

Abstract
Thrombospondin-1 (TSP-1) has been proposed to have both pro-metastatic and anti-metastatic properties. To elucidate its role in breast cancer metastasis, we compared tumor progression in the polyomavirus middle T antigen (Pyt) transgenic mouse and the TSP-1-null Pyt transgenic mouse. We characterized the tumors in these mice at 45, 60 and 90 days of age. Tumor size, areas of necrosis, macrophage infiltration, levels of active and total TGF-β, vessel morphology, and lung and blood metastasis were measured in these mice. Mammary tumors were larger in the TSP-1-null mouse, and vessels were larger, but fewer in number in these tumors. The level of total TGF-β was significantly higher in the Pyt tumors at 90 days of age. Importantly, significantly fewer metastases were observed in the lungs of the TSP-1-null/Pyt mouse. Primary Pyt tumor cells were more migratory than TSP-1-null Pyt tumor cells on collagen. Treatment of Pyt mice with recombinant proteins that contain the type-1 repeats of TSP-1 resulted in decreased primary tumor growth and metastasis. Sequences that are involved in CD36 binding and those required for TGF-β activation mediated the inhibition of primary tumor growth. Thus, TSP-1 in the mammary tumor microenvironment inhibits angiogenesis and tumor growth, but promotes metastasis to the lung in the Pyt transgenic mouse. The ability of TSP-1 to support metastasis correlates with its ability to promote tumor cell migration.
Keywords: Metastasis, Migration, Polyomavirus middle T antigen, TGF-β, Transgenic mouse
Introduction
The thrombospondins comprise a family of homologous proteins that serve to regulate cellular phenotype and extracellular matrix structure during tissue genesis and remodeling [1]. Thrombospondin-1 (TSP-1), the first member identified, has been shown to be a potent inhibitor of angiogenesis and tumor progression [2]. In tumor cells, changes in the activity of oncogenes and tumor suppressor genes result in decreased expression of TSP-1 and the acquisition of an angiogenic phenotype [3–6]. By contrast, TSP-1 is highly expressed by stromal fibroblasts, macrophages and endothelial cells in breast cancer [7]. In the tumor microenvironment, TSP-1 interacts with a wide range of other proteins to affect (1) the level of active transforming growth factor β (TGFβ) [8, 9], (2) the level of vascular endothelial cell growth factor (VEGF) [10, 11], (3) the composition of the extracellular matrix and the activity of extracellular proteases [10, 12], and (4) the survival and migration of endothelial cells [13–15]. In addition, TSP-1 may inhibit tumor angiogenesis by down-regulating circulating endothelial cell progenitors [16, 17]. The inhibition of endothelial cell migration and the induction of endothelial apoptosis are reportedly mediated by the interaction of the type 1 repeats of TSP-1, designated TSRs, with CD36 [18, 19].
Whereas the role of TSP-1 as an inhibitor of primary tumor growth is well documented, its role in tumor metastasis is controversial. TSP-1 expression correlates with a non-metastatic phenotype in two clones that were derived from the MDA-MB-435 cell line, which were originally thought to be a breast cancer cell line, but it is now considered to be a melanoma cell line [20]. The nonmetastatic cell line NM-2C5 displays a 15-fold higher level of TSP-1 expression as compared to the metastatic cell line M-4A4 [20]. In another study, transfection of full-length TSP-1 into MDA-MB-435 cells decreased the growth of experimental tumors in the mammary gland and decreased the number of pulmonary metastases by approximately 50% [21]. Interestingly, this activity is lost if the last eighty amino acids of the C-terminal domain are removed from the protein [22]. These residues fall in the region that has the highest degree of homology between the members of the thrombospondin gene family [23]. However, in vitro studies suggest that TSP-1 promotes MDA-MB-435 and MDA-MB-231 cell migration and invasion [24, 25]. Over-expression of TSP-1 results in an approximately 50% increase in cell-associated plasmin. TSP-1 also increases the migration of invasive breast cancer cell lines through collagen gels, but does not affect the migration of a noninvasive cell line [26]. A similar correlation is seen with squamous cancer cells [27–29]. Migration of these cells in response to TSP-1 is inhibited by an antibody to the C-terminal domain [28, 30].
Because the thrombospondins interact with a wide range of other proteins, their in vivo function can be difficult to interpret with in vitro assays or orthotopic tumor models. To examine the role of TSP-1 in tumor metastasis, we have crossed the TSP-1-null strain to a transgenic model of breast cancer, designated Pyt [31]. In this model, expression of the polyomavirus middle T antigen (Pyt) is driven by the murine mammary tumor virus long terminal repeat [31]. Primary tumors are palpable at approximately 60 days of age and micrometastases can be detected by histology in the lung by 90 days of age [32, 33] [31]. The Pyt mouse breast cancer model recapitulates key aspects of the progression of the human disease [33]. By 4−6 weeks of age, hyperplasia is observed in the mammary glands. At 7−9 weeks of age, tumors resemble florid ductal epithelial breast hyperplasia in humans and are considered advanced, premalignant lesions (termed Adenoma/Min) [33]. By 8−12 weeks of age, the tumors are in the initial stages of malignant transition and are early carcinomas (or human ductal carcinoma in situ with early stromal invasion) [33]. At 10 weeks of age, 50% of the tumors are late carcinomas and resemble poorly differentiated invasive ductal carcinoma in humans. Like in the human disease, the Pyt tumors express less estrogen receptor alpha and progesterone receptor as they progress in malignancy [33]. Moreover, there is an increase in expression of ErbB2/Neu and cyclinD1 as tumors progress from hyperplasia to late carcinomas [33]. These expression patterns are also found in the human disease [33].
The Pyt and TSP-1-null/Pyt mice were characterized at two time-points, 60 days of age (8 weeks) and 90 days of age (12 weeks). As expected, the primary tumors grew more rapidly in the TSP-1-null background compared to wild-type/Pyt control mice. Consistent with previously published studies, these tumors displayed a tendency toward increased angiogenesis. Importantly, we have found that the level of metastasis to the lung was significantly lower in the absence of TSP-1. Treatment of Pyt mice with TSR proteins decreased the level of primary tumor growth and metastasis.
Materials and methods
Mice
Generation of the TSP-1-null mouse on the 129Sv background was carried out previously in the lab [34]. These mice were backcrossed to wild type FVB mice (Taconic Farms) eight times and then intercrossed to produce TSP-1-null mice on the FVB background. A male mouse with the mouse tumor virus long terminal repeat polyomavirus middle T antigen (Pyt) transgene was purchased from Jackson lab (Bar Harbor, Maine) [31] and bred to either a wild-type FVB female mouse or a TSP-1-null/FVB female mouse to generate the wild type/Pyt mouse or a TSP-1-null heterozygote/Pyt mouse, respectively. A male TSP-1 heterozygote null/Pyt mouse was subsequently bred with a TSP-1-null female mouse to generate the TSP-1-null/Pyt mice. The male mice are used to carry the Pyt gene because expression of this gene in females renders them unable to lactate and sustain a litter of pups [31]. Tail snips were taken from the mice to isolate genomic DNA, which was used in PCR reactions to determine the genotype. The following oligos were used to determine expression of the Pyt gene: MT3 (5′-AGTCACTGCTACTGCA CCCAG-3′) AND MT4 (5′-CTCTCCTCAGTTCTTCGCTCC-3′). The oligos used to determine expression of the TSP-1 gene has been previously reported [5]. All animal experiments were approved by the Beth Israel Deaconess Medical Center's Institutional Animal Care and Use Committee.
Materials
TGF-β ELISA kit was purchased from R&D Systems (Minneapolis, MN). The recombinant forms of the type 1 repeats (TSRs), designated TSR2 ± RFK were produced in Drosophila insect cells and purified as described previously [35].
Tumor volume
Mice were euthanized by carbon dioxide inhalation at 60 days and 90 days of age, and all 10 mammary glands were removed. At 60 days of age, the tumors from all 10 mammary glands were pooled and weighed. At 90 days of age, each mammary tumor was measured using a caliper, and tumor volume was calculated using the following equation: 4/3π (1/2 smaller diameter)2 × (larger diameter) [36].
Lung metastasis
To measure the level of metastasis in the lungs, the lungs were removed from either 45 or 90 day-old mice, washed in PBS and then flash frozen in liquid nitrogen and stored at −80°C. Lung RNA was isolated using Qiagen RNAeasy Kit (Valencia, CA) and 0.5 μg was subsequently used in reverse-transcription/real-time PCR reactions (Taqman Reverse Transcription Reagents and Taqman Fast Universal PCR Master Mix from Applied Biosystems, Foster City, CA). Taqman Rodent GAPDH Control reagents were purchased from Applied Biosystems (Foster City, CA), and primers against parts of the Pyt gene were purchased from Integrate DNA Technologies, Inc. (Coralville, IA). The primer sequences are Pyt For (5′-AGCCCGATGACAG CATATCC-3′). PyT Rev (5′-GGTCTTGGTCGCTTTC TGGA-3′) and PyT Probe (5′ 56FAM CGGACCCCCC CAGAACTCCTGT 36-TAMSp 3′). All reactions were carried out in duplicate and repeated once to confirm accuracy. Level of metastasis corresponds to expression of the Pyt gene since this gene would not be expressed in the lung unless mammary metastases were present. The ΔCT (differential cycle threshold) value was calculated by subtracting the sample CT value from the GAPDH CT value. The ΔCT value is an indirect indicator of when the Pyt gene product is first detected during the real-time PCR reaction. The lower the number, the earlier the product is detected suggesting a higher expression level of the gene. The experimental group ΔCT value was then normalized by dividing it by the ΔCT of the control mice. This number was subtracted from 1 to determine a level of metastasis.
In a separate method to determine levels of lung metastasis, we also sectioned formalin fixed, paraffin embedded lungs, stained the sections with hematoxylin and eosin and then counted metastasis using a 20× objective on a microscope. The number of metastasis found in the lungs from 8 mice was averaged.
Blood metastasis
Mice were euthanized and approximately 1 ml of blood was obtained by cardiac puncture. RNA was isolated using a kit for RNA isolation from blood (GentraSystem/Qiagen, Valencia, CA). The isolated blood RNA was reversed-transcribed and amplified as described for lung RNA to obtain level of expression of the Pyt gene in the blood.
CD31 Staining, F4/80 staining and area of necrosis
Tumors were embedded in OCT medium, slowly frozen in liquid nitrogen and 5 micron sections were cut and stained with a rat anti-CD31 antibody (BD Pharmingen, Franklin Lakes, NJ) as previously described [36] or with a rat anti-F4/ 80 antibody (Serotec, Oxford, UK) using manufacturer's guidelines. F4/80 is specific for macrophages [37]. Images were viewed using a Nikon E-600 microscope (Nikon, Melville, NY) with a 20× objective, captured using a spot digital camera (Diagnostic Instruments, Sterling Heights, IL) and analyzed using the IP LAB software (Scanalytics, Inc., Fairfax, VA). To calculate area of necrosis, sections were stained with hematoxylin and eosin using standard techniques and areas of necrosis were calculated using the IP LAB software. Tumor tissue that failed to stain with hematoxylin was labeled necrotic since non-viable cells were present.
Isolation of tumor cells from tumors
Thoracic tumors from mammary glands number 1, 2, 3, 6, 7 and 8, and from 60 day old mice were aseptically removed and washed twice in wash buffer (F12; 50 μg/ml gentamycin; 5% FBS) to remove excess red blood cells. The tumors were minced and suspended in 5 ml of enzymatic solution [2.5 mg/ml trypsin (Sigma, St. Louis MO); 5 mg/ml albumin (Sigma, St. Louis MO); and 850 units/ml collagenase type II (Worthington, Lakewood NJ.) in PBS] and incubated in a sterile 50 ml tube at a 45° angle at 37°C for 20 min under continuous shaking at 120 rpm. The liquid fraction was then removed and the undigested tissue was incubated for an additional 20 min with fresh enzymatic solution under the same conditions. Enzyme action was stopped by adding wash buffer and spinning at 1500 rpm for 10 min. Epithelial cells and fibroblasts were separated by a modification of the sedimentation technique as described by Lanari et al. [38]. Briefly, the cells were re-suspended in another 20 ml of wash buffer and allowed to settle for 20 min in the laminar flow hood. The upper 15 ml were plated in 100 mm culture dishes while cells in the sediment were re-suspended in another 20 ml of wash buffer, allowed to settle for 20 min, and the procedure was repeated approximately 10 times. The final epithelial pellet (tumor cells) was plated in 100 mm tissue culture dishes in growth media (F12:DMEM; 5 μg/ml insulin; 1 μg/ml hydrocortisone; 5 ng/ml EGF; 50 μg/ml gentamycin; 10 U/ml Pen/Strep; 5% FBS). After 24 h, the medium was removed and replaced by fresh growth media.
Solid-phase binding assay
The solid-phase binding assay was performed on Nunc Maxisorb plates designed for fluorescence. The wells were incubated overnight at 4°C with 200 μl of various proteins at 50 μg/ml dissolved in coating buffer (pH 9.6) containing 15 mM Na2CO3, 35 mM NaCO3 and 0.02% NaN3. The wells were washed three times with 300 μl of assay buffer (pH 7.6) containing 10 mM Tris, 0.14 M NaCl, 0.02% NaN3 and 0.05% Tween 20. The recombinant CLESH (CD36, LIMPII, Emp sequence homology; amino acids 95−143 of human CD36) domain of CD36 was expressed in S2 cells and fluorescently labeled using the EZ-Label Fluorescein Protein Labeling Kit from Pierce. One hundred microliters of varying concentrations of labeled protein were incubated overnight in the wells at 4°C. The wells were washed twice with assay buffer and the fluorescence was read with a plate reader (Tecan Group LTD). Known amounts of labeled CLESH were included in each assay to construct a standard curve. Wells were coated with BSA as a negative control.
Migration assay
Wild-type and TSP-1 null primary tumor cells were plated at different densities in 60 mm tissue culture dishes and allowed to adhere and proliferate for two days. Cells that were at similar density and had similar cellular morphology were used in the assay. Corning Transwell Permeable Supports 8.0 μm (Fisher Scientific, Pittsburgh, PA) were coated on both sides with collagen type 1 the previous night, blocked with 2% BSA/PBS for 1 h and washed with PBS before being placed in a 24 well plate. Cells were re-suspended in 0.2%BSA/DMEM media at a concentration of 106 cell/ml, and 100 μl was placed in the top chamber of the insert. In the bottom chamber, 3T3 conditioned media was placed, and the cells were allowed to migrate for 18 h at 37°C. Cells that remained in the top chamber were removed with a Q-tip and the cells in the bottom chamber were fixed and stained with 2% ethanol/0.2%crystal violet for 10 min. Inserts were washed in deionized water and allowed to dry for 1 h. The membranes were cut out of the inserts with a razor blade and placed on slides, covered with mineral oil and coverslipped. Cells were counted using a 40× objective and four fields were chosen per membrane with two membranes per cell line.
Cells were stained with alpha-smooth muscle cell actin (Labvision/Neomarker; Fremont, CA) and cytokeratin (Research Diagnostic, Inc., Concord, MA) antibodies to determine the relative proportion of tumor epithelial cells and fibroblasts in the primary cell lines. In both cell lines that were used in this manuscript, the tumor cell preparations contained approximately 30% fibroblasts. To verify the genotype of the cell lines, DNA was isolated from cells not used in the assay using the Qiamp DNA Mini Kit (Valencia, CA), and PCR amplification was carried out using oligos that have been previously described [34]. In all cases, the cells were the correct genotype.
Statistical analysis
Statistics were carried out using the two-tailed student t-test available on Microsoft Excel.
Results
Endogenous TSP-1 inhibits tumor growth
When examining the tumors derived from both the wild-type Pyt and TSP-1-null/PyT transgenic mice, it was found that on average, the tumors from the TSP-1-null mice were larger and more advanced in regards to tumor progression, as defined by Lin and colleagues [33] in their characterization of this mouse model of breast cancer (Fig. 1a, b). Within each genotype and at both time-points, there exists a range in both tumor size and malignancy; however, in the TSP-1-null tumors there was a noticeable increase in areas containing solid sheets of epithelial cells with little ascinar and ductal structure, but with cystic, non-cellular areas. These acellular areas were not as prevalent at 90 days of age, presumably because the tumor cells grew into this space; however, an increase in hematoxylin-poor areas was observed, suggesting an increase in necrosis in the TSP-1-null tumors. Indeed at 90 days, we saw an increase in necrotic area though the difference between the wild-type and TSP-1-null/Pyt mice was not statistically significant (Fig. 1c; P-value = 0.175). In summary, the TSP-1-null tumors were in general more advanced in malignancy than the wild type tumors. These results are similar to those obtained with the c-neu mouse, another murine transgenic model of breast cancer [10]. At 168 days, tumors from the TSP-1-null/c-neu mice were more than twice as big as the wildtype/c-neu tumors.
Fig. 1.
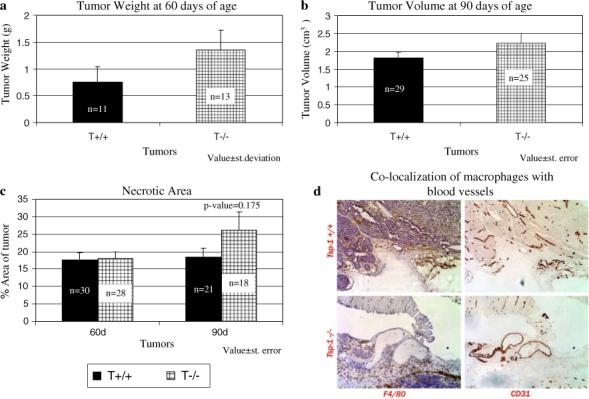
Characterization of mammary tumors in the wild-type and TSP-1-null Pyt transgenic mice. (a) Tumor weight is higher in the TSP-1-null tumors at 60 days of age. Value ± standard deviation. (b) tumor volume is higher in the TSP-1-null mice at 90 days of age. Value ± standard error. (c) Whereas increased necrosis is seen between the TSP-1-null and wild-type tumors at 90 days of age, the difference was not statistically significant Tumors were stained with hematoxylin and eosin and areas of necrosis were measured using the IP Lab software. Value ± standard error. (d) Co-localization of macrophages and blood vessels are seen in the tumors. Tumors were stained with CD31 and F4/80 antibodies which are specific for endothelial cells and macrophages, respectively. Photos were taken using a 20× objective
Because others have reported that tumor associated macrophages promote tumor progression and regulate the angiogenic switch in this mouse model of breast cancer [39–41], we stained 90 day tumor sections with an antibody against the protein F4/80 which is known to be expressed by macrophages [37]. In both the TSP-1 null/Pyt and wild-type Pyt tumors, we observed macrophages on the outer borders of the tumor, within the tumors as well as in cystic area of the tumors, which is not surprising considering the fact that this is a glandular organ and necrosis is occurring as early as 60 days of age. We compared macrophage infiltration at 90 days of age between the two groups by grading the degree of macrophage infiltration on a scale from 0−3. Comparable levels of F4/80-positive cells were observed in the wild-type Pyt and TSP-null/Pyt tumors. When the staining pattern for F4/80 was compared with that for CD31, an endothelial cell marker, we found equivalent areas where large populations of macrophages were in close proximity to blood vessels in the wild-type Pyt and TSP-1-null/Pyt tumors (Fig. 1d).
Blood vessel size and area
Because TSP-1 has been shown to be an inhibitor of angiogenesis, we next analyzed the blood vessels in the tumors and found that the number of vessels was fewer in the TSP-1-null/Pyt mice (Fig. 2a) at both 60 and 90 days of age though the difference was statistically significant at only 60 days (Fig. 2a; P-value < 0.001 at 60 days and P-value = 0.066 at 90 days). By contrast, the average vessel area was larger in the tumors from TSP-1-null/Pyt mice as compared to those from the wild-type Pyt mice (Fig. 2b; P-value < 0.001 for both time points). Therefore, even though the TSP-1-null/Pyt tumors had fewer blood vessels, these blood vessels were much larger compared to the wild-type Pyt tumors. Our findings at the 90 day time-point and results that we have published previously with this model [42] are in agreement with those of Iruela-Arispe's group who found that the vessel density did not change between the TSP-1-null/c-neu tumors and the c-neu wild-type tumors; however, the vessels were significantly larger in the TSP-1-null/c-neu mice [10].
Fig. 2.
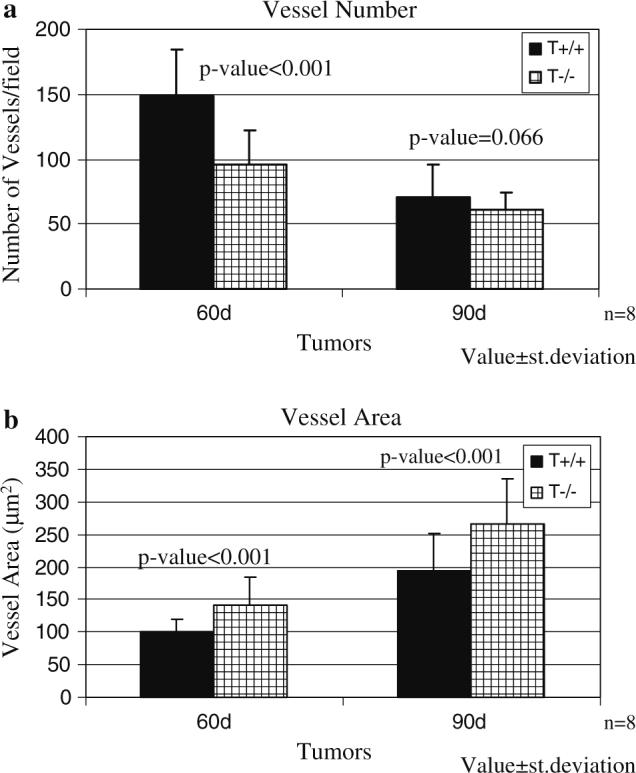
Vessel morphology differs between the TSP-1-null tumors and wild-type tumors. (a) Fewer vessels are present in the TSP-1-null mice. (b) Larger size vessels are present in the TSP-1-null mice. Bars represent value ± standard deviation
Levels of TGF-β in tumor tissue
We have previously shown that expression of a recombinant protein, designated TSR2 + RFK, that includes the second TSR of TSP-1 inhibits tumor growth through inhibition of angiogenesis and activation of TGF-β [35, 36]. Expression of TSR2 + RFK in A431 cells resulted in increases in the levels of both total and active TGF-β in subcutaneous tumors formed by these cells [36]. However, the effect of endogenous TSP-1 on TGFβ levels in autochthonous tumors has not been reported. To determine whether endogenous TSP-1 affects TGF-β levels in the mammary tumor microenvironment, the level of active and total TGF-β in the spontaneous tumors that formed in the wild-type Pyt and TSP-1-null/PyT mice were measured using an ELISA assay. The level of active and total TGF-β at 60 days of age did not differ between the two groups; however, at 90 days of age, the levels of active and total TGF-β were higher in the wild-type Pyt tumors than the TSP-1-null /Pyt tumors (Fig. 3a, b). Whereas the differences in total TGF-β reached statistical significance (n = 6; P-value = 0.02), the difference in active TGF-β did not, in part due to the fact that the levels of active TGF-β in the tumor tissue are low and difficult to detect (n = 6; P-value = 0.176).
Fig. 3.
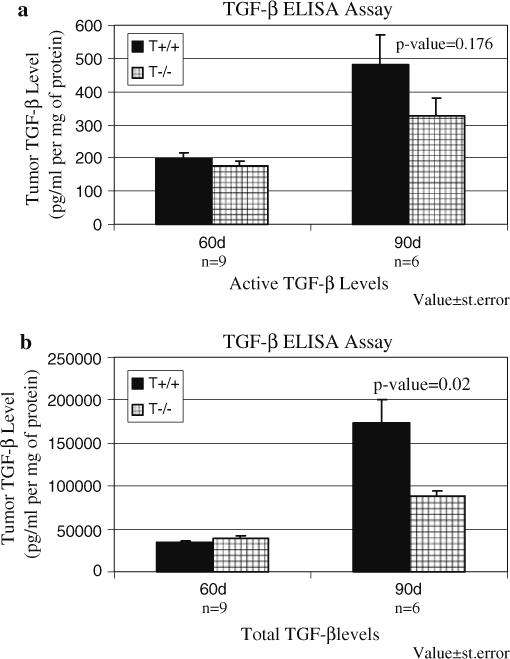
(a) Active TGF-β levels do not differ statistically at both 60 and 90 days of age between the two groups (n = 9 at 60 days; n = 6 at 90 days). (b) Total TGF-β levels are higher in the wild-type tumors at 90 days of age. Protein lysates were extracted from the tumors and used in an ELISA assay. Value ± standard error
Endogenous TSP-1 promotes metastasis
Next, we determined the effect of TSP-1 on advance mammary tumor progression, and specifically, the effect of TSP-1 on lung metastasis. At 90 days of age, the number of mice with surface lung metastasis, as seen by the naked eye, was higher in the wild-type Pyt mice, though not statistically significant (4/23 or 17.4% vs. 3/31 or 9.7%; P-value = 0.269). Looking at hematoxylin/eosin stained lung sections, more lung metastases were counted in wild-type Pyt lungs as compared to TSP-1-null/Pyt lungs (average of 3.2 lung metastasis longitudinal section for wild-type vs. 1.4 lung metastasis longitudinal section for the TSP-1-null; Fig. 4a; P-value = 0.015).
Fig. 4.
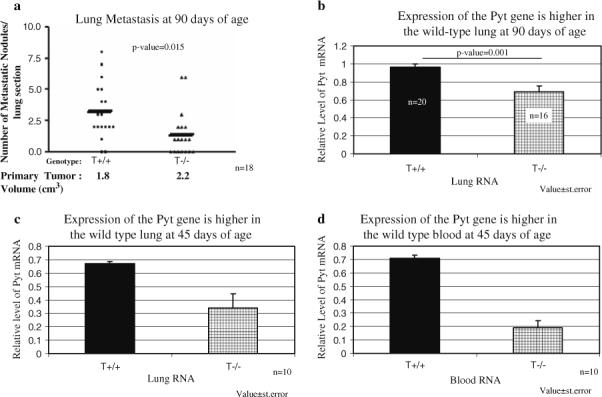
Lung metastasis is greater in the wild-type mice. (a) The lungs of 90 day-old mice were fixed in formalin, embedded in paraffin, and stained with hematoxylin and eosin. Longitudinal lung sections were viewed under a 20× objective microscope and metastatic nodules were counted. The data from Fig. 1b was added below the graph to demonstrate that tumor burden does not correlate with increased metastasis. (b) Expression of the Pyt gene is higher in wild-type lung tissue at 90 days of age. (c) Wild-type lung tissue at 45 days of age also expresses higher Pyt gene expression. (d) Higher levels of Pyt gene expression are detected in wild-type blood at 45 days of age. RNA was extracted from lung tissue or blood and subject to reverse transcription and real-time PCR using oligos against the polyomavirus middle T gene. Pyt Values were normalized to GAPDH to obtain relative levels of Pyt expression.Value ± standard deviation
To further quantify the level of metastasis in the lungs, real-time PCR was utilized on RNA isolated from the lungs. The expression level of the Pyt mRNA corresponds to the level of metastasis in the lung since the Pyt gene should not be expressed by the lung cells but only by the tumor cells that have metastasized to the lung. Lungs from non-transgenic PyT mice were used as controls. At 90 days of age, the relative level of Pyt mRNA was 0.96 in the wild-type mice and 0.69 in the TSP-1-null/Pyt mice (P-value < 0.001; n = 20 and 16, respectively; Fig. 4b). To determine how early metastasis was occurring, RNA was isolated from the lungs of 45 day-old mice and subjected to real-time PCR. A significant difference in Pyt mRNA expression level was observed between the wild-type Pyt and TSP-1-null/Pyt lungs (0.67 vs. 0.34; P-value < 0.001; n = 11; Fig. 4c). Finally, RNA was isolated from the blood of 45 day old mice and a higher level of expression of the Pyt mRNA was also observed in the wild-type/Pyt mice, indicating that even when the tumors are at the hyperplastic stage, tumor cells are able to intravasate (0.71 vs. 0.19; P-value = 0.006; n = 10; Fig. 4d). This observation is consistent with the presence of localized breakdown of basement membrane at this stage in this model [33].
TSP-1 may promote metastasis by affecting the behavior of the individual tumor and stromal cells that comprise the tumor and/or by affecting the overall tissue architecture. To determine if the absence of TSP-1 affects tumor cell behavior in ways that may promote metastasis, we isolated primary cells from the tumors and assayed their ability to migrate on collagen towards 3T3 conditioned media. On average, the wild-type Pyt tumor cells were able to migrate more efficiently over an 18 h period of time (Fig. 5; P-value < 0.001). Twice as many wild-type Pyt tumor cells were counted on the bottom of the permeable membrane as compared to the TSP-1-null cells. Comparable levels of cells of each genotype were found on the top of the permeable membrane after thirty minutes indicating that the difference in migration was not due to a difference in the ability of the cells to adhere to the membrane. These results indicate that endogenous TSP-1 expression affects the migratory phenotype of the tumor cells.
Fig. 5.
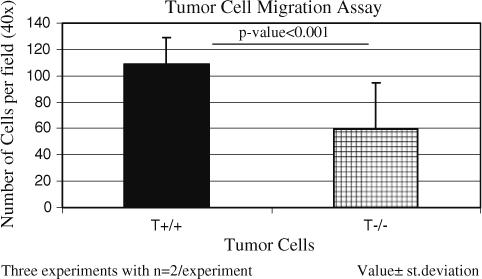
Wild-type tumor cells migrate faster than TSP-1-null tumor cells. Cells were allowed to migrate toward 3T3 conditioned media over collagen-coated permeable membranes for 18 h at 37°C. Values ± standard deviation. Data represents three experiments with n = 2 per experiment
TSR2 ± RFK inhibits tumor growth and metastasis
Multiple therapeutic strategies for the inhibition of tumor angiogenesis by increasing the level of TSP-1 have been proposed [43]. In addition, the anti-angiogenic compound ABT-510 is currently in clinical trials for the treatment of cancer [44, 45]. ABT-510 is derived from the sequence, GVTIRIR, found in the second type 1 repeat (designated TSR2) of TSP-1 [45]. In light of the observation that endogenous TSP-1 promotes metastasis, it is important to ask if treatment with a recombinant version of TSR2 or its related protein TSR2 + RFK promotes metastasis. In TSR + RFK, the sequence has been extended at the N-terminus to include the RFK sequence that has been shown to be essential for the activation for TGF-β [8, 46–52]. Mice were treated every other day with TSR2 or TSR2 + RFK at 2 mg/kg between days 30 and 90 of age. A comparable level of inhibition of primary tumor growth was observed with the two recombinant proteins at 60 days; however, the TSR2 + RFK protein was more active at 90 days of age (Fig. 6a). These data indicate that the ability to activate TGF-β can contribute to the tumor suppressive activity of these recombinant proteins in this model. When we looked at the effect of TSR2 and TSR2 + RFK on lung metastasis using real time PCR, we found that the relative mRNA level decreased 2-fold with 30 days of treatment (ΔCT values of Control = 2.51 ± 1.64; TSR2 = 4.29 ± 1.15; P-value = 0.0198; TSR2 + RFK = 4.49 ± 0.89; P-value = 0.0195) and decreased 4 to 6-fold with treatment for 60 days (ΔCT values of Control = 0.66 ± 3.16; TSR2 = 2.36 ± 2.00; P-value = 0.169; TSR2 + RFK = 3.68 ± 2.16; P-value = .020). These data indicate that treatment with these recombinant proteins suppresses metastasis, and that the RFK sequence is more effective beyond 30 days of treatment.
Fig. 6.

Recombinant proteins that contain the type 1 repeats (TSR) of TSP-1 inhibit tumor growth. (a) The TSR2 ± RFK and TSR2 peptides inhibit tumor growth in the polyomavirus middle T transgenic mice. Peptides were injected interperioteneal in the mice at a concentration of 2 mg/kg every other day for 30 days (n = 7). Values ± standard error. (b) Mutation of the arginine residues in TSR2 + RFK significantly diminishes binding to the CLESH domain. TSR2 + RFK (n = 7) and TSR2 + RFKR440QR442S n = 2) were allowed to adhere to the wells of a 96 well plate overnight. Fluorescently labeled, recombinant CLESH (CD36, LIMPII, Emp sequence homology; amino acids 95−143 of human CD36) domain of CD36 was added to the wells, allowed to bind to the proteins overnight and non-bound proteins were washed off. Fluorescence intensity was read with a plate reader. Values ± standard deviation. (c) Mutation in the arginine residues in TSR2 abolishes the inhibition of tumor growth while these mutations have no effect on TSR2 + RFK's ability to inhibit tumor growth. The mice received interperioteneal injections every other day between 30 and 60 days of age with recombinant proteins at a concentration of 2 mg/kg. Values ± standard error
The observation that the TSR2 and TSR2 + RFK peptides have comparable activity and effect on tumor growth at 60 days of age suggests that the activation of TGFβ by TSR2 + RFK is either (1) not a significant component of the inhibition of tumor growth at this time point, or (2) functionally overlapping with the anti-angiogenic effect. To distinguish these possibilities, we mutated arginines 440 and 442, to serine and glutamine residues, respectively. These two arginine residues contribute positive charge to the front face of the TSRs and we propose that this site mediates CD36 binding [53]. This conclusion is consistent with the observation that arginine 442 is essential for the activity of ABT-510 [54]. A solid-phase binding assay was used to establish that mutation of arginines 440 and 442 affects TSR + RFK binding to CD36. In this assay, plates are coated with the recombinant TSR + RFK proteins and varying amounts of a recombinant version of the CLESH (CD36, LIMPII, Emp sequence homology; amino acids 95−143 of human CD36) domain of CD36, which is fluorescently labeled, are incubated in the plates. The CLESH domain has been shown by others to mediate the binding of TSP-1 to CD36 [55]. As shown in Fig. 6b, the CLESH domain binds to TSR2 + RFK in a saturable fashion. By contrast, little to no binding to wells coated with BSA is observed. The binding of fluorescently labeled CLESH domain to TSR2 + RFK can be inhibited by non-labeled CLESH domain indicating that the observed binding is not an artifact of the labeling procedure. Double reciprocal plots indicate that the CLESH domain has comparable affinity for TSR2 and TSR2 + RFK (data not shown). Mutation of arginine 440 and 442 markedly decreases the ability of the CLESH domain to bind to TSR2 + RFK (Fig. 6b).
These mutations were made in both the TSR2, designated TSR2(R440S/R442Q), and TSR2 + RFK, designated TSR2 + RFK(R440S/R442Q), proteins. Treatment of mice between 30 and 60 days of age with the TSR2(R440S/ R442Q) protein did not result in any inhibition of tumor growth (Fig. 6b). Interestingly, tumor size was significantly larger (P-value = 0.048) between mice treated with this protein and saline treated control mice. This observation indicates that the inhibition with this protein can be attributed to an anti-angiogenesis activity that is mediated by CD36. By contrast, TSR2 + RFK(R440S/R442Q) is still capable of inhibiting tumor growth after 30 days of treatment as compared to control mice (Fig. 6b; P-value = 0.025). The data indicate that the inhibition of angiogenesis and the activation of TGF-β are overlapping activities for inhibition of tumor growth at this early time point. At later time points, both activities are required for optimal inhibition of tumor growth (Fig. 6a).
Discussion
In this study, we have characterized the effect of endogenous levels of TSP-1 on spontaneous breast cancer growth and metastasis and shown for the first time that endogenous TSP-1 promotes breast cancer metastasis. The effect of TSP-1 on primary tumor growth in the Pyt model are comparable to other subcutaneous, orthotopic and spontaneous models in that primary tumors grow more rapidly in the TSP-1-null background, and the vessels are larger [10, 36]. We have also shown for the first time in a transgenic mouse model of breast cancer that TSP-1 can modulate the level of TGF-β in the tumors. We have shown that higher levels of both active and total TGF-β are present in tumors of 90-day-old wild-type mice. Increased levels of active and total TGF-β may cause a decrease in tumor progression since we have found in this study and others that TSR2 + RFK, which activates TGF-β, inhibits tumor growth more effectively than TSR2, which does not activate TGFβ [35, 36].
Our data indicate that endogenous TSP-1 promotes tumor metastasis to the lung. We first observed an increase in lung metastasis in the wild type mice by looking by eye at the lung surface and counting the number of mice with visible metastases. These results were validated by sectioning the lungs and counting metastatic nodules. To quantify the amount of lung metastases, we used real-time PCR to detect the expression of the Pyt gene. Since normal lungs do not express the Pyt gene, the level of its expression corresponds to the extent of invasion of metastatic tumor cells to the lung. We took care not to contaminate the lungs with tumor tissue by removing this organ first, and by cleaning our instruments between mice. We also used normal mice as our control, to which we compared the expression of the Pyt gene. Increased expression of Pyt in the lung of wild type Pyt mice was detected at 90 days of age and as early as 45 days of age. Higher expression of the Pyt gene was also detected in the blood of 45 day-old wild-type mice, suggesting that metastasis was occurring at a very early stage of tumor progression, consistent with the human disease [56–58]. In our study, we compared mice at specific ages, but since the tumors grew more rapidly in the TSP-1-null/Pyt mice, the decreased metastasis in these mice was occurring in the presence of a greater primary tumor burden. One could predict that the difference in the level of metastasis between the wild-type Pyt and TSP-1-null/Pyt mice would be greater when the primary tumors are of comparable size.
TSP-1 is a member of the family of matricellular proteins, which also includes tenascin and osteopontin [59]. These proteins are components of the provisional extra-cellular matrix that is formed during the tissue remodeling that is associated with development, wound healing and neoplasia. Tenascin and osteopontin serve to regulate cellular proliferation, migration and invasion, and support metastasis [60, 61]. To determine whether endogenous TSP-1 also promotes tumor cell migration, we tested the ability of primary tumor cells to migrate on collagen toward 3T3 conditioned media, and found that TSP-1 null/PyT cells migrated poorly as compared to the TSP-1 expressing tumor cells, with a two-fold difference in cell numbers between the two groups. These data suggest that a tumor cell autonomous effect of TSP-1 leads to increased cellular migration and metastasis to the lung. The ability of TSP-1 to confer a greater migratory phenotype was also seen in smooth muscle in that these cells migrated better after carotid injury in the wild-type mice as compared to the TSP-1 null mice [62]. The decreased migratory response of TSP-1-null tumor cells is consistent with published data indicating that exogenous TSP-1 promotes the migration of tumor cell lines. Tuszynski and colleagues have shown that TSP-1's ability to increase the formation of plasmin and to serve as an adhesive protein can create an environment that promotes cell invasion in vitro [24–26, 30, 63, 64]. Interestingly, the regulation of plasmin activity was reportedly mediated by TGF-β in breast cancer cells [25, 30]. Thus, the activation of TGF-β by TSP-1 that we have observed in the tumor microenvironment may promote tumor cell migration, invasion and metastasis. We are currently examining the activity of various proteases in the wild-type and TSP-1-null tumors.
Because of the development of TSP-1-based therapeutics as anti-cancer drugs, it was important to ask if they would promote metastasis. We treated wild-type Pyt mice with two recombinant TSP-1 proteins that contain the amino acid sequence that have previously been shown to inhibit melanoma, squamous cell carcinoma and pancreatic cancer [35, 36, 65]. Both of these proteins include the amino acids that comprise ABT-510. These two proteins differed by three amino acid residues, RFK, which has been shown to activate TGF-β [8, 49–51]. We found that both of these peptides were able to inhibit tumor growth and inhibit lung metastasis. The effect on lung metastasis may be secondary to the inhibition of primary tumor growth. Whereas it is difficult to establish the direct effect of TSR-containing proteins on metastasis because of their effect on primary tumor growth, the data presented indicate that systemic treatment with these proteins does not promote metastasis. These studies are the first to show that TSR2 and TSR2 + RFK are potent inhibitors of autochthonous tumor growth.
The crystallographic structure of the TSRs revealed the presence of a positively charged groove in a surface that we designated as the front face [53]. Arginines 440 and 442 project their side chains into this groove to contribute to the positive charge. In this study, we show that mutation of these arginines significantly reduces the ability of the TSRs to bind to the CLESH domain of CD36. In addition, mutation of these argi-nines abolishes the ability of TSR2, but not TSR2 + RFK, to inhibit tumor growth. These data, in conjunction with the observation that arginine 442 is required for the activity of ABT-510, indicate that the positively charged groove does in fact represent the CD36 binding site. The observation that TSR2 + RFK(R440SR442Q) retains anti-tumor activity indicates that the ability to activate TGF-β contributes to the inhibition of mammary tumor growth. At this time, we can not exclude the possibility that TSR2(R440SR442Q) is inactive because it is less stable in circulation. However, the activity of TSR2 + RFK(R440SR442Q) indicates that this mutation does not significantly reduce the stability of the protein. The data presented here has important implications for the development of TSR-based anti-tumor therapeutics. The use of a recombinant protein that contains all of the activity of the TSRs appears to have greater activity than peptides that solely inhibit angiogenesis.
The results presented here help to reconcile some of the controversial data regarding the role of TSP-1 in metastasis. Since TSP-1 is a potent inhibitor of angiogenesis, its expression can suppress metastasis secondarily to the inhibition of primary tumor growth. By contrast, TSP-1 within the tumor microenvironment appears to promote tumor cell migration and intravasation, and this effect may involve the activation of TGF-β. TSP-1 could also confer a protective effect on tumor cells in the circulation. Plasma TSP-1 levels are higher in advanced breast cancer patients versus early breast cancer patients, and these levels are significantly higher than normal controls [66]. Furthermore, patients with lymph node metastasis have higher level of plasma TSP-1. The overall effect of TSP-1 on metastasis may vary with tumor type and may depend upon the balance of the anti-angiogenic effect on endothelial cells and the pro-migratory effect on tumor cells.
Acknowledgements
The authors would like to thank fellow lab members Eric Galardi for assistance with the mice and Xuefeng Zhang for helpful discussion on the project. We also would like to thank Ricky Sanchez for sectioning the frozen tumors. Karen Yee was supported by a fellowship from Aid for Cancer Research (Newton, MA). Raymond Washington is supported by a training grant from the National Institute of Health (T32HL007893). This work was supported by grants from the National Institute of Health (CA92644 and HL68003).
References
- 1.Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 2.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 4.Watnick RS, Cheng YN, Rangarajan A, Ince TA, Weinberg RA. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell. 2003;3:219–231. doi: 10.1016/s1535-6108(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 5.Lawler J, Miao WM, Duquette M, Bouck N, Bronson RT, Hynes RO. Thrombospondin-1 gene expression affects survival and tumor spectrum of p53-deficient mice. Am J Pathol. 2001;159:1949–1956. doi: 10.1016/S0002-9440(10)63042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volpert OV, Pili R, Sikder HA, Nelius T, Zaichuk T, Morris C, Shiflett CB, Devlin MK, Conant K, Alani RM. Id1 regulates angiogenesis through transcriptional repression of thrombospondin-1. Cancer Cell. 2002;2:473–483. doi: 10.1016/s1535-6108(02)00209-x. [DOI] [PubMed] [Google Scholar]
- 7.Brown LF, Guidi AJ, Schnitt SJ, Van De Water L, Iruela-Arispe ML, Yeo TK, Tognazzi K, Dvorak HF. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res. 1999;5:1041–1056. [PubMed] [Google Scholar]
- 8.Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-beta. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- 9.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci USA. 2001;98:12485–12490. doi: 10.1073/pnas.171460498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta K, Gupta P, Wild R, Ramakrishnan S, Hebbel RP. Binding and displacement of vascular endothelial growth factor (VEGF) by thrombospondin: effect on human microvascular endothelial cell proliferation and angiogenesis. Angiogenesis. 1999;3:147–158. doi: 10.1023/a:1009018702832. [DOI] [PubMed] [Google Scholar]
- 12.Bein K, Simons M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J Biol Chem. 2000;275:32167–32173. doi: 10.1074/jbc.M003834200. [DOI] [PubMed] [Google Scholar]
- 13.Volpert OV, Zaichuk T, Zhou W, Reiher F, Ferguson TA, Stuart PM, Amin M, Bouck NP. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002;8:349–357. doi: 10.1038/nm0402-349. [DOI] [PubMed] [Google Scholar]
- 14.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 15.Nor JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res. 2000;37:209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- 16.Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, Rawn K, Voskas D, Dumont DJ, Ben-David Y, Lawler J, Henkin J, Huber J, Hicklin DJ, D'Amato RJ, Kerbel RS. Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis; Implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell. 2005;7:101–111. doi: 10.1016/j.ccr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 17.Rafii DC, Psaila B, Butler J, Jin DK, Lyden D. Regulation of vasculogenesis by platelet-mediated recruitment of bone marrow derived cells. Arterioscler Thromb Vasc Biol. 2008;28:217–222. doi: 10.1161/ATVBAHA.107.151159. [DOI] [PubMed] [Google Scholar]
- 18.Magnetto S, Bruno-Bossio G, Voland C, Lecerf J, Lawler J, Delmas P, Silverstein R, Clezardin P. CD36 mediates binding of soluble thrombospondin-1 but not cell adhesion and haptotaxis on immobilized thrombospondin-1. Cell Biochem Funct. 1998;16:211–221. doi: 10.1002/(SICI)1099-0844(199809)16:3<211::AID-CBF788>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 19.Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. 1997;138:707–717. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urquidi V, Sloan D, Kawai K, Agarwal D, Woodman AC, Tarin D, Goodison S. Contrasting expression of thrombospondin-1 and osteopontin correlates with absence or presence of metastatic phenotype in an isogenic model of spontaneous human breast cancer metastasis. Clin Cancer Res. 2002;8:61–74. [PubMed] [Google Scholar]
- 21.Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res. 1994;54:6504–6511. [PubMed] [Google Scholar]
- 22.Yabkowitz R, Mansfield PJ, Dixit VM, Suchard SJ. Motility of human carcinoma cells in response to thrombospondin: relationship to metastatic potential and thrombospondin structural domains. Cancer Res. 1993;53:378–387. [PubMed] [Google Scholar]
- 23.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albo D, Rothman VL, Roberts DD, Tuszynski GP. Tumour cell thrombospondin-1 regulates tumour cell adhesion and invasion through the urokinase plasminogen activator receptor. Br J Cancer. 2000;83:298–306. doi: 10.1054/bjoc.2000.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albo D, Berger DH, Wang TN, Hu X, Rothman V, Tuszynski GP. Thrombospondin-1 and transforming growth factor-beta l promote breast tumor cell invasion through up-regulation of the plasminogen/plasmin system. Surgery. 1997;122:493–499. doi: 10.1016/s0039-6060(97)90043-x. discussion 499–500. [DOI] [PubMed] [Google Scholar]
- 26.Wang TN, Qian X, Granick MS, Solomon MP, Rothman VL, Berger DH, Tuszynski GP. Thrombospondin-1 (TSP-1) promotes the invasive properties of human breast cancer. J Surg Res. 1996;63:39–43. doi: 10.1006/jsre.1996.0219. [DOI] [PubMed] [Google Scholar]
- 27.Albo D, Berger DH, Tuszynski GP. The effect of thrombospondin-1 and TGF-beta 1 on pancreatic cancer cell invasion. J Surg Res. 1998;76:86–90. doi: 10.1006/jsre.1998.5299. [DOI] [PubMed] [Google Scholar]
- 28.Albo D, Arnoletti JP, Castiglioni A, Granick MS, Solomon MP, Rothman VL, Tuszynski GP. Thrombospondin (TSP) and transforming growth factor beta 1 (TGF-beta) promote human A549 lung carcinoma cell plasminogen activator inhibitor type 1 (PAI-1) production and stimulate tumor cell attachment in vitro. Biochem Biophys Res Commun. 1994;203:857–865. doi: 10.1006/bbrc.1994.2262. [DOI] [PubMed] [Google Scholar]
- 29.Wang TN, Qian XH, Granick MS, Solomon MP, Rothman VL, Tuszynski GP. The effect of thrombospondin on oral squamous carcinoma cell invasion of collagen. Am J Surg. 1995;170:502–505. doi: 10.1016/s0002-9610(99)80340-7. [DOI] [PubMed] [Google Scholar]
- 30.Arnoletti JP, Albo D, Granick MS, Solomon MP, Castiglioni A, Rothman VL, Tuszynski GP. Thrombospondin and transforming growth factor-beta 1 increase expression of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 in human MDA-MB-231 breast cancer cells. Cancer. 1995;76:998–1005. doi: 10.1002/1097-0142(19950915)76:6<998::aid-cncr2820760613>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 31.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic Polyoma middle-T mice model premalignant mam-mary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- 33.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miao WM, Seng WL, Duquette M, Lawler P, Laus C, Lawler J. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-beta-dependent and -independent mechanisms. Cancer Res. 2001;61:7830–7839. [PubMed] [Google Scholar]
- 36.Yee KO, Streit M, Hawighorst T, Detmar M, Lawler J. Expression of the type-1 repeats of thrombospondin-1 inhibits tumor growth through activation of transforming growth factor-beta. Am J Pathol. 2004;165:541–552. doi: 10.1016/s0002-9440(10)63319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoue T, Plieth D, Venkov CD, Xu C, Neilson EG. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005;67:2488–2493. doi: 10.1111/j.1523-1755.2005.00358.x. [DOI] [PubMed] [Google Scholar]
- 38.Lanari C, Luthy I, Lamb CA, Fabris V, Pagano E, Helguero LA, Sanjuan N, Merani S, Molinolo A. Five novel hormone-responsive cell lines derived frommurine mamnary ductal carcinomas: in vivo and in vitro effects of estrogens and progestins. Cancer Res. 2001;61:293–302. [PubMed] [Google Scholar]
- 39.Lin EY, Pollard JW. Macrophages: modulators of breast cancer progression. Novartis Found Symp. 2004;256:158–168. discussion 168–172, 259–169. [PubMed] [Google Scholar]
- 40.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 41.Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67:5064–5066. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- 42.Yee KO, Connolly CM, Pines M, Lawler J. Halofuginone inhibits tumor growth in the polyoma middle T antigen mouse via a thrombospondin-1 independent mechanism. Cancer Biol Ther. 2006;5:218–224. doi: 10.4161/cbt.5.2.2419. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res. 2007;74:90–99. doi: 10.1016/j.mvr.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson JC, Grammer JR, Wang W, Nabors LB, Henkin J, Stewart JE, Jr, Gladson CL. ABT-510, a modified type 1 repeat peptide of thrombospondin, inhibits malignant glioma growth in vivo by inhibiting angiogenesis. Cancer Biol Ther. 2007;6:454–462. doi: 10.4161/cbt.6.3.3630. [DOI] [PubMed] [Google Scholar]
- 45.Isenberg JS, Yu C, Roberts DD. Differential effects of ABT-510 and a CD36-binding peptide derived from the type 1 repeats of thrombospondin-1 on fatty acid uptake, nitric oxide signaling, and caspase activation in vascular cells. Biochem Pharmacol. 2008;75:875–882. doi: 10.1016/j.bcp.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harpel JG, Schultz-Cherry S, Murphy-Ullrich JE, Rifkin DB. Tamoxifen and estrogen effects on TGF-beta formation: role of thrombospondin-1, alphavbeta3, and integrin-associated protein. Biochem Biophys Res Commun. 2001;284:11–14. doi: 10.1006/bbrc.2001.4922. [DOI] [PubMed] [Google Scholar]
- 47.Ribeiro SM, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J Biol Chem. 1999;274:13586–13593. doi: 10.1074/jbc.274.19.13586. [DOI] [PubMed] [Google Scholar]
- 48.Ribeiro SM, Schultz-Cherry S, Murphy-Ullrich JE. Heparin-binding vitronectin up-regulates latent TGF-beta production by bovine aortic endothelial cells. J Cell Sci. 1995;108(Pt 4):1553–1561. doi: 10.1242/jcs.108.4.1553. [DOI] [PubMed] [Google Scholar]
- 49.Schultz-Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, Roberts DD, Murphy-Ullrich JE. Regulation of transforming growth factor-beta activation by discrete sequences of thrombospondin 1. J Biol Chem. 1995;270:7304–7310. doi: 10.1074/jbc.270.13.7304. [DOI] [PubMed] [Google Scholar]
- 50.Schultz-Cherry S, Ribeiro S, Gentry L, Murphy-Ullrich JE. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J Biol Chem. 1994;269:26775–26782. [PubMed] [Google Scholar]
- 51.Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy-Ullrich JE, Schultz-Cherry S, Hook M. Transforming growth factor-beta complexes with thrombospondin. Mol Biol Cell. 1992;3:181–188. doi: 10.1091/mbc.3.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, Lawler J, Wang JH. Crystal structure of the TSP-1 type 1 repeats: a novel layered fold and its biological implication. J Cell Biol. 2002;159:373–382. doi: 10.1083/jcb.200206062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dawson DW, Volpert OV, Pearce SF, Schneider AJ, Silverstein RL, Henkin J, Bouck NP. Three distinct D-amino acid substitutions confer potent antiangiogenic activity on an inactive peptide derived from a thrombospondin-1 type 1 repeat. Mol Pharmacol. 1999;55:332–338. doi: 10.1124/mol.55.2.332. [DOI] [PubMed] [Google Scholar]
- 55.Simantov R, Silverstein RL. CD36: a critical anti-angiogenic receptor. Front Biosci. 2003;8:s874–s882. doi: 10.2741/1168. [DOI] [PubMed] [Google Scholar]
- 56.Nakagawa T, Martinez SR, Goto Y, Koyanagi K, Kitago M, Shingai T, Elashoff DA, Ye X, Singer FR, Giuliano AE, Hoon DS. Detection of circulating tumor cells in early-stage breast cancer metastasis to axillary lymph nodes. Clin Cancer Res. 2007;13:4105–4110. doi: 10.1158/1078-0432.CCR-07-0419. [DOI] [PubMed] [Google Scholar]
- 57.Alix-Panabieres C, Muller V, Pantel K. Current status in human breast cancer micrometastasis. Curr Opin Oncol. 2007;19:558–563. doi: 10.1097/CCO.0b013e3282f0ad79. [DOI] [PubMed] [Google Scholar]
- 58.Lang JE, Hall CS, Singh B, Lucci A. Significance of micrometastasis in bone marrow and blood of operable breast cancer patients: research tool or clinical application? Expert Rev Anticancer Ther. 2007;7:1463–1472. doi: 10.1586/14737140.7.10.1463. [DOI] [PubMed] [Google Scholar]
- 59.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 60.Orend G, Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 61.Tuck AB, Chambers AF, Allan AL. Osteopontin overexpression in breast cancer: knowledge gained and possible implications for clinical management. J Cell Biochem. 2007;102:859–868. doi: 10.1002/jcb.21520. [DOI] [PubMed] [Google Scholar]
- 62.Moura R, Tjwa M, Vandervoort P, Cludts K, Hoylaerts MF. Thrombospondin-1 activates medial smooth muscle cells and triggers neointima formation upon mouse carotid artery ligation. Arterioscler Thromb Vasc Biol. 2007;27:2163–2169. doi: 10.1161/ATVBAHA.107.151282. [DOI] [PubMed] [Google Scholar]
- 63.Tuszynski GP, Gasic TB, Rothman VL, Knudsen KA, Gasic GJ. Thrombospondin, a potentiator of tumor cell metastasis. Cancer Res. 1987;47:4130–4133. [PubMed] [Google Scholar]
- 64.Wang TN, Qian XH, Granick MS, Solomon MP, Rothman VL, Berger DH, Tuszynski GP. Inhibition of breast cancer progression by an antibody to a thrombospondin-1 receptor. Surgery. 1996;120:449–454. doi: 10.1016/s0039-6060(96)80322-9. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Galardi E, Duquette M, Delic M, Lawler J, Parangi S. Antiangiogenic treatment with the three thrombospondin-1 type 1 repeats recombinant protein in an orthotopic human pancreatic cancer model. Clin Cancer Res. 2005;11:2337–2344. doi: 10.1158/1078-0432.CCR-04-1900. [DOI] [PubMed] [Google Scholar]
- 66.Byrne GJ, Hayden KE, McDowell G, Lang H, Kirwan CC, Tetlow L, Kumar S, Bundred NJ. Angiogenic characteristics of circulating and tumoural thrombospondin-1 in breast cancer. Int J Oncol. 2007;31:1127–1132. doi: 10.3892/ijo.31.5.1127. [DOI] [PubMed] [Google Scholar]