functional groups other than the intended amine are t-boc protected.
Reaction of 5–10 mg (5.1 or 10.2 μmol) TPPF20 with 10 equivalents of a primary amine in 1–2 mL N-methylpyrrolidone (NMP) at 60–70 °C overnight leads to mixtures of six different TPPF20 derivatives. However, the same amounts of porphyrin and amine in 100μL of NMP and 10 minutes MW irradiation yields only the tetra substituted products. The reaction time was dramatically reduced (see Table 1 and abstract). The nucleophilic aromatic substitution of the para-fluoro group by primary amines take 10–30 minutes in this procedure compared to 1–2 days with conventional heating.11 A significant added advantage for the MW reaction, is that it tolerates functional groups that can decompose under extended heating.
Table 1.
Comparison of Conventional versus Microwave Heating in the Synthesis of Protected Porphyrin Derivativesa
| porphyrin derivative | solvent | Conventional heatingb | yield | Microwave irradiation | yield |
|---|---|---|---|---|---|
|
|
NMP | 2 days | ~50% | 12 min | 94 ± 1% |
|
|
NMP | 1 day | ~50% | 10 min | 94 ± 2% |
|
|
NMP | 20 h | 72% | 30 min | 77 ± 5% |
Reaction conditions: 5.1 μLmol of TPPF20, 10 equivalent of amine in 100 μL of N-methylpyrrolidone were irradiated for the times indicated at 2 min intervals. The solvent was removed in vacuo, the residue was purified by silica gel column chromatography.
In an oil bath maintained at 60–70 °C.
The improved yields of the tetra-substituted derivatives are concomitant with the reduced amounts of incompletely reacted products. Also, prolonged heating in DMF results in the fragmenting of the PEG moieties (data not shown). A 95% yield of the Por-PEG4 product indicates that each reaction on the porphyrin proceeds with ~99% efficiency. In general, the protected acids are used because these are more soluble in the NMP and are found to result in greater yields. The deprotection of these groups is readily accomplished using literature procedures. For the EDA and the PEG the amide esters are cleaved using HCl in MeOH. For the Lys derivative, 6N HCl is used to cleave both the acetamide and methyl ester groups.
These conditions are specific for para fluoride substitution by primary amines, because under similar reaction conditions secondary amines exhibit no reactivity. Refluxing of TPPF20 in DMF is known to yield dimethylamino substitution on the para position, thus previous methods require temperatures <60 °C and extended reaction times. When 5 mg TPPF20 in DMF is heated in the microwave for 10 minutes, several dimethylamino derivatives are observed. Procedures in other solvents, such as toluene, DMSO and morpholine generally result in low reactivity or significantly diminished yields compared to NMP.
All porphyrins were characterized by electrospray ionization mass spectrometry (ESI-MS, see Supporting Information). For all porphyrins, the parent [M+H+] was seen and in some cases the sodium adduct. The electronic absorption spectra of the porphyrins were characterized at 1μM concentrations and were found to be typical for non-aggregated porphyrins. Substitution of the electron withdrawing fluorine with the electron donating amine results in a 7–9 nm red shift in the Soret. Table 2 summarizes the UV-Visible spectra of these porphyrins in methanol. All the derivatives show similar spectra, including the B band or Soret band at 417–419 nm, the Q-band at 508–509 nm together with three vibronic bands at 543–545 nm, 585–587 nm, and 643–649 nm. A singlet for the pyrrole H in the 1H-NMR indicates the presence of the four para substituents, whereas incomplete substitution results in multiplets for this proton.
Table 2.
ESI-MS, UV-Vis and Emission data of Protected Porphyrin Derivatives
| derivative | massa | UV-visb | emissionb |
|---|---|---|---|
| TPPF20 | 975 | 406, 502 (536, sh), 580 (634, sh) | 640, 707 |
| Por-PEG4 | 2176 | 418, 509, 545, 586, 643 | 652, 714 |
| Por-EDA4 | 1535 | 417, 508, 543, 585, 644 | 647, 713 |
| Por-Lys4 | 1703 | 419, 509, 545 587, 649 | 652, 711 |
[M+H]+
Absorption and emission in and methanol.
The X-ray crystal structure of Por-EDA4 derivative was elucidated (Fig. 1). This derivative was crystallized from layered hexane over toluene at room temperature over several weeks. There are two different EDA conformations, ‘bent’ and extended; the bent conformation organized by an intramolecular H-bond. The most interesting aspect of the structure is that it forms a two-dimensional lattice (Fig. 2) organized by intermolecular N-H· · · ·O=C hydrogen bonds. The bent EDA moieties form a cyclic structure mediated intramolecular hydrogen bonds. The extended EDA moieties on opposite sides of the porphyrin H-bond to the bent EDA moieties on adjacent porphyrins. (See packing diagram Fig. 2). There is a surprising lack of porphyrin pi-stacking in the crystal structure, and the layers are organized by dispersion and packing forces. The intramolecular and intermolecular hydrogen-bonding motifs were not expected, and may explain the aggregation of these compounds at unexpectedly low concentrations.
Figure 1. X-ray Crystal Structure of Por-EDA4.
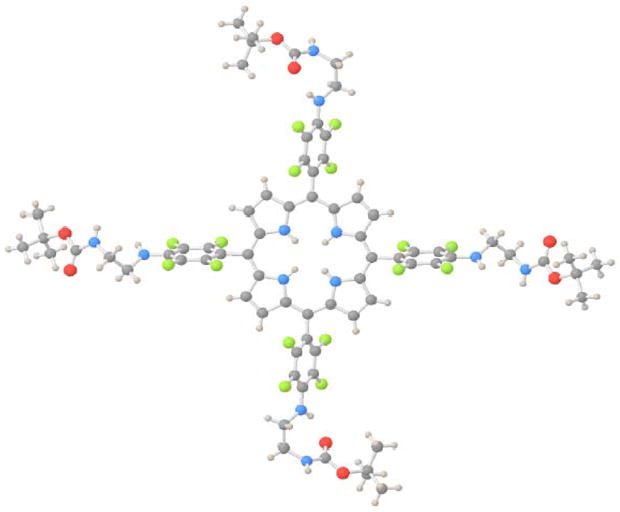
H white; C grey; N blue; O red; F green.
Figure 2. X-ray Crystal Packing Structure of Por-EDA4 Illustrating the Inter- and Intra- molecular H-bonding.
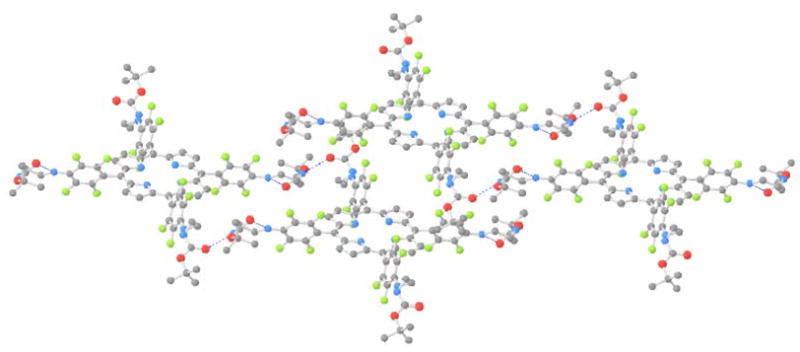
H white; C grey; N blue; O red; F green
One of the immediate goals for these porphyrin derivatives is to evaluate their uptake into cancer cells. Thus the solubility of these protected derivatives in an aqueous cell culture medium (Dulbecco’s modified eagle medium without phenol red or phosphate buffered saline) must be considered. The absorption spectra of Por-EDA4 in this medium show a red shift and substantial broadening of the Soret band, from 417 nm to 431 nm (supporting information), and similar spectral changes are observed for the other derivatives. These changes in the electronic spectra are indicative of aggregate formation.
The fluorescence emission of these derivatives in methanol and aqueous solvents was also recorded. For Por-EDA4 in methanol, excitation at 417 nm in the Soret band, resulted in a strong fluorescence emission at 647–652 nm with a Stokes shift of 7–12 nm, which is typical of porphyrins. In the cell culture medium, the ground state spectra are red shifted by 6–10 nm, thus there is a similar red shift in the emission bands (Table 2).
Based on the above results for primary amines we hypothesized that combinatorial libraries of porphyrins can be created by this method as long as the individual reagents react with the TPPF20 with similar rates and efficiencies. As a proof-of-principle experiment, a 1:1:1 mixture of the three amine reagents were used in a solution phase combinatorial reaction. The total amount of the three primary amines were present in the reaction at the same 10:1 ratio with the porphyrin as used above. Statistically this should yield 21 compounds, but ESI-MS analysis indicated incomplete reactivity as a significant number of compounds with one or more unsubstituted fluorines were observed. Sequential addition of the less reactive amine followed by the more reactive amines also yields inconsistent results.
In summary, protected amine conjugates of tetrapentafluorophenyl porphyrin with polyethylene glycol, ethylenediamine, and lysine groups were synthesized by microwave irradiation. The protocol described allows for significant reduction in reaction times yet results in high yields and limits by-product formation compared to earlier reports.11 The procedure uses commercially availabe reagents at ca. 10-fold greater concentrations in an environmentally friendly solvent. In general the MW method works for primary nucleophiles significantly better than secondary, and better for amines than for sulfides. 12
In future studies, the prepared protected and deprotected porphyrin conjugates will be assessed for their photodynamic efficiency using MDA-MB-231 cancer cells. The crystal structure indicates these types of porphyrins may self-organize into photonic materials.
Supplementary Material
Experimental procedures and full spectral data for new compounds and x-ray data. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH SCORE (GM60654), NSF CHE-0554703 and IGERT DGE-9972892. DS acknowledges NSF LS-AMP and MAGNET graduate fellowships. Hunter College Chemistry infrastructure is supported by the NSF, NIH, including the RCMI program (G12-RR-03037), and the City University of New York. We thank Dr. Mikki Vinodu of Hunter College of the City University of New York for careful reading of the manuscript.
References
- 1.(a) Benaglia M, Danelli T, Fabris F, Sperandio D, Pozzi G. Org Lett. 2002;4:4229. doi: 10.1021/ol0267230. [DOI] [PubMed] [Google Scholar]; (b) Yang J, Weinberg R, Breslow R. Chem Commun. 2000:531. [Google Scholar]
- 2.Drain CM, Hupp JT, Suslick KS, Wasielewski MR, Chen X. J Porphyrins Phthalocyanines. 2002;6:243. [Google Scholar]
- 3.(a) Chen X, Hui L, Foster DA, Drain CM. Biochemistry. 2004;43:10918. doi: 10.1021/bi049272v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sylvain I, Zerrouki R, Granet R, Huang YM, Lagorce JF, Guilloton M, Blais JC, Krausz P. Bioorg Med Chem. 2002;10:57. doi: 10.1016/s0968-0896(01)00255-3. [DOI] [PubMed] [Google Scholar]; (c) Pasetto P, Chen X, Drain CM, Franck RW. Chem Commun. 2001:82. [Google Scholar]
- 4.Sauer DR, Kalvin D, Phelan KM. Org Lett. 2003;5:4721. doi: 10.1021/ol0358915. [DOI] [PubMed] [Google Scholar]
- 5.Yoon DS, Han Y, Stark TM, Haber JC, Gregg BT, Stankovich SB. Org Lett. 2004;6:4775. doi: 10.1021/ol047919y. [DOI] [PubMed] [Google Scholar]
- 6.(a) Liu MO, Tai CH, Wang WY, Chen JR, Hu AT, Wei TH. J Organomet Chem. 2004;689:1078. [Google Scholar]; (b) Liu MO, Hu AT. J Organomet Chem. 2004;689:2450. [Google Scholar]; (c) Boufatah N, Gellis A, Maldonado J, Vanelle P. Tetrahedon. 2004;60:9131. [Google Scholar]
- 7.Drain CM, Gong X. Chem Commun. 1997:2117. [Google Scholar]
- 8.(a) Chaouchi M, Loupy A, Marque S, Petit A. Eur J Org Chem. 2002:1278. [Google Scholar]; (b) Mojtahedi MM, Saidi MR, Shirzi JS, Bolourtchian M. Syn Commun. 2002;32:851. [Google Scholar]
- 9.(a) Shaw SJ, Elgie KJ, Edwards C, Boyle RW. Tetrahedron Lett. 1999;40:1595. [Google Scholar]; (b) Suzuki M, Shimizu S, Shin JY, Osuka A. Tetrahedron Lett. 2003;44:4597. [Google Scholar]; (c) Battioni P, Brigaud O, Desvaux H, Mansuy D, Traylor TG. Tetrahedron Lett. 1991;32:2893. [Google Scholar]
- 10.(a) Molineux G. Pharmacotherapy. 2003;23:3S. doi: 10.1592/phco.23.9.3s.32886. [DOI] [PubMed] [Google Scholar]; (b) Hamblin MR, Miller JL, Rizvi I, Ortel B, Maytin EV, Hasan T. Cancer Res. 2001;61:7155. [PubMed] [Google Scholar]
- 11.Battioni P, Brigaud O, Desvaux H, Mansuy D, Traylor TG. Tetrahedron Lett. 1991;32:2893. [Google Scholar]
- 12.General Procedure for the Preparation of Amine Derivatives of TPPF20 by Microwave Irradiation. Into a 3.4 mL vial, 5 mg of 5,10,15,20-tetrakispentafluorophenylporphyrin (5.1 μmol) and 10 equivalents of mono-N-t-boc EDA (Quanta BioDesign) in NMP (0.1 mL) was added. The closed vial was irradiated using a domestic microwave oven (1100 W, Samsung MW4250W) at 2 min intervals until no starting material was visualized by TLC (10 min). After the vial was cooled, the solvent (NMP) was removed in vacuo. The reaction mixture was purified on a silica gel prep TLC plate (hexane : ethyl acetate : methanol, 2 : 1.5 : 0.5). The porphyrin was redissolved in methanol and evaporated under reduced pressure to afford >90 % yield.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and full spectral data for new compounds and x-ray data. This material is available free of charge via the Internet at http://pubs.acs.org.