

Abstract
Multiple Sclerosis (MS) is an autoimmune inflammatory disease that presents clinically with a range of symptoms including motor, sensory, and cognitive dysfunction as well as demyelination and lesion formation in brain and spinal cord. A variety of animal models of MS have been developed that share many of the pathological hallmarks of MS including motor deficits (ascending paralysis), demyelination and axonal damage of central nervous system (CNS) tissue. In recent years, neuropathic pain has been recognized as a prevalent symptom of MS in a majority of patients. To date, there have been very few investigations into sensory disturbances in animal models of MS. The current work contains the first assessment of hind paw mechanical allodynia (von Frey test) over the course of a relapsing-remitting myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis (MOG-EAE) rat model of MS and establishes the utility of this model in examining autoimmune induced sensory dysfunction. We demonstrate periods of both decreased responsiveness to touch that precedes the onset of hind limb paralysis, and increased responsiveness (allodynia) that occurs during the period of motor deficit amelioration traditionally referred to as symptom remission. Furthermore, we tested the ability of our recently characterized anti-inflammatory IL-10 gene therapy to treat the autoimmune inflammation induced behavioral symptoms and tissue histopathological changes. This therapy is shown here to reverse inflammation induced paralysis, to reduce disease associated reduction in sensitivity to touch, to prevent the onset of allodynia, to reverse disease associated loss of body weight, and to suppress CNS glial activation associated with disease progression in this model.
Introduction
Multiple sclerosis (MS) is an autoimmune disease affecting at least 1 in 1000 people in the US and is anticipated to become substantially more prevalent in coming years (Hirtz et al., 2007). This debilitating disease involves an attack by the immune system against antigens of the central nervous system (CNS), especially antigens derived from oligodendrocytes that constitute the myelin sheaths surrounding axons. Initiating factors that cause the disease are poorly understood and are thought to involve a genetic predisposition (Kenealy et al., 2003) as well as environmental influences (Marrie, 2004).
In addition to the auto-aggressive T-cells that characterize MS (Martin et al., 1992), marked activation of glia (microglia and astrocytes) occurs in MS patients in both the spinal cord and brain (Petzold et al., 2002; Gray et al., 2008). This results in inflammation involving pro-inflammatory cytokines and chemokines (Ozenci et al., 2002; Szczucinski and Losy, 2007). These inflammatory molecules may profoundly change signaling properties of neurons (Katsuki et al., 1990; Cunningham et al., 1996; Viviani et al., 2003) and lead to demyelination and axonal loss, two hallmarks of MS (Bruck, 2005; Hendriks et al., 2005).
Inflammation induced disruption of neuronal signaling in MS patients leads to a diverse array of motor, cognitive, and sensory symptoms. Motor dysfunctions are striking and include spasticity, loss of normal gait, paresis, and progressive ascending paralysis. Decreased sensitivity to touch is a common early sensory symptom that often leads to the initial diagnosis of MS. In addition, neuropathic pain has been documented to occur in a majority of patients (Osterberg et al., 2005; Kenner et al., 2007).
Several experimental animal models, termed Experimental Autoimmune Encephalomyelitis (EAE), have been developed that produce anatomical and behavioral symptoms that closely mimic those observed in MS. These models have been used in order to study disease development and progression as well as for pre-clinical testing of potential therapeutics. Many of these models, including the model utilized in the current studies, induce autoimmune inflammation by generating an adaptive immune cell mediated attack against antigens contained in the myelin sheaths surrounding axons in CNS. Some of these EAE models, including the one utilized in the current studies, create a relapsing/remitting course of symptomology, similar to that exhibited in humans.
We have recently developed and characterized a powerful non-viral gene therapy paradigm that targets CNS inflammation associated with a variety of nerve injury and toxicity models (Milligan et al., 2006; Ledeboer et al., 2007a). This therapy utilizes 2 successive intrathecal injections of plasmid DNA (pDNA) containing the interleukin-10 (IL-10) gene with a point mutation (pDNA-IL-10F129S; see methods) into the intrathecal space surrounding the lumbar spinal cord. This paradigm leads to prolonged reversal of neuropathic pain. These initial studies made use of a control plasmid DNA (not containing the IL-10 gene) and it was determined that control DNA induced a transient increase in endogenous IL-10 protein lasting for several days (Sloane et al., Manuscript in review). The effects of control DNA were short lived and it was determined that DNA containing the IL-10 gene was required for long term therapeutic efficacy. IL-10 is a potent endogenous regulator of pro-inflammatory activity that works both via the suppression of the expression or activity of several important pro-inflammatory molecules (Ledeboer et al., 2000; Moore et al., 2001; Mu et al., 2005b). There is great interest in developing effective, enduring treatments to control the chronic inflammation thought to underlie MS symptom expression. Thus, here we test whether this pDNA-IL-10F129S gene therapy may be able to impact the behavioral and/or anatomical consequences of autoimmune inflammation in a relapsing-remitting myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis (MOG-EAE) rat model of MS (Storch et al., 1998). In addition to testing the efficacy of pDNA-IL-10F129S gene therapy in the treatment of autoimmune disease, we also characterize sensory changes associated with hind paw sensitivity to touch (mechanical allodynia) that have not previously been described in an animal model of MS.
Materials and Methods
Rats
Viral-free adult male Dark Agouti rats (250-300g; Harlan Labs) were used in all experiments. Rats were housed in temperature (23°C ± 3°C) and light (12:12 light:dark; lights on at 0700 hr) controlled rooms with standard rodent food and water available ad libitum. Behavioral testing was performed during the first half of the light cycle. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
EAE induction
Relapsing-remitting experimental autoimmune encephalomyelitis was induced as previously described (Ledeboer et al., 2003). Recombinant protein corresponding to the N-terminal sequence of rat myelin oligodendrocyte glycoprotein (MOG; amino acids 1–125) was expressed in Escherichia coli and purified to homogeneity by Ni-chelate chromatography using a 6-His tag. The purified protein was dissolved in 6 M urea and dialyzed against 10 mM of sodium acetate buffer (NaAc; pH 3.0) then stored at −80 °C. EAE was induced via injection of 35 ug MOG in 0.01M NaAc (pH 3.0) emulsified in incomplete Freund's adjuvant (IFA; Sigma, St. Louis, MO; in a 1:1 ratio of MOG (35 ug in 50 ul) : IFA (50 ul)). Rats were briefly anaesthetized with isoflurane and given a single intra-dermal injection (100 ul total volume) into the dorsal skin (intra-dermal) just rostral to the base of the tail. Rats were monitored daily for body weight changes, motor symptoms, and sensory symptoms. During periods of motor symptom expression, the rats' bladders were expressed manually to help control bladder infections. Rats that lost greater than 25% body weight from baseline (N=3 from group receiving vehicle therapy, N=2 from group receiving IL-10 therapy), or that displayed symptoms of paralysis severe enough to include upper limb paralysis were euthanized and excluded from analysis.
Motor score
Motor deficits were scored on a scale from 0-7 based on the degree of ascending paralysis. The scores correspond to the following deficits: 0 = no symptom expression; 1 = partial tail paralysis; 2 = full tail paralysis; 3 = hindlimb weakness (unsteady gait while walking); 4 = partial hindlimb paralysis (no weight bearing but observable movement of the limb); 5 = full hindlimb paralysis; 6 = partial forelimb paralysis; 7 = euthanasia due to disease progression. Motor scores were recorded during the first few hours of the light cycle.
Body weight
Body weights were recorded daily during the first few hours of the light cycle on a standard triple beam scale.
von Frey test
Rats were allowed to habituate to the testing room and conditions (low intensity red light and room temperature of 82-84° F) on four occasions prior to the initial von Frey test. Allodynia was assessed by randomly applying a von Frey filament (Stoelting, Wood Dale, IL) to the rear portion of the plantar surface of the left and right hindpaws using a testing protocol modified from previously published methods (Milligan et al., 2006). Each presentation of the von Frey filaments was performed by moving the filament into position without making skin contact for six seconds. The filament was then applied to the hindpaw for six seconds. If no response was observed, the filament was removed from the skin but held nearby for an additional six seconds before moving away. Testing began with two presentations of the 2.83 filament and proceeded until 3 consecutive responses at a given level of stimulation were observed. If one or more non-responses were observed at a level of stimulation, then the next filament in the series of increasing stimulation was tested. von Frey measurements were taken prior to surgery and every few days following surgery for the duration of the experiment, with the exception of rats exhibiting a motor deficit including unsteady hindlimb gait (score of 3 or greater). Behavioral measures were performed blind with respect to treatment group. Data are presented as a 50% withdrawal threshold (stimulation required to elicit a paw withdrawal in 50% of trials) calculated from the measured absolute withdrawal threshold (stimulation that elicits paw withdrawal up to 3 consecutive withdrawals). von Frey scores were recorded during the first few hours of the light cycle.
Intrathecal injection
pDNA-IL-10F129S and vehicle injections were acutely administered intrathecally as previously described (Milligan et al., 2006) and initiated at the onset of motor symptoms. Briefly, rats were anesthetized with isoflurane (2.5% in O2) and an 18-ga 1.5 inch hypodermic needle, Beckton Dickinson & CO, Franklin Lakes, NJ) was removed from its hub and percutaneously inserted between lumbar (L) vertebrae L5 and L6. A catheter of PE-10 tubing (Beckton Dickinson & CO, Sparks, MD) was then threaded through this 18-ga. guide 7.3-cm rostrally from the exterior end of the guide, allowing for the tip of the catheter to be placed at the level of the lumbosacral enlargement. Rats receiving pDNA-IL-10 were administered 100 ug DNA in 17 ul on the first injection and 25 ug in 4.5 ul on the second injection. Rats receiving vehicle intrathecal injections were administered equivolume vehicle (3% sucrose in DPBS; Dulbecco's Phosphate-Buffered Saline 0.1mM pore-filtered, pH 7.2; Gibco, Grand Island, NY). Control DNA injected intrathecally at these doses has been previously determined in our lab to induce a transient increase in endogenous IL-10 protein lasting for several days (Sloane et al., Manuscript in review). Given the daily changes in symptom expression that are observed in the current EAE model, use of control DNA would pose serious confounds. Therefore, the present studies utilize a PBS vehicle to avoid the induction of IL-10 protein in both the control and experimental groups.
Injections were administered over approximately 20 sec. The PE-10 catheter and 18-ga. needle were then removed and anesthesia discontinued. Rats were anesthetized for an average of 2-5 min during injections.
pDNA-IL-10F129S
The plasmid construct coding for rat interleukin-10 (pDNA-IL-10F129S) was identical to that previously described (Milligan et al., 2006). Briefly, this plasmid contains cDNA encoding rat IL-10 under the control of a hybrid cytomegalovirus (CMV) enhancer/chicken beta actin promoter. A point mutation resulting in an amino acid change (F129S), has been identified in the rat IL-10 gene expressed in these studies (Milligan et al., 2005). This mutation lies outside of identified receptor binding regions. The effect of this point mutation on IL-10 bioactivity in vitro as well as a comparison of wildtype IL-10 and the F129S plasmid construct is currently under investigation. Preliminary results with macrophages and B cells in vitro suggest differences between wild type and variant (Busha and Chavez, personal communications). However, this construct has previously been documented to exert anti-inflammatory activity in vivo across diverse models of inflammation including models of chronic renal disease (Mu et al., 2005a), aortic tissue transplantation (Storch et al., 1998), spinal nerve transection (Storek et al., 2008), and sciatic nerve ligation (Milligan et al., 2005). Plasmid was grown up in SUREII cells (Stratagene, La Jolla, CA; cat#200152). Isolation and purification of the plasmid were performed using Qiagen Giga Endotoxin-Free kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. All purified plasmid was suspended in a vehicle of 0.2 micron filtered 3% sucrose solution in Dulbecco's Phosphate-Buffered Saline (DPBS; 0.1mM pore-filtered, pH 7.2; Gibco, Grand Island, NY) and stored at -20°C until thawed on ice prior to injection. pDNA injections were delivered at a concentration of 5.9 ug/ul as determined by optical density at 260/280 nm.
Tissue collection
Tissues from 3 rats per treatment group were collected 30 days following MOG injection. Rats were deeply anesthetized with sodium pentobarbital prior to transcardial perfusion with ice cold 0.9% heparinized saline for 5 min followed by ice cold 4% paraformaldehyde in 0.1M PB for 5 min. Each spinal cord was exposed via laminectomy and carefully removed from the vertebral column. A 6 cm section of tissue centered around the lumbar enlargement (site of intrathecal injections) was collected. Tissue was post-fixed in 4% paraformaldehyde at 4°C overnight and subsequently stored in a 30% sucrose solution at 4°C. Paraffin embedding of tissue and mounting of 5 micron thick slices onto slides was then conducted by Comparative Biosciences Inc., (Sunnyvale, CA) and stored until processed for immunohistochemical analyses.
Immunohistochemistry
Paraffin embedded, slide mounted tissue was deparaffinized and stained according to standard procedures as follows. Deparaffinization was achieved by two 30 min incubations in Histoclear (xylene substitute) followed by two 5 min incubations in 100% EtOH, incubations of 1 min each in 95% EtOH and 70% EtOH, and finally by a 5 min incubation in distilled water. Slides were then loaded into a Sequenza Immunostaining Center (Shandon; Pittsburgh, PA) slide holder for staining. Slides were washed with 2 ml 0.01M PBS, cleared with a 0.9% H2O2 0.9% Triton-X solution for 15 min. This was followed by another 2 ml PBS wash and overnight incubation in primary detection antibody (mouse anti-rat ED1, a classic marker of macrophage/microglia activation; AbD Serotec; Raleigh, NC; 1:200 dilution for final concentration of 2.5 ug/ml; mouse anti-rat glial fibrillary acidic protein (GFAP), a marker of astrocyte activation; MP Biomedicals LLC; Solon, OH; 1:100 dilution). The next day, a 2 ml PBS wash was followed by a 2 hr incubation in secondary antibody (Alexa Fluor goat anti-mouse 594 secondary; Invitrogen; Eugene, OR; 1:200 dilution for final concentration of 10 ug/ml). A final 2 ml PBS wash was performed prior to coverslipping with Vectashield fluorescence mounting medium containing the nuclear stain DAPI (Vector Laboratories, Inc.; Burlingame, CA). Immunohistochemistry procedures were conducted simultaneously on tissue from both treatment groups for each detection antigen. Specificity of primary antibodies were assessed by omission of primary antibody during staining of selected slides from each treatment group. This omission resulted in a complete lack of signal.
Microscopy quantification
Lumbar spinal cord tissue was analyzed subsequent to IHC antigen detection. Tissue from 3 animals per group (control and IL-10 treated), 8 sections per animal, and 2 images per section were included in the analysis. Tissue images were captured by an investigator blinded to treatment group designation at 20× magnification using a fluorescence microscope (Olympus BX61; Melville, NY) to capture multiple channel images (Olympus DP70 3.0 camera). Quantification of phenotype markers was achieved using Microsuite Biological Suite 2.6 software. The red channel, corresponding to the 594 secondary antibody excitation wavelength, was extracted from each color composite image and converted to grayscale. A small area of the photo was analyzed to determine background threshold levels individually for each image. A larger area of tissue was then analyzed for average gray value. Following investigator blinded quantification of all images, statistical analysis demonstrates no difference between groups in the tissue area analyzed (ED1 marker, p=0.27; GFAP marker, p=0.73), or in the amount of background threshold subtracted from each treatment group (ED1 marker, p=0.53; GFAP marker, p=0.31).
Statistical Analysis
All statistical comparisons of motor deficit score and immunohistochemical analysis data were computed using the statistical software package R (R: A Language and Environment for Statistical Computing; http://www.R-project.org). von Frey and body weight data were analyzed using Statview 5.0.1 for Macintosh. Motor deficit scores were analyzed using a non-parametric Wilcoxon rank sum test. Data from the von Frey test were analyzed as the interpolated 50% threshold (absolute threshold) in log base 10 of stimulus intensity (monofilament stiffness in milligrams × 10). Time course measures for body weight and the von Frey test were analyzed by repeated measures ANOVAs followed by Fisher's protected least significant difference posthoc comparisons, where appropriate. The repeated measures ANOVA statistic examines group effects across the timepoints tested throughout the experiments. Immunohistochemistry markers were analyzed by two tailed t-test statistic.
Results
Motor scores
All rats were administered intradermal myelin oligodendroctye glycoprotein to induce experimental autoimmune encephalomyelitis (MOG-EAE), a previously established model of multiple sclerosis (MS) (Storch et al., 1998; Ledeboer et al., 2003). After MOG immunization, but prior to intrathecal injection with either vehicle or IL-10 gene therapy, no difference in motor deficits was observed between groups (Fig.1). Intradermal MOG induced classic EAE symptoms of ascending motor paralysis in a relapsing-remitting pattern in rats receiving intrathecal vehicle after symptom onset (n=6/group; Fig.1). Intrathecal IL-10 gene therapy (pDNA-IL-10F129S; n=5/group) was likewise initiated at the onset of motor symptoms (first signs of motor weakness at tip of tail), to allow for a normal induction of the auto-aggressive immune response associated with EAE prior to induction of IL-10. The IL-10 gene therapy group exhibited no further progression of paralysis, an extended period of motor symptom remission, and suppressed paralysis relapse as compared to rats receiving vehicle therapy (Fig.1). Beginning one day after the initial intrathecal injection, rats that received pDNA-IL-10F129S showed significantly less motor deficits than rats receiving intrathecal vehicle during the initial period of motor symptoms (Wilcoxon rank sum, p<0.03). Rats that received intrathecal vehicle spent approximately 3 days in remission compared to an average time of 8 days spent in remission for the rats that received intrathecal pDNA-IL-10F129S (Fig.1). Motor deficits were significantly milder in the pDNA-IL-10F129S group at the peak of initial symptom expression (average score=1.3, sem=0.44) as compared to the vehicle group (average score=3.3, sem=0.4; Wilcoxon rank sum test: p<0.02). Both groups entered the first period of symptom remission 4 days following initial symptom onset though a more complete maximal recovery of motor function during the first symptom remission was observed in the pDNA-IL-10 group (average score=0.2, sem=0.2) compared to rats receiving intrathecal vehicle (average score=1.33, sem=0.36) as demonstrated by Wilcoxon rank sum test (p=0.04). Rats receiving pDNA-IL-10F129S displayed fewer motor deficits at the 2nd peak of symptom expression (average score=1.0, sem=0.41) as compared to the vehicle groups (average score=5.1, sem=1.0) as demonstrated by Wilcoxon rank sum test (p<0.01). On each day following the second intrathecal injection (occurring 3 days after motor symptom onset and first intrathecal injection) the group receiving pDNA-IL-10F129S showed significantly fewer motor deficits than the group receiving intrathecal vehicle (Wilcoxon rank sum test, p<0.05).
Figure 1.
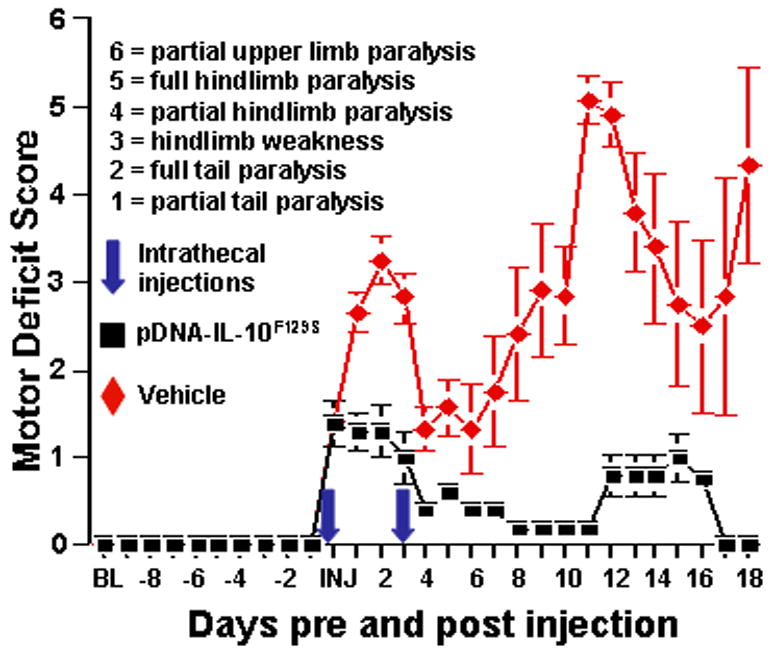
pDNA-IL-10F129S therapy reduces EAE induced motor paralysis. Motor deficits were scored prior to and every day following MOG injection. The time from MOG injection to initial motor deficits observed varied from 10 to 18 days. Here, scores are displayed relative to the first day that motor deficits were observed (score of 1 or greater). Black squares represent the group receiving pDNA-IL-10F129S (N=5) with standard error bars. Red diamonds represent the group receiving intrathecal vehicle (N=6). Intrathecal (intrathecal) injections of 100 ug pDNA-IL-10F129S or equivolume vehicle (3% sucrose PBS) occurred on the first day that motor deficits were apparent and the 2nd injection occurred 3 days later with either 25 ug pDNA-IL-10F129S or equivolume vehicle. A significant reduction in the severity of motor paralysis was observed following pDNA-IL-10F129S therapy.
Assessment of hindpaw sensitivity to touch (mechanical allodynia)
No differences in responsivity to calibrated mechanical stimuli (von Frey test) were observed between groups either prior to MOG injection (repeated measures ANOVA, F(1,18)=.17, p=.69) or after MOG but prior to the first intrathecal injection (repeated measures ANOVA, F(1,144)=2.2, p=.16) (Fig.2). All rats displayed a transient allodynia, observed as a decrease in response thresholds days 3 through 5 after intradermal MOG, as compared to baseline values (repeated measures ANOVA, F(5,126)=58, p<.0001), at which point withdrawal thresholds were equivalent to baseline. On days 5, 4, 3, and 2 prior to first intrathecal injection as well as on the day of injection, von Frey response thresholds were significantly higher than baseline values (repeated measures ANOVA, F(10,226)=12.4, p<.0001), reflecting a reduction in sensitivity to touch. Rats were not assessed for mechanical responsivity during periods of hind limb weakness or paralysis as defined by a motor deficit score of 3 or higher. The mechanical response thresholds of rats that received intrathecal pDNA-IL-10F129S did not differ from their baseline thresholds, beginning the first day following the initial pDNA-IL-10F129S injection and lasting through the remainder of the time course (repeated measures ANOVA, F(16,48)=1.24, p=.27). In contrast, at 24 hr following i.t. injection, animals receiving vehicle still demonstrated reduced hindpaw sensitivity compared to baseline, suggesting a restoration of sensitivity due to IL-10 gene therapy. The group that received intrathecal vehicle was significantly more allodynic as compared to rats receiving intrathecal pDNA-IL-10F129S during the first period of symptom remission (repeated measures ANOVA, F(1,14)=30, p<.0001) as well as during the second period of symptom remission (repeated measures ANOVA, F(1,10)=6.4, p=.03).
Figure 2.
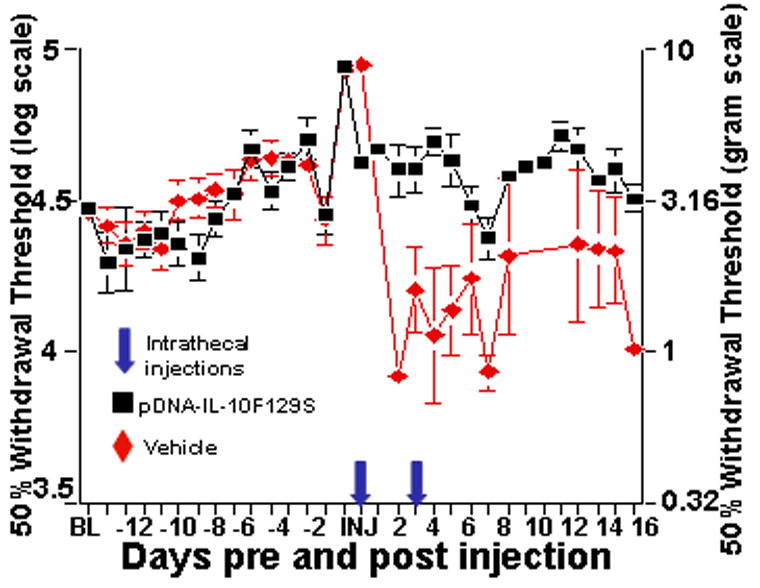
pDNA-IL-10F129S therapy improves EAE induced reduced sensitivity to touch and allodynia. These are the same rats (pDNA-IL-10F129S, black squares: n=5; vehicle, red diamonds: n=6) whose motor scores are presented in Figure 1. von Frey measures were assessed prior to and every day following MOG injection with the exception of rats that displayed a motor deficit score of 3 (unsteady hind limb gait) or higher on a given day. Data are displayed relative to the day of first intrathecal injection of pDNA-IL-10 or vehicle (onset of paralysis). Prior to the onset of hind limb paralysis, a significant reduction in hind limb sensitivity to touch is observed. This is reversed by pDNA-IL-10F129S therapy. During the period of motor symptom remission, a significant increase in hind limb sensitivity to touch is observed that is prevented by pDNA-IL-10F129S therapy.
Body weight
No differences in body weight were observed between groups prior to MOG injection or through 14 days following the first intrathecal injection (repeated measures ANOVA, F(1,16)=1.24, p=.28). On days 15 through 18 following the first intrathecal injection, the group receiving pDNA-IL-10F129S displayed significant recovery of body weight (average weight = 265 g, sem = 2.6) as compared to rats receiving intrathecal vehicle (average weight = 244 g, sem = 2.3; repeated measures ANOVA, F(1,18)=40.5, p<.0001; Fig.3).
Figure 3.
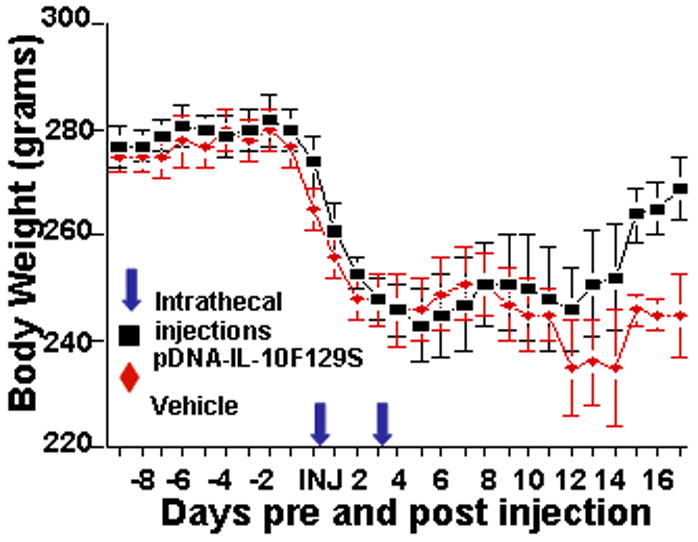
pDNA-IL-10F129S therapy reverses EAE induced loss of body weight. These are the same rats (pDNA-IL-10F129S, black squares: n=5; vehicle, red diamonds: n=6) whose motor and pain scores are presented in Figure 1 and 2 respectively. Baseline body weight is reported as the average of values on 2 consecutive days prior to MOG injection. Rats were then weighed every day following MOG injection for the remainder of the timecourse. A significant reduction in body weight was observed in parallel with the onset of behavioral symptoms in both treatment groups. Beginning on day 15 following initiation of pDNA-IL-10F129S therapy, this group demonstrated a significant recovery of lost body weight as compared to the group receiving vehicle therapy.
Histopathology
Tissues from 3 randomly selected rats in each group were collected 34 days following MOG injection and stained for markers of astrocytes and microglial/macrophage activation. At the time of tissue harvest, the group receiving intrathecal vehicle after intradermal MOG had an average motor deficit score of 5.67 (sem 0.33) and the group receiving pDNA-IL-10F129S after MOG had an average motor score of 0.33 (sem 0.33), demonstrating significantly less paralysis (p<0.001).
Glial fibrillary acidic protein (GFAP, a cytoskeletal protein that is upregulated specifically in astrocytes during inflammatory) is used here to quantify astrocyte activation. As seen in Fig.4A, pDNA-IL-10F129S therapy significantly suppressed GFAP immunoreactivity in lumbar spinal cord as compared to vehicle therapy (t-test, p<0.001). Representative photos of lumbar spinal cord are shown from rats receiving pDNA-IL-10F129S therapy (Fig.4B) or vehicle therapy (Fig.4C).
Figure 4.
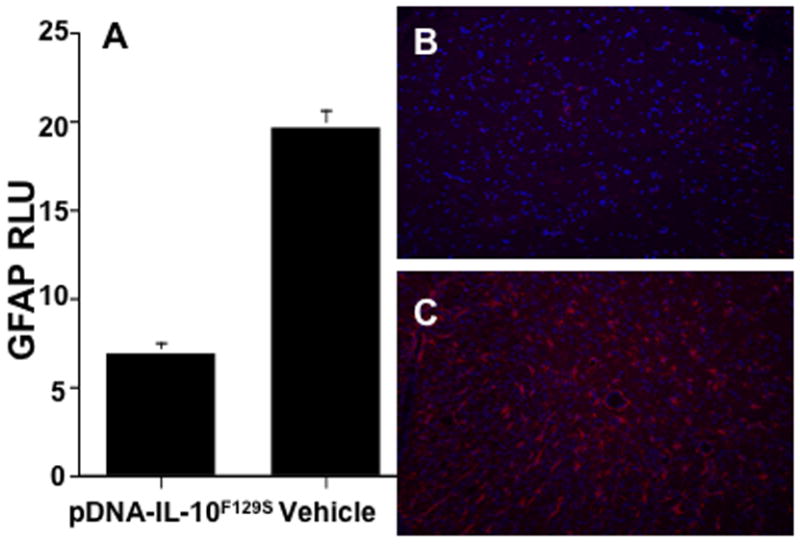
pDNA-IL-10F129S therapy suppresses EAE induced astrocyte activation. Panel A represents average signal density + sem from lumbar tissue in rats receiving pDNA-IL-10F129S therapy compared to vehicle therapy following immunohistochemical detection of the astrocytic marker glial fibrillary acidic protein (see methods). GFAP phenotype marker is shown in red and the nuclear stain DAPI is shown in blue in panel B and C. Panel B is a representative photo of lumbar tissue following pDNA-IL-10F129S therapy (tissue from N=3 animals, average motor deficit score 0.33). Panel C is a representative photo of lumbar tissue following vehicle therapy (tissue from N=3 animals, average motor deficit score 5.67).
In addition to astrocyte activation, we examined the expression level of ED1 protein (a lysosomal protein that is upregulated during inflammatory conditions in cells of monocytic origin including macrophages and microglia). Fig.5A demonstrates a suppression of ED1 immunoreactivity in lumbar spinal cord following pDNA-IL-10F129S therapy as compared to vehicle therapy (t-test, p<0.001). Representative photos of lumbar spinal cord are shown from rats receiving pDNA-IL-10F129S therapy (Fig.5B) or vehicle therapy (Fig.5C). This figure demonstrates pDNA-IL-10F129S induced suppression of microglia/macrophage activation.
Figure 5.
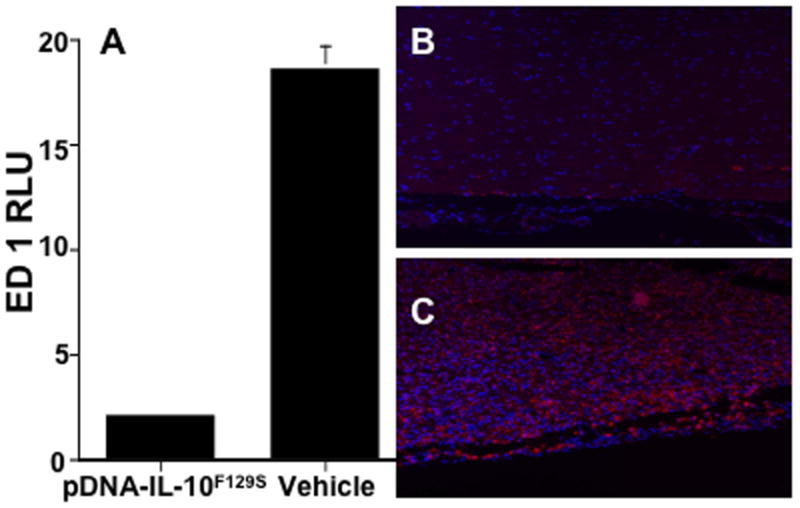
pDNA-IL-10F129S therapy suppresses EAE induced microglial/macrophage activation. Panel A represents average signal density + sem from lumbar tissue in rats receiving pDNA-IL-10F129S therapy compared to vehicle therapy following immunohistochemical detection of the microglial/macrophage activation marker ED1 (see methods). ED1 phenotype marker is shown in red and the nuclear stain DAPI is shown in blue in panel B and C. Panel B is a representative photo of lumbar tissue following pDNA-IL-10F129S therapy (tissue from N=3 animals, average motor deficit score 0.33). Panel C is a representative photo of lumbar tissue following vehicle therapy (tissue from N=3 animals, average motor deficit score 5.67).
Discussion
The current study had 2 concurrent purposes. It has been well established in the literature that model of autoimmune disease used in these studies is an inflammatory neuropathy of the central nervous system (CNS). Inflammatory mediators involved in autoimmune inflammation have previously been implicated in the expression of sensory symptoms of other neuropathies including the expression of chronic pain. To date, however, the types of sensory changes associated with models of CNS autoimmune disease have not been adequately characterized. The current experiments document previously uncharacterized sensory disturbances including both reductions in sensitivity to touch and allodynia of the hind paws associated with a myelin oligodendroctye glycoprotein induced experimental autoimmune encephalomyelitis (MOG-EAE) model of multiple sclerosis (MS). Prior work has demonstrated the therapeutic utility of controlling inflammatory mediators in neuropathies to control expression of neuropathic pain. The current study examines the effect on sensory disturbances present in the MOG-EAE model of controlling inflammation via a previously characterized anti-inflammatory gene therapy. An anti-inflammatory cytokine gene therapy (pDNA-IL-10F129S) to control autoimmune inflammation was demonstrated to improve both motor (paralysis), and sensory dysfunction (reduced sensitivity to touch and allodynia), decrease glial/immune activation in lumbar spinal cord, as well as decrease MOG-EAE-induced body weight loss.
Symptoms common across neuropathies, including hypoalgesia (reduced pain sensitivity) and reduced sensitivity to touch, have long been associated with MS, and are amongst the earliest symptoms detected (Calabresi, 2004). In recent years, however, symptoms of neuropathic pain, including allodynia (perception of normally non-painful stimuli as painful) and thermal and mechanical hyperalgesia (exaggerated pain sensitivity) have become increasingly recognized as common symptoms of MS (Kenner et al., 2007). A majority of patients experience a painfully enhanced sensitivity to thermal and tactile stimuli at some point during the course of their disease (Hadjimichael et al., 2007).
The recent recognition of the prevalence of sensory dysfunction in MS begs an examination of parallel symptoms in models of this disease. Few studies have examined sensory changes that occur in animal models of MS. Several reports exist in current literature addressing sensory changes, though this is the first description of a rat model of MS. Additionally, the model used in these studies has the advantage of following a relapsing-remitting course of symptom expression similar in nature to the clinical disease. Chronic or neuropathic pain is often associated with changes in sensitivity to both thermal and tactile stimuli in animal models as well as in the clinic. Thermal and tactile information is relayed from the periphery to the CNS via distinct pathways and involve distinct sets of receptors and ion channels, which may be differentially affected by neuropathies including autoimmune inflammatory neuropathies such as EAE. The current study describes changes in tactile sensitivity, which has not been previously described in any model of MS. Aicher et al. (Aicher et al., 2004) characterized changes in mouse tail thermal sensitivity over the course of two chronic-relapsing models. Aicher and colleagues reported a marked thermal hypoalgesia beginning several days prior to the onset of motor deficits. This was followed during motor symptom remission by a thermal hyperalgesia. An earlier study in two acute EAE models in rat (Pender, 1986) reported an ascending sensory loss prior to onset of motor symptoms, as measured by tail pinch-evoked vocalization. This basic pattern of reduced sensitivity prior to the onset of motor symptoms, followed in turn by an exaggerated sensitivity to stimuli during remission of motor symptoms was again observed in the present study regarding tactile stimuli applied to rat hind paws.
MOG-EAE shares many of the behavioral and anatomical hallmarks of clinical MS and has been used to examine the pathology of disease progression as well as to test the potential therapeutic value of experimental disease treatments (Sunnemark et al., 2005; Balatoni et al., 2007; Graaf et al., 2008). It appears that sensory changes including both hypoalgesia and hyperalgesia in models characterized to date may parallel sensory changes seen clinically. This finding not only helps to validate the use of these animal models to study the complexities of MS, but offers a new front on which to examine the pathology caused by autoimmune inflammation as well as test potential therapeutic interventions.
The response of CNS glial cells (including microglia and astrocytes) and the crucial contribution of these cells to symptoms of inflammatory neuropathies has been well characterized (Watkins and Maier, 2002; Milligan et al., 2003; Watkins et al., 2003). Direct activation via immunogenic stimuli or to intense neuronal activity causes microglia and astrocytes to undergo a phenotypic change characterized by altered morphology and the production of diverse signaling molecules including cytokines, chemokines, and reactive oxygen species (Benveniste, 1997; Gonzalez-Scarano and Baltuch, 1999; Milligan et al., 2001; Kim and Vellis, 2005). Activated glia respond to substances released by pre-synaptic neurons (excitatory amino acid neurotransmitters, chemokines, and cytokines) and in turn release signaling molecules that can alter the firing rates of post-synaptic neurons (Araque et al., 1999; Araque and Perea, 2004). Thus, much focus has been directed towards detailing ways in which glia participate in synaptic scaling, or the fine tuning of neuronal firing properties. This intimate relationship between glial and neuronal activity has been termed the “tetrapartite synapse” (DeLeo et al., 2006).
Widespread glial activation is commonly reported over the course of MS (Petzold et al., 2002; Gray et al., 2008). Data presented here demonstrate high levels of both astrocytic and microglia/macrophage activation in rats receiving MOG followed by intrathecal vehicle. Astrocytic activation was noted throughout the lumbar spinal cord of these rats. Microglia/macrophage activation was noted primarily in the white matter in these tissues. Treatment with pDNA-IL-10F129S significantly suppressed this activation. It is hypothesized that inflammation underlies the expression of the range of symptoms in MS and other inflammatory neuropathies. Controlling this inflammation, as has been demonstrated following pDNA-IL-10F129S, ameliorates symptoms of paralysis as well as reduced sensitivity to touch and pain expression. Wide ranging benefits may thus be achieved by targeting pathological inflammation.
Sensory disturbances arising from inflammatory neuropathies often include symptoms of decreased sensitivity sensitivity to touch. Diabetic neuropathy, a disease which activates CNS glia and leads to demyelination (Malik, 1997; Zeng et al., 2000; Sharma et al., 2002), is characterized in late stage disease by decreased thermal and tactile sensitivity (Calcutt et al., 2006; Pabbidi et al., 2008). Decreased sensitivity also occurs in cases of chronic chemotherapy induced neuropathy (Bois et al., 1999), traumatic spinal cord injury (Jensen et al., 2007), and CNS infections (Rowbotham and Fields, 1996; Macleod et al., 2002). Many neuropathies that lead to a loss of sensitivity also produce neuropathic pain. These include diabetes (Daulhac et al., 2006; Pabbidi et al., 2008), chemotherapeutic neuropathy (Polomano and Bennett, 2001; Ledeboer et al., 2007b), spinal cord injury (Jensen et al., 2007), and CNS infections (Rowbotham and Fields, 1996; Macleod et al., 2002). We here demonstrate this EAE model shares characteristic symptoms of both reduced sensitivity to touch and mechanical allodynia seen in other neuropathies. The striking observation that this diverse array of insults results in similar sensory symptoms as well as similar glial activation and demyelination suggests that glial activation is a fundamental event in response to injury. Glial activation has been known to involve the production of inflammatory mediators that exert diverse effects on neuronal cells. Many of these factors acting alone as well as in conjunction with each other could alter neuronal activity possibly leading to symptoms of either increased activity (allodynia) or decreased activity (reduced mechanical sensitivity). Ultimately, the effects of inflammation on neuronal signaling likely results from a complex interaction of the inflammatory mediators. Additionally, a great deal of evidence suggests a pivotal role for glial activation in the expression of sensory symptoms, highlighting the importance of these cells in a therapeutic context.
MS is an inflammatory disease involving widespread activation of innate and adaptive immune mechanisms. The immune system is composed of a complex network of cell types and signals and therefore poses a difficult therapeutic problem. Inflammatory signaling is carried out by a large number of different mediators ranging from relatively large proteins to small peptides, gases, and protons. Moreover, inflammatory processes often involve multiple redundant signaling systems. Thus, treatment strategies that target the activity of one specific inflammatory mediator may not adequately suppress pathological inflammation. The immune system contains several powerful regulatory molecules that are able to exert widespread anti-inflammatory actions. IL-10 is one of these endogenous anti-inflammatory cytokines that has proven effective in suppressing inflammation in a variety of contexts. IL-10 is multifaceted and potent in activity and is able to block the activation of immune cells, including glia, following stimulation with pro-inflammatory cytokines, various injuries, or pathogen (viral or bacterial) exposure both in vitro and in vivo (Moore et al., 2001). The mechanisms by which IL-10 reduces glial activation include both transcriptional and translational actions including suppression of pro-inflammatory gene expression, upregulation of inflammatory cytokine decoy receptors, and reduction in the activation of transcription factors responsible for pro-inflammatory gene activation. The powerful anti-inflammatory actions of IL-10 make it an attractive therapeutic candidate for controlling pathological inflammation, including pathological CNS glial activation. The therapeutic utility of IL-10 has been demonstrated in a number of autoimmune diseases (Xiao et al., 1998; Jung et al., 2004; Raz et al., 2005; Schif-Zuck et al., 2006) including amelioration of relapsing-remitting EAE following CNS delivery of adenoviral vector containing IL-10 gene (Hutchins et al., 2001). There are several clinical drugs in use as disease modifying agents in MS that function as anti-inflammatory agents or that induce anti-inflammatory mediators. Interferon beta (IFNβ) is one of these common drugs thought to provide therapeutic benefit via suppression of pro-inflammatory mediators and induction of proteins such as IL-10. Glatiramer Acetate is another commonly prescribed drug that is thought to work via induction of subsets of T cells that produce IL-10 among other anti-inflammatory mediators (Arnon and Aharoni, 2004).
Ample evidence from basic, as well as clinical, research suggests that pro-inflammatory processes drive autoimmune pathology. Therefore, a logical therapeutic approach has been to counteract this inflammation, as is the approach using anti-inflammatory cytokine gene therapy demonstrated in these studies. Currently approved therapies for MS achieves anti-inflammtory ends via direct interference with inflammatory signaling with the use of neutralizing antibodies or receptor antagonists. This general therapeutic approach, however, is not without potential problems. Fundamentally, non-pathological inflammation initiates wound healing. Interference with this process in the context of MS, for example with a neutralizing antibody against TNF (tumor necrosis factor; pro-inflammatory cytokine), has been shown to worsen MS symptoms (Arnason et al., 1999). A specific area of therapeutic interest in MS is the question of processes of demyelination and remyelination. Several studies have shown a requirement for pro-inflammatory signaling in the differentiation of oligodendrocyte progenitor cells during remyelination (Zawadzka and Franklin, 2007). It is important to consider potential negative impacts of interfering with the wound healing process when developing anti-inflammatory therapies. Anti-inflammatory therapies, however, are not limited to the passive blockade of pro-inflammatory factors but also include the active induction of counter-regulatory mediators. IL-10, for example, not only suppresses the action of inflammatory cytokines, but also activates a variety of transcription factors including some activated following growth factor signaling such as STAT3 (Akira, 1997; Donnelly et al., 1999). Regulatory T cell populations are also characterized by the production of many cytokines, chemokines, and growth factors that may control inflammation (Wilczynski et al., 2008).
In sum, inflammatory neuropathies such as MS are associated with a wide range of motor and sensory symptoms. The current studies provide the first description of mechanical allodynia in a model of MS. Animal models of MS have proven useful in the development of therapeutic strategies. The MOG-EAE model employed here exhibits symptoms of inflammatory neuropathy including both decreased and enhanced sensitivity to touch. This model may prove appropriate for examining mechanisms of disease induced pain as well as for testing potential therapeutic agents. To that end, the anti-inflammatory cytokine gene therapy employed in these studies demonstrates the ability to improve the behavioral and anatomical consequences caused by autoimmune inflammation.
Acknowledgments
This work was supported by grants from Avigen Inc. and NIH grants DA018156, DA015642, DA015656, and HL5510. Tissue embedding and slicing for analysis was conducted by Comparative Biosciences Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Aicher S, Silverman M, Winkler C, B B., Jr Hyperalgesia in an animal model of multiple sclerosis. Pain. 2004;110:560–570. doi: 10.1016/j.pain.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Akira S. IL-6-regulated Transcription Factors. Int J Biochem Cell Biol. 1997;29:1401–1418. doi: 10.1016/s1357-2725(97)00063-0. [DOI] [PubMed] [Google Scholar]
- Araque A, Perea G. Glial modulation of synaptic transmission in culture. Glia. 2004;47:241–248. doi: 10.1002/glia.20026. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri R, Haydon P. Tripartite synapsees: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Arnason B, Jacobs G, Hanlon M, Clay B, Noronha A, Auty A, Davis B, Nath A, Bouchard J, Belanger C, Gosselin F, Thibault M, Duquette P, Bourgoin P, DuBois R, Girard M, Ebers G, Rice G. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53:457–465. [PubMed] [Google Scholar]
- Arnon R, Aharoni R. Mechanism of action of glatiramer acetate in multiple sclerosisand its potential for the development of new applications. PNAS. 2004;101:14593–14598. doi: 10.1073/pnas.0404887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatoni B, Storch M, Swoboda E, Schonborn V, Koziel A, Lambrou G, Hiestand P, Weissert R, Foster C. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Research Bulletin. 2007;74:307–316. doi: 10.1016/j.brainresbull.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Benveniste E. Cytokines: influence on glial cell gene expression and function. Neuroimmunoendocrinology. 1997;69:31–75. doi: 10.1159/000058653. [DOI] [PubMed] [Google Scholar]
- Bois Ad, Schlaich M, Luck HJ, Mollenkopf A, Wechsel U, Rauchholz M, Bauknecht T, Meerpohl HG. Evaluation of neurotoxicity induced by paclitaxel second-line chemotherapy. Support Care Caner. 1999;7:354–361. doi: 10.1007/s005200050275. [DOI] [PubMed] [Google Scholar]
- Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol. 2005;252:V/3–V/9. doi: 10.1007/s00415-005-5002-7. [DOI] [PubMed] [Google Scholar]
- Calabresi P. Diagnosis and Management of Multiple Sclerosis. Am Fam Physician. 2004;70:1935–1944. [PubMed] [Google Scholar]
- Calcutt N, Freshwater J, Hauptmann N, Taylor E, Mizisin A. Protection of sensory function in diabetic rats by Neotrofin. Eur J Pharmacol. 2006;534:187–193. doi: 10.1016/j.ejphar.2006.01.047. [DOI] [PubMed] [Google Scholar]
- Cunningham A, Murray C, O'Neill L, Lynch M, O'Connor J. Interleukin-1beta (IL-1beta) and tumor necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neuroscience Letters. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat A, Eschalier A, Fialip J. Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-D-aspartate-dependent mechanisms. Mol Pharmacol. 2006;70:1246–1254. doi: 10.1124/mol.106.025478. [DOI] [PubMed] [Google Scholar]
- DeLeo J, Tawfik V, LaCroix-Fralish M. The tetrapartite synapse: Path to CNS sensitization and chronic pain. Pain. 2006;122:17–21. doi: 10.1016/j.pain.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Donnelly R, Dickensheets H, Finbloom D. The Interleukin-10 Signal Transduction Pathway and Regulation of Gene Expression in Mononuclear Phagocytes. Journal of Interferon & Cytokine Research. 1999;19:563–573. doi: 10.1089/107999099313695. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Graaf Kd, Barth S, Herrmann M, Storch M, Wiesmuller K, Weissert R. Characterization of the encephalitogenic immune response in a model of multiple sclerosis. Eur J Immunol. 2008;38:299–308. doi: 10.1002/eji.200737475. [DOI] [PubMed] [Google Scholar]
- Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated Activity and Microglial Expression of Myeloperoxidase in Demyelinated Cerebral Cortex in Multiple Sclerosis. Brain Pathology. 2008;18:86–95. doi: 10.1111/j.1750-3639.2007.00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjimichael O, Kerns R, Rizzo M, Cutter G, Vollmer T. Persistent pain and uncomfortable sensations in persons with multiple sclerosis. Pain. 2007;127:35–41. doi: 10.1016/j.pain.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Hendriks JJ, Teunissen CE, Vries HEd, Dijkstra CD. Macrophages and neurodegeneration. Brain Research Reviews. 2005;48:185–195. doi: 10.1016/j.brainresrev.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Hirtz D, Thurman D, Gwinn-Hardy K, Mohamed M, Chadhuri A, Zalutsky R. How common are the “common” neurologic disorders? Neurology. 2007;68:326–337. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- Hutchins DCB, LaFace D, Stohlman S, Coffman R. Central nervous system expression of IL-10 inhibits autoimmune encephalomyelitis. J Immunol. 2001;166:602–608. doi: 10.4049/jimmunol.166.1.602. [DOI] [PubMed] [Google Scholar]
- Jensen M, Kuehn C, Amtmann D, Cardenas D. Symptom Burden in Persons With Spinal Cord Injury. Arch Phys Med Rehab. 2007;88:638–645. doi: 10.1016/j.apmr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Siglienti I, Grauer O, Magnus T, Scarlato G, Toyka K. Induction of IL-10 in rat peritoneal macrophages and dendritic cells by glatiramer acetate. Journal of Neuroimmunology. 2004;148:63–73. doi: 10.1016/j.jneuroim.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M. Interleukin-1b inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharm. 1990;181:323–326. doi: 10.1016/0014-2999(90)90099-r. [DOI] [PubMed] [Google Scholar]
- Kenealy SJ, Pericak-Vance MA, Haines JL. The genetic epidemiology of multiple sclerosis. Journal of Neuroimmunology. 2003;143:7–12. doi: 10.1016/j.jneuroim.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Kenner M, Menon U, Elliott D. Multiple Sclerosis as a painful disease. Int Rev Neurobiol. 2007;79:303–321. doi: 10.1016/S0074-7742(07)79013-X. [DOI] [PubMed] [Google Scholar]
- Kim S, Vellis Jd. Microglia in Health and Disease. Journal of Neuroscience Research. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Breve J, Poole S, Tilders F, Dam AV. Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia. 2000;30:134–142. doi: 10.1002/(sici)1098-1136(200004)30:2<134::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Wierinckx A, Bol J, Floris S, Lavalette Cd, Vries HD, Berg Tvd, Dijkstra C, Tilders F, Dam AV. Regional and temporal expression patterns of interleukin-10, interleukin-10 receptor and adhesion molecules in the rat spinal cord during chronic relapsing EAE. Journal of Neuroimmunology. 2003;136:94–103. doi: 10.1016/s0165-5728(03)00031-6. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Jekich B, Sloane E, Mahoney J, Langer S, Milligan E, Martin D, Maier S, Johnson K, Leinwand L, Chavez R, Watkins L. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain, Behavior, and Immunity. 2007a;21:686–698. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Liu T, Shumilla J, Mahoney J, Vijay S, Gross M, Vargas J, Sultzbaugh L, Claypool M, Sanftner L, Watkins L, Johnson K. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol. 2007b;2:279–291. doi: 10.1017/S1740925X0700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod M, Stewart G, Zeidler M, Will R, Knight R. Sensory features of variant Creutzfeldt-Jakob disease. J Neurol. 2002;249:706–711. doi: 10.1007/s00415-002-0696-2. [DOI] [PubMed] [Google Scholar]
- Malik R. The Pathology of Human Diabetic Neuropathy. Diabetes. 1997;46:S50–S53. doi: 10.2337/diab.46.2.s50. [DOI] [PubMed] [Google Scholar]
- Marrie RA. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurology. 2004;3:709–718. doi: 10.1016/S1474-4422(04)00933-0. [DOI] [PubMed] [Google Scholar]
- Martin R, McFarland H, McFarlin D. Immunological Aspects of Demyelinating Diseases. Annual Review of Immunology. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- Milligan E, O'Connor K, Nguyen K, Armstrong C, Twining C, Gaykema R, Holguin A, Martin D, Maier S, Watkins L. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci. 2001;21:2808–2819. doi: 10.1523/JNEUROSCI.21-08-02808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan E, Twining C, Chcur M, Biedenkapp J, O'Connor K, Poole S, Tracey K, Martin D, Maier S, Watkins L. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan E, Sloane E, Langer S, Cruz P, Chacur M, Spataro L, Wieseler-Frank J, Hammack S, Maier S, Flotte T, Forsayeth J, Leinwand L, Chavez R, Watkins L. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol Pain. 2005;25 doi: 10.1186/1744-8069-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan E, Sloane E, Langer S, Hughes T, Jekich B, Frank M, Mahoney J, Levkoff L, Maier S, Cruz P, Flotte T, Johnson K, Mahoney M, Chavez R, Leinwand L, Watkins L. Repeated intrathecal injections of plasmid DNA encoding interleukin-10 produce prolonged reversal of neuropathic pain. Pain. 2006;126:294–308. doi: 10.1016/j.pain.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Moore K, Malefyt R, Coffman R, O'Garra A. Interleukin-10 and the Interleukin-10 Receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Mu W, Ouyang X, Agarwal A, Zhang L, Long DA, Cruz PE, Roncal CA, Glushakova OY, Chiodo VA, Atkinson MA, Hauswirth WW, Flotte TR, Rodriguez-Iturbe B, Johnson RJ. IL-10 Suppresses Chemokines, Inflammation, and Fibrosis in a Model of Chronic Renal Disease. J Am Soc Nephrol. 2005a;16:3651–3660. doi: 10.1681/ASN.2005030297. [DOI] [PubMed] [Google Scholar]
- Mu W, Ouyang X, Agarwal A, Zhang L, Long D, Cruz P, Roncal C, Glushakova O, Chiodo V, Atkinson M, Hauswirth W, Flotte T, Rodriguez-Iturbe B, Johnson R. IL-10 Suppresses Chemokines, Inflammation, and Fibrosis in a Model of Chronic Renal Disease. J Am Soc Nephrol. 2005b;16:3651–3660. doi: 10.1681/ASN.2005030297. [DOI] [PubMed] [Google Scholar]
- Osterberg A, Boivie J, Thuomas K. Central pain in multiple sclerosis - prevalence and clinical characteristics. European Journal of Pain. 2005;9:531–542. doi: 10.1016/j.ejpain.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Ozenci V, Kouwenhoven M, Link H. Cytokines in multiple sclerosis: methodological aspects and pathogenic implications. Mult Scler. 2002;8:396–404. doi: 10.1191/1352458502ms837rr. [DOI] [PubMed] [Google Scholar]
- Pabbidi R, Yu S, Peng S, Khardori R, Pauza M, Premkumar L. Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol Pain. 2008;4 doi: 10.1186/1744-8069-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender M. Ascending impairment of nociception in rats with experimental allergic encephalomyelitis. J Neurol Sci. 1986;75:317–328. doi: 10.1016/0022-510x(86)90079-1. [DOI] [PubMed] [Google Scholar]
- Petzold A, Eikelenboom M, Gveric D, Keir G, Chapman M, Lazeron R, Cuzner M, Polman C, Uitdehaag B, Thompson E, Giovannoni G. Markers for different glial cell responses in multiple sclerosis: clinical and pathological correlations. Brain Pathology. 2002;125:1462–1473. doi: 10.1093/brain/awf165. [DOI] [PubMed] [Google Scholar]
- Polomano R, Bennett G. Chemotherapy-evoked painfulperipheral neuropathy. Pain Med. 2001;2:8–14. doi: 10.1046/j.1526-4637.2001.002001008.x. [DOI] [PubMed] [Google Scholar]
- Raz I, Eldor R, Naparstek Y. Immune modulation for prevention of type 1 diabetes mellitus. Trends in Biotechnology. 2005;23:128–134. doi: 10.1016/j.tibtech.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Rowbotham M, Fields H. The relationship of pain, allodynia, and thermal sensation in post-herpetic neuralgia. Brain. 1996;119:347–354. doi: 10.1093/brain/119.2.347. [DOI] [PubMed] [Google Scholar]
- Schif-Zuck S, Wildbaum G, Karin N. Coadministration o plasmid DNA constructs encoding an encephalitogenic determinant and IL-10 elicits regulatory T cell-mediated protective immunity in the central nervous system. J Immunol. 2006;177:8241–8247. doi: 10.4049/jimmunol.177.11.8241. [DOI] [PubMed] [Google Scholar]
- Sharma K, Cross J, Farronay O, Ayyar D, Sherbert R, Bradley W. Demyelinating neuropathy in diabetes mellitus. Arch Neurol. 2002;59:758–765. doi: 10.1001/archneur.59.5.758. [DOI] [PubMed] [Google Scholar]
- Storch M, Stefferl A, Brehm U, Weissert R, Wallstrom E, Kerschensteiner M, Olsson T, Linington C, Lassmann H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathology. 1998;8:681–694. doi: 10.1111/j.1750-3639.1998.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storek B, Reinhardt M, Wang C, Janssen WG, Harder NM, Banck MS, Morrison JH, Beutler AS. Sensory neuron targeting by self-complementary AAV8 via lumbar puncture for chronic pain. PNAS. 2008;105:1055–1060. doi: 10.1073/pnas.0708003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnemark D, Eltayeb S, Nilsson M, Wallstrom E, Lassmann H, Olsson T, Berg A, Ericsson-Dahlstrand A. CX3CL1 (fractalkine) and CX3CR1 expressionin myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflammation. 2005;2 doi: 10.1186/1742-2094-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczucinski A, Losy J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta Neurol Scand. 2007;115:137–146. doi: 10.1111/j.1600-0404.2006.00749.x. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens M, Bartfai T, Binaglia M, Corsini E, Luca MD, Galli C, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins L, Maier S. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- Watkins L, Milligan E, Maier S. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1–21. [PubMed] [Google Scholar]
- Wilczynski J, Radwan M, Kalinka J. The characterization and role of regulatory T cells in immune reactions. Front Biosci. 2008;13:2266–2274. doi: 10.2741/2840. [DOI] [PubMed] [Google Scholar]
- Xiao B, Bai X, Zhang G, Link H. Suppression of acute and protracted-relapsing experimental allergic encephalomyelitis by nasal administration of low-dose IL-10 in rats. Journal of Neuroimmunology. 1998;84:230–237. doi: 10.1016/s0165-5728(97)00264-6. [DOI] [PubMed] [Google Scholar]
- Zawadzka M, Franklin R. Myelin regeneration in demyelinating disorders: new developments in biology and clinical pathology. Curr Opin Neurol. 2007;20:294–298. doi: 10.1097/WCO.0b013e32813aee7f. [DOI] [PubMed] [Google Scholar]
- Zeng X, Ng Y, Ling E. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci. 2000;17:463–471. doi: 10.1017/s0952523800173122. [DOI] [PubMed] [Google Scholar]