

Abstract
ABCG2 is a half-transporter initially described in multidrug-resistant cancer cells and lately identified as an important factor in the pharmacokinetics of its substrates. Q141K is by far the most intensively studied single nucleotide polymorphism of ABCG2 with potential clinical relevance. Here we used stably transfected HEK cells to study the Q141K polymorphism together with the deletion of amino acids 315–316, which were recently reported to coexist in two cancer cell lines (A549 and SK-OV-3). Functional studies confirmed our previous report that when normalized to surface expression, Q141K has impaired transport of mitoxantrone. This result was extended to include the ABCG2-specific substrate pheophorbide a. While we found no functional consequence of deleting amino acids 315 and 316, we did find that the deletion mutant is no longer recognized by the BXP-21 antibody. We conclude that amino acids 315 and 316 form part of the epitope for the BXP-21 antibody.
Keywords: ABCG2, ABC transporter, Multidrug resistance, Single nucleotide polymorphism, Mitoxantrone, Pheophorbide a
Introduction
It has long been known that large interindividual differences exist in plasma levels, efficacy, tolerance, and adverse effects of drugs, and pharmacogenomics is among one of the fastest growing medical fields in this century. To explore and address these differences is crucial in cancer chemotherapy, where patients could benefit from targeted drugs administered at effective doses and with minimized side effects. Some of the interindividual differences can be attributed to single nucleotide polymorphisms (SNPs) of drug metabolizing enzymes and efflux transporters. ATP-binding cassette (ABC) transporters are a group of membrane proteins that use the energy of ATP hydrolysis to transport a wide variety of substrates across biologic membranes, and thus play a crucial role in the absorption, distribution, and excretion of drugs [1]. ABCG2 is a member of the G subfamily of human ABC transporters. The protein is a half-transporter that requires homodimerization for function [2]. ABCG2 was first identified in multidrug-resistant carcinoma cell lines and transports several chemotherapeutic agents such as topotecan, mitoxantrone, flavopiridol, irinotecan, methotrexate, gefitinib, and imatinib [3–9]. However, the transporter’s role in clinical multidrug resistance has yet to be evaluated [10]. Meanwhile, the list of ABCG2 substrates is rapidly growing, including drugs from multiple pharmacological groups, such as antibiotics, HMG-CoA reductase inhibitors, antivirals, flavonoids, and porphyrins [11]. This broad substrate specificity together with the large number of known inhibitors of ABCG2 suggests a major role for the protein in pharmacology [12].
To date, more than 50 SNPs of the ABCG2 gene have been identified, some of which were shown to affect protein expression levels and function. The most extensively studied of these SNPs with potential clinical relevance is 421 C>A resulting in a glutamic acid to lysine substitution (Q141K) in the protein. The Q141K SNP was identified with varying frequencies in different ethnic groups and was found to be the most prevalent in the Japanese population (∼30%). Q141K has been associated with lower levels of protein expression and impaired transport in vitro, though some controversies exist in the publications characterizing this SNP [13–17]. The polymorphism has also been studied in vivo; patients carrying the SNP were found to have elevated plasma levels of gefitinib and diflomotecan, and increased bioavailability of oral topotecan [18–20]. Further, Q141K was associated with a higher incidence of diarrhea in non-small cell lung cancer patients treated with gefitinib [21]. Sequencing the ABCG2 gene in the 60 cancer cell lines maintained and used for screening in the National Cancer Institute by various groups revealed that ten of the cell lines carried the Q141K polymorphism. Interestingly, A549 and SK-OV-3 cells were reported to have an additional polymorphism, a splicing variant occurring at the mRNA level, resulting in the deletion of amino acids 315 and 316 [16]. This splicing variant was also identified in MCF7 and HT-29 cells, which are wild type at residue 141. In the present study, we expanded our previous functional studies on the Q141K SNP [13] using pheophorbide a, a fluorescent compound that was recently reported by our group as an ABCG2-specific substrate [22]. In addition, we examined the effect of the Δ315–316 variant with or without the Q141K polymorphism on ABCG2 function.
Materials and methods
Cell culture
Human embryonic kidney (HEK) 293 cells (ATCC, Manassas, VA) were maintained in Minimal Essential Medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Invitrogen), 2 mM glutamine (BioFluids, Rockville, MD), and 100 units/L penicillin/streptomycin (BioFluids) at 37°C in 5% CO2. Stably transfected cell lines were maintained in 2 mg/ml G418 (Invitrogen).
Mutagenesis and transfection
The Δ315–316 and the Q141K/D315–316 mutants were generated by site-directed mutagenesis in the pcDNA3.1/Myc-HisA(-) vector (Invitrogen) as previously described [23]. The wild type (R482), Q141K, R482G, and empty vector-transfected (pcDNA) cells were previously reported [13]. All mutations were confirmed by sequencing the plasmids. The full-length ABCG2 insert was also sequenced from genomic DNA from one representative clone of each stable transfectant generated in HEK 293 cells. Transfections were performed using TransFast transfection reagent (Promega, Madison, WI). Colonies were selected in 2 mg/ml G418 with frequent removal of dead cells and were expanded prior to study.
Membrane preparation and immunoblotting
Microsomal membrane preparation and immunoblotting were performed as described previously [23]. Briefly, cells were disrupted by nitrogen cavitation (Parr Instrument, Moline, IL) in a hypotonic lysis buffer, and membranes were obtained by ultracentrifugation at 40,000 rpm. Membrane proteins were loaded onto precast 7.5% (w/v) SDS-polyacrylamide gels (Bio-Rad, Hercules, CA), subjected to electrophoresis, and electrotransferred onto PVDF membranes (Millipore, Bedford, MA). Blots were probed with a 1:250 dilution of the monoclonal anti-ABCG2 antibody BXP-21 (Kamiya Biomedical, Seattle, WA), or a 1:1000 dilution of the 405 polyclonal antibody, followed by incubation in anti-mouse IRDye800® and anti-rabbit IRDye680® secondary antibodies (LI-COR Biosciences, Lincoln, NE) and visualized with the Odyssey Imaging System (LI-COR). Membranes were stained with 0.1% Ponceau S (Sigma, St. Louis, MO) and checked for comparable loading.
Flow cytometry
Flow cytometry with the anti-ABCG2 antibody, 5D3 (eBioscience, San Diego, CA), was performed as previously described [23]. Briefly, following trypsinization, cells were incubated with phycoerythrin-conjugated 5D3 and control antibodies for 30 min at room temperature, washed twice with DPBS, and kept in the dark until analysis. For transport studies, cells were trypsinized, resuspended in complete media containing 20 μM mitoxantrone (Sigma) or 10 μM pheophorbide a (Frontier Scientific, Logan, UT) with or without 10 μM of the ABCG2-blocker, Fumitremorgin C (FTC), and incubated for 30 min at 37°C in 5% CO2. (FTC was synthesized by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, National Institutes of Health, Bethesda, MD.) Cells were then incubated for 1 h at 37°C in substrate-free media, continuing with or without 10 μM FTC, centrifuged and resuspended in DPBS prior to analysis on a FACSort flow cytometer, equipped with both a 488 nm argon laser and a 635 nm red diode laser.
Data analysis
For statistical analysis, data generated in flow cytometry experiments with two or three clones of each mutant and the wild type were used. Experiments were repeated eight times with mitoxantrone and five times with pheophorbide a with parallel measurement of ABCG2 surface expression with the 5D3 antibody each time. The FTC-inhibitable mitoxantrone and pheophorbide a efflux was determined as the mean channel number difference between the efflux with FTC and efflux without FTC histograms. ABCG2 surface expression was determined from the difference between the mean channel numbers for 5D3 and negative control histograms. To determine the transport efficacy of each clone, the FTC-inhibitable efflux was divided by the ABCG2 surface expression, generating the ratios plotted in Fig. 4.
Fig. 4.
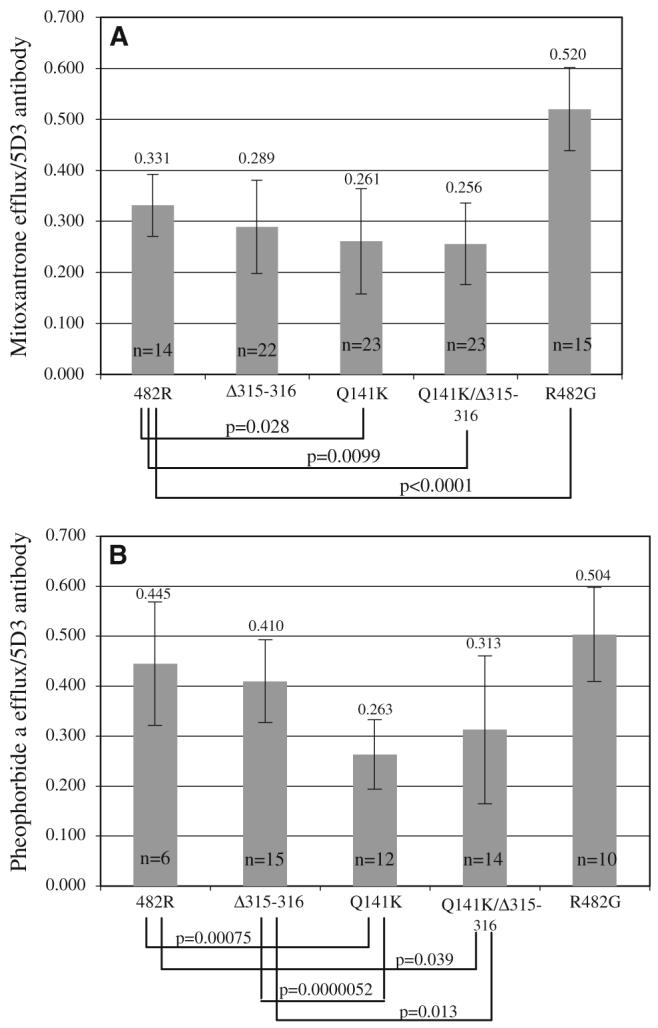
Efflux normalized to cell surface expression. The mean FTC-inhibitable mitoxantrone (panel A) and pheophorbide a (panel B) efflux was plotted for the wild type (482R) and each mutant (Q141K, Δ315–316, aQ141K/Δ315–316, and R482G). For each measurement, efflux by flow cytometry was normalized to simultaneously determined ABCG2 surface expression by the 5D3 antibody. Two or three clones for each mutant and the wild type were used; the experiments were repeated multiple times and the number of measurements used to create the average is indicated (n). Differences with significant unadjusted P-values (P<0.05) are indicated. The numbers above the bars represent the mean values plotted
First, we used a Spearman correlation analysis to determine that within one clone none of the efflux and surface expression values showed strong inverse correlation. Once strong inverse correlations were ruled out, then a Kruskal—Wallis test was used to determine if the three clones of particular mutant, or a Wilcoxon rank sum test if the two clones of a mutant, were significantly different from one another. Following the determination that the clones were not significantly different, a Q-test was performed on the transport efficacy ratios created as described above for each clone in order to determine if outliers indicative of invalid results were present. Based on the results of the Q-test, five of the data points were removed from the analysis. Finally, a Wilcoxon rank sum test was used to compare the ratios for the mutants to that of the wild type and to each other. All P-values are two-tailed and presented without formal adjustment for multiple comparisons.
Results
Given the importance of understanding the impact of genetic variability on the pharmacology of anticancer agents, the role of polymorphisms in ABCG2 has been widely studied. A recent report added a deletion of amino acids 315–316 to the list of ABCG2 polymorphisms. To determine the effect that deleting the alanine at 315 and the threonine at 316 has on the transport activity of ABCG2, we generated stable transfectants in HEK 293 cells (Δ315–316). We have previously used the same expression system to study non-synonymous SNPs, such as Q141K, V12M, and D620N, and found that Q141K results in impaired function [13]. Given that Q141K and Δ315–316 were found to coexist in the A549 and SK-OV-3 carcinoma cell lines, we also generated mutants carrying both the deletion and the Q141K SNP (Q141K/Δ315–316). First, flow cytometry with the 5D3 monoclonal antibody recognizing a yet unidentified extracellular epitope of ABCG2 was performed on non-permeabilized cells to analyze surface expression. We selected three clones of both the Δ315–316 and the Q141K/Δ315–316 mutants that displayed significant levels of surface expression (Fig. 1) and expanded these clones for further studies.
Fig. 1.

ABCG2 surface expression in the mutants. Flow cytometry with the 5D3 monoclonal antibody recognizing an extracellular epitope of ABCG2 is shown for the wild type and three clones of both mutants (Δ315–316 and Q141K/Δ315–316, with clone numbers indicated). Stably transfected HEK 293 cells were trypsinized and incubated for 30 min in phycoerythrin-labeled negative control antibody (solid line) or 5D3 antibody (dashed line) and analyzed in a FACSsort flow cytometer. The distance between the solid and dashed lines is representative of the amount of ABCG2 expressed on the cell surface
Crude membranes extracted from the Δ315–316 and the Q141K/Δ315–316 clones and from the wild type and Q141K transfectants previously generated in our lab were subjected to immunoblotting with the commercially available BXP-21 monoclonal antibody. Surprisingly, despite sufficient surface expression levels seen on flow cytometry, none of the Δ315–316 and Q141K/Δ315–316 clones were detectable on immunoblot with the BXP—21 antibody (Fig. 2), while the wild type and the Q141K mutant clones were readily detected. The exact epitope of the BXP-21 antibody has so far been unknown; the immunogen used to generate the antibody was a fusion protein composed of E. coli maltose binding protein and a peptide containing amino acids 271–396 of ABCG2 (Kamiya Biomedical Company data Sheet, 2006). Since amino acids 315 and 316 are included in the peptide, we surmised that they are part of the BXP-21 epitope and that the antibody no longer binds to the ABCG2 protein when these two amino acids are deleted. To prove this, we used the 405 polyclonal antibody created in our laboratory [24] on the immunoblot previously probed with the BXP-21 antibody and found that with this antibody the Δ315–316 and the Q141K/Δ315–316 clones were also detectable, though the 405 antibody gives a weaker signal when compared to BXP-21 (Fig. 2). This result suggests that residues 315 and 316 are indeed critical residues in the epitope of the BXP-21 antibody.
Fig. 2.

The Δ315–316 mutant is not detected on immunoblot with the BXP-21 antibody. Membrane proteins extracted from HEK 293 cells stably transfected with the wild type (482R) and the mutants (Q141K, Δ315–316, and Q141K/D315–316) were separated on SDS/PAGE (18 μg/lane), transferred onto a PVDF membrane, and incubated overnight with the mouse monoclonal anti-ABCG2 antibody BXP-21 (upper panel) or the 405 rabbit polyclonal anti-ABCG2 antibody (lower panel). Two clones are shown for the Q141K mutant and three clones for the Δ315–316 and Q141K/Δ315–316 mutants, with clone numbers indicated. Membranes were stained with 0.1% Ponceau S and checked for comparable loading (not shown)
We next utilized a flow cytometry-based assay measuring the efflux of mitoxantrone and pheophorbide a to determine the activity of the mutants. In this assay, cells are incubated with the fluorescent compounds with or without the ABCG2-inhibitor FTC. Figure 3 shows a representative example of surface expression, mitoxantrone and pheophorbide a efflux for each mutant compared to the wild type and the R482G mutant which is a previously characterized, so-called gain-of-function mutant with a wider substrate specificity than wild type ABCG2 [25]. We previously demonstrated that transport correlates with surface protein expression in cells carrying the wild type protein [26]. For the Q141K variant, where a reduced surface expression has been confirmed [13, 16, 17], our previous studies also demonstrated impaired transport when mitoxantrone efflux was normalized to surface expression [13]. Figure 4 shows that this impairment in transport for mitoxantrone was confirmed in the present study and extended to the substrate pheophorbide a. FTC-inhibitable efflux was determined as the mean channel number difference between the efflux with FTC and the efflux without FTC histograms. Surface expression was defined as the difference between the mean channel numbers for the 5D3 and negative control histograms. The FTC-inhibitable efflux value was normalized by the surface expression for each experiment. The average of the ratios calculated in this manner for each mutant and the wild type is presented in Fig. 4. A Q-test was performed on the ratios for each clone separately to identify outliers that were excluded from further analysis. Using a Wilcoxon rank sum test, differences with unadjusted P-values <0.05 were found when the wild type was compared to the Q141K and Q141K/Δ315–316 mutants for both mitoxantrone and pheophorbide a. In agreement with previous reports, the transport efficacy of the R482G mutant was found to be significantly higher than that of the wild type for mitoxantrone [13]. The average ratio calculated for pheophorbide a for the Δ315–316 mutant was significantly higher than that for the Q141K and the Q141K/Δ315–316 mutants. In summary, the mutant carrying the deletion was similar in transport efficacy to the wild type for both substrates, while any mutant carrying the Q141K was impaired.
Fig. 3.

Mitoxantrone and pheophorbide a efflux. First column:▶ ABCG2 surface expression with the 5D3 antibody for one representative clone of each mutant (as described in Fig. 1). Second and third columns: mitoxantrone and pheophorbide a efflux for the same mutants, respectively. For the efflux experiments, stably transfected cells were trypsinized and incubated for 30 min in complete media containing 20 μM mitoxantrone or 1 μM pheophorbide a with or without 10 μM of the ABCG2-blocker FTC. Efflux is represented by the shift between the histograms with (dashed line) and without (solid line) FTC
Discussion
ABCG2 is an ABC half-transporter initially described in multidrug-resistant cancer cells, but now understood to be an important determinant of oral bioavailability, the blood-brain barrier, and the maternal-fetal barrier with a wide range of substrates and inhibitors, including several drugs currently in clinical use. Of the numerous SNPs identified so far in the ABCG2 gene, 421 C>A, resulting in a mutant protein with a glutamine to lysine substitution at residue 141 (Q141K), is the most extensively studied and is suggested to have impaired transport function. Interestingly, some cell lines of the NCI-60 panel were reported to have the Q141K SNP together with a splicing variant resulting in the deletion of amino acids 315 and 316. Here, we investigated the functional consequences of the 315–316 deletion with/or without the Q141K mutation in stably transfected HEK 293 cells using a flow cytometry-based assay to analyze mitoxantrone and pheophorbide a efflux. After normalizing the measured efflux values to ABCG2 expression by the 5D3 monoclonal antibody, we have found that the Δ315–316 mutant has equivalent efflux capacity to the wild type, while the double mutant (Q141K/Δ315–316) was similar to the Q141K SNP. Additionally, the experiments reconfirmed that the Q141K SNP has significantly impaired function compared to the wild type protein when normalized by surface expression.
This latter observation is built upon the ability of the 5D3 antibody to recognize an external epitope of ABCG2. Some investigators have recently shown that recognition of ABCG2 by 5D3 at low antibody concentrations is subject to conformational changes in the protein [27], a phenomenon that could have an effect on ABCG2 levels measured in our study. However, at the high antibody dilutions used here, we have shown that measuring expression of ABCG2 with the 5D3 antibody correlated well (r2 = 0.87) with pheophorbide a transport in a series of drug-selected cell lines [22], validating our use of 5D3 surface staining to normalize transport function by surface expression.
Finally, it should be noted that the gain-of-function feature of the R482G mutant was again demonstrated, although the latter was only significant when mitoxantrone was used. While mutation of the arginine at position 482 of the ABCG2 protein to a threonine or glycine has been shown to result in the acquisition of rhodamine 123 and anthracyline transport and increased mitoxantrone compared to wild type protein [25, 28], the mutation does not universally affect all substrates. We have previously shown that SN-38 transport is only weakly affected by the 482 mutation [28] and it has also been demonstrated that transport of Hoechst 33342 did not differ in cells expressing mutant or wild type ABCG2 [29]. Similarly, Miwa and colleagues transfected PA317 mouse fibroblast cells with ABCG2 harboring 15 different mutations at position 482 and reported that, while most mutations conferred increased resistance to mitoxantrone compared to cells expressing wild type protein, SN-38 resistance was largely unaffected [30]. The mechanism underlying this gain-of-function has not been elucidated. Particularly perplexing is the observation that almost any amino acid substitution results in a protein that demonstrates increased transport efficiency for rhodamine and mitoxantrone [30,31]. Interestingly, it has been reported that mutations at amino acid 482 do not affect drug binding, as rhodamine 123, doxorubicin, and the photoaffinity label [125I]-iodoarylazidoprazosin have been shown to bind similarly to wild type and mutant ABCG2 [31, 32].
Residues alanine 315 and threonine 316 are localized in the so-called linker region of the ABCG2 protein between the nucleotide-binding domain (NBD) and the transmembrane domain (TMD), which is thought to be responsible for the communication between the two domains. This part of the protein is not conserved in the ABCG subfamily. When we generated a computer model of ABCG2 based on available crystal structure of ABC transporters [33], a stretch of approximately 60 amino acids in the linker region could not be modeled due to lack of homologous regions in those structures.
Given that the BXP-21 monoclonal antibody is the most frequently used antibody for detection of ABCG2 on immunoblot and in immunohistochemistry in the literature, our finding that it does not recognize the protein when amino acids 315 and 316 are absent has significance and it might explain some discrepancies in laboratory studies, since the cell lines carrying this splicing variant would falsely be identified as negative for ABCG2 protein expression using these methods. This would also be true for tumors from patients with the same polymorphism. However, review of several publicly available SNP databases (Entrez SNP, JSNP, Genome Variation Server, HapMap) revealed that the Δ315–316 polymorphism has not yet been reported in individuals. This could mean that similar to the R482G mutation [34] this splicing variant will be found only in cell culture. Note, however, that most of the data entered into these databases are obtained from sequencing genomic DNA, and a splicing variant such as the 315–316 deletion would not be detected. Further, the splicing variant may also not be prevalent in a given individual or cell type. Indeed, we could not detect the Δ315–316 in sequencing several cell lines, including A549 and MCF-7, in our laboratory. A definitive test of the frequency of this variant would go beyond the scope of our work.
In conclusion, the results presented here confirm our previous finding that the clinically relevant, frequently occurring Q141K SNP impairs transport of the chemotherapeutic agent mitoxantrone by ABCG2 [13]. In addition, we report similar results with the chlorophyll catabolite pheophorbide a, a known ABCG2 substrate. We believe the Q141K mutation results in decreased surface expression due to impaired trafficking of ABCG2 [13], which, combined with impaired transporter function, effects a clinically detectable reduction in transporter function [19]. On the other hand, deleting amino acids 315 and 316 of the ABCG2 protein did not have any functional consequence. Finally, we have identified these two residues as part of the epitope for the commercially available BXP-21 antibody. These studies add to our general understanding of ABCG2, a transporter of major interest for its role in clinical pharmacology and normal tissue protection.
Acknowledgments
The authors would like to thank Elizabeth Finley for help with the flow cytometry experiments.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 2.Ozvegy C, Litman T, Szakacs G, Nagy Z, Bates S, Varadi A, Sarkadi B. Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem Biophys Res Commun. 2001;285:111–117. doi: 10.1006/bbrc.2001.5130. doi:10.1006/bbrc.2001.5130
- 3.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–5339. [PubMed] [Google Scholar]
- 4.Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, Brangi M, Greenberger L, Dean M, Fojo T, Bates SE. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- 5.Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y, Ross DD, Bates SE, Kruh GD. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63:4048–4054. [PubMed] [Google Scholar]
- 6.Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–4563. [PubMed] [Google Scholar]
- 7.Ozvegy-Laczka C, Hegedus T, Varady G, Ujhelly O, Schuetz JD, Varadi A, Keri G, Orf L, Nemet K, Sarkadi B. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol. 2004;65:1485–1495. doi: 10.1124/mol.65.6.1485. doi: 10.1124/mol.65.6.1485
- 8.Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, Senderowicz AM, Ross DD, Bates SE. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (MXR/BCRP/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001;7:145–152. [PubMed] [Google Scholar]
- 9.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. doi:10.1073/pnas.95.26.15665
- 10.Polgar O, Bates SE. ABC transporters in the balance: is there a role in multidrug resistance? Biochem Soc Trans. 2005;33:241–245. doi: 10.1042/BST0330241. doi:10.1042/BST0330241
- 11.Krishnamurthy P, Schuetz JD. Role of abcg2/bcrp in biology and medicine. Annu Rev Pharmacol Toxicol. 2006;46:381–410. doi: 10.1146/annurev.pharmtox.46.120604.141238. doi:10.1146/annurev.pharmtox.46.120604.141238
- 12.Hardwick LJ, Velamakanni S, van Veen HW. The emerging pharmacotherapeutic significance of the breast cancer resistance protein (ABCG2) Br J Pharmacol. 2007;151:163–174. doi: 10.1038/sj.bjp.0707218. doi: 10.1038/sj.bjp.0707218
- 13.Morisaki K, Robey RW, Ozvegy-Laczka C, Honjo Y, Polgar O, Steadman K, Sarkadi B, Bates SE. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol. 2005;56:161–172. doi: 10.1007/s00280-004-0931-x. doi:10.1007/s00280-004-0931-x
- 14.Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109:238–246. doi: 10.1002/ijc.11669. doi:10.1002/ijc.11669
- 15.Kondo C, Suzuki H, Itoda M, Ozawa S, Sawada J, Kobayashi D, Ieiri I, Mine K, Ohtsubo K, Sugiyama Y. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res. 2004;21:1895–1903. doi: 10.1023/b:pham.0000045245.21637.d4. doi:10.1023/B:PHAM.0000045245.21637.d4
- 16.Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, Miki Y, Sugimoto Y. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–616. [PubMed] [Google Scholar]
- 17.Tamura A, Wakabayashi K, Onishi Y, Takeda M, Ikegami Y, Sawada S, Tsuji M, Matsuda Y, Ishikawa T. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 2007;98:231–239. doi: 10.1111/j.1349-7006.2006.00371.x. doi:10.1111/j.1349-7006.2006.00371.x
- 18.Li J, Cusatis G, Brahmer J, Sparreboom A, Robey RW, Bates SE, Hidalgo M, Baker SD. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–438. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- 19.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, Twelves C, Verweij J, McLeod HL. Diflomotecan pharmacokinetics in relation to ABCG2 421 C>A genotype. Clin Pharmacol Ther. 2004;76:38–44. doi: 10.1016/j.clpt.2004.03.003. doi:10.1016/j.clpt. 2004.03.003
- 20.Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, Nooter K, Gelderblom H. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol Ther. 2005;4:650–658. doi: 10.4161/cbt.4.6.1731. doi:10.1158/1535-7163.MCT-04-0238
- 21.Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, Ludovini V, Villa E, Hidalgo M, Sparreboom A, Baker SD. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–1742. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- 22.Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–1246. doi: 10.1158/0008-5472.can-03-3298. doi: 10.1158/0008-5472.CAN-03-3298
- 23.Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE, Ambudkar SV, Tarasova N, Bates SE. Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry. 2004;43:9448–9456. doi: 10.1021/bi0497953. doi:10.1021/bi0497953
- 24.Litman T, Jensen U, Hansen A, Covitz K, Zhan Z, Fetsch P, Abati A, Hansen P, Horn T, Skovsgaard T, Bates S. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim Biophys Acta. 2002;1565:6–16. doi: 10.1016/s0005-2736(02)00492-3. doi:10.1016/S0005-2736(02)00492-3
- 25.Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, Litman T, Dean M, Bates SE. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–6639. [PubMed] [Google Scholar]
- 26.Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, Bates SE. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2) Biochim Biophys Acta. 2001;1512:171–182. doi: 10.1016/s0005-2736(01)00308-x. doi:10.1016/S0005-2736(01)00308-X
- 27.Ozvegy-Laczka C, Varady G, Koblos G, Ujhelly O, Cervenak J, Schuetz JD, Sorrentino BP, Koomen GJ, Varadi A, Nemet K, Sarkadi B. Function-dependent conformational changes of the ABCG2 multidrug transporter modify its interaction with a monoclonal antibody on the cell surface. J Biol Chem. 2005;280:4219–4227. doi: 10.1074/jbc.M411338200. doi:10.1074/jbc.M411338200
- 28.Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, Poruchynsky MS, Bates SE. Mutations at amino acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–1978. doi: 10.1038/sj.bjc.6601370. doi:10.1038/sj.bjc.6601370
- 29.Ozvegy C, Varadi A, Sarkadi B. Characterization of drug transport, ATP hydrolysis and nucleotide trapping by the human ABCG2 multidrug transporter: modulation of substrate specificity by a point mutation. J Biol Chem. 2002;277:47980–47990. doi: 10.1074/jbc.M207857200. doi: 10.1074/jbc.M207857200
- 30.Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. Int J Cancer. 2003;107:757–763. doi: 10.1002/ijc.11484. doi:10.1002/ijc.11484
- 31.Ejendal KF, Diop NK, Schweiger LC, Hrycyna CA. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006;15:1597–1607. doi: 10.1110/ps.051998406. doi:10.1110/ps.051998406
- 32.Pozza A, Perez-Victoria JM, Sardo A, Ahmed-Belkacem A, Di Pietro A. Purification of breast cancer resistance protein ABCG2 and role of arginine-482. Cell Mol Life Sci. 2006;63:1912–1922. doi: 10.1007/s00018-006-6159-7. doi:10.1007/s00018-006-6159-7
- 33.Li YF, Polgar O, Okada M, Esser L, Bates SE, Xia D. Towards understanding the mechanism of action of the multidrug resistance-linked half-ABC transporter ABCG2: a molecular modeling study. J Mol Graph Model. 2007;25:837–851. doi: 10.1016/j.jmgm.2006.08.005. doi: 10.1016/j.jmgm.2006.08.005
- 34.Honjo Y, Morisaki K, Huff LM, Robey RW, Hung J, Dean M, Bates SE. Single-nucleotide polymorphism (SNP) analysis in the ABC half-transporter ABCG2 (MXR/BCRP/ABCP1) Cancer Biol Ther. 2002;1:696–702. doi: 10.4161/cbt.322. [DOI] [PubMed] [Google Scholar]