

Abstract
Calpains are intracellular cysteine proteases that catalyze the cleavage of target proteins in response to Ca2+ signaling. When Ca2+ homeostasis is disrupted, calpain over-activation causes unregulated proteolysis, which can contribute to diseases such as post-ischemic injury and cataract formation. Potent calpain inhibitors exist, but of these many cross-react with other cysteine proteases and will need modification to specifically target calpain. Here, we present crystal structures of rat calpain 1 protease core (μI–II) bound to two α-ketoamide-based calpain inhibitors containing adenyl and piperazyl primed-side extensions. An unexpected aromatic-stacking interaction is observed between the primed-side adenine moiety and the Trp298 side chain. This interaction increased the potency of the inhibitor towards μI–II and heterodimeric m-calpain. Moreover, stacking orients the adenine such that it can be used as a scaffold for designing novel primed-side address regions, which could be incorporated into future inhibitors to enhance their calpain specificity.
In response to Ca2+ signaling, the calpain family of intracellular cysteine proteases catalyzes the limited cleavage of target proteins, resulting in changes to processes such as gene expression, cytoskeleton remodeling and apoptosis.1 Problems arise following ischemic or cerebral injury, when cells lose their ability to regulate Ca2+ influx to the cytoplasm. The elevated Ca2+ concentration leads to calpain hyperactivation, which causes uncontrolled proteolysis and irreversible cell damage. Since their overactivation has been linked to the development of pathological conditions such as stroke, Alzheimer disease, Duchenne muscular dystrophy and cataractogenesis, calpains represent an important class of targets for pharmacological inhibition.2,3
To date, all known calpain isoforms are multidomain enzymes,4 with a catalytic cleft located at the interface between domains I and II.5 These two domains, which encompass the enzyme’s proteolytic core, must each bind one Ca2+ ion to facilitate the rearrangement of the catalytic triad and substrate binding pocket into an active conformation.6 Although the various other domains also contribute somewhat to calpain activation, the susceptibility of full-length calpain to autolysis, subunit dissociation and aggregation following Ca2+ activation has complicated its study in the full-length form.7 The protease core though, remains resistant to autolysis and maintains its Ca2+-dependent activity, albeit, at a significantly reduced level.8 In addition, because of the relative ease with which they can be expressed in E. coli and crystallized, these protease cores have become an invaluable tool for the structure-based design of calpain inhibitors.9 While two structures have been reported for the Ca2+-activated human protease core,10,11 in our hands, the rat protease core has been much easier to purify and crystallize. The sequences for the protease cores of rat and human calpains 1 and 2 show a high degree of identity (87% between rat and human calpains 1 and 70% between rat calpain 1 and human calpain 2). Furthermore, because the active site clefts are particularly well conserved, the rat calpain 1 structure remains a suitable model for designing and studying inhibitors of calpain.
Of the reversible inhibitors that have been developed to target calpains, most are peptide analogues containing an electrophilic warhead group to covalently modify calpain’s active site thiol.9,12,13 Although aldehyde and α-ketoamide functional groups have been widely used as warheads, the latter has emerged as the superior form with respect to both metabolic stability and cell permeability.12 However, the poor specificity of α-ketoamide inhibitors continues to limit their applicability as potential therapeutic agents.2 Consequently, there has been an increasing focus on designing peptidyl “address regions” flanking the warhead to target the inhibitor to the calpain active site. To improve specificity, these “address regions” are designed to correspond with calpain’s residue preferences at each position in a peptide substrate. For instance, μ-calpain’s protease core (μI–II) demonstrates a preference for hydrophobic residues on the N-terminal (unprimed) side of the scissile bond,14 specifically phenylalanine and leucine at the P1 and P2 positions, respectively. The crystal structure of μI–II in complex with 3 (SNJ-1945),15 a peptidyl α-ketoamide containing this optimized selection, shows each of the two side chains interacting with the substrate binding cleft, thus showing how this unprimed “address region” can target the warhead to calpain’s active site. By itself though, this unprimed “address region” is insufficient to confer specificity towards calpain since the P2 leucyl side chain is also accommodated by a hydrophobic pocket in other cysteine proteases.16 Hence, there is an advantage to developing an additional optimal “address region” on the C-terminal (primed) side of the warhead. If the “address regions” on both the unprimed and primed sides can be incorporated into a single inhibitor, it would possess a substantially improved ability to specifically target calpain.
Previous studies on calpain inhibitors have shown that the extension of an inhibitor into the primed region can increase the inhibitor potency. For instance, Li et al17. showed that the inclusion of an arylalkyl primed-side substituent often improved the potency towards both calpains 1 and 2 as well as cathepsin B. In particular, the best primed-side substituent on the α-keto amide was a (CH2)3-Phe group. In another study, Chattergee et al.18 examined the effect of different primed-side extending biaryl sulfonamides and found that the inclusion of a spacer between the two aryl motifs reduced the inhibitory activity. Donkor et al.19 studied a series of inhibitors containing polar substituents in the P1′ position within the linker to a phenyl group. They modeled the polar group as interacting with Glu261, which we later showed to reside in a flexible loop.15 The common feature amongst these reports though, is that an aromatic moiety within the primed side was potentially beneficial to inhibitory activity. However, without crystal structures of these inhibitors bound to calpain, it is difficult to identify the interactions that these primed-side substituents might make with the active site.
Here, we present the crystal structures of μI–II bound to two α-ketoamide inhibitors 1,2 (Figure 1), each possessing substituents that extend deeper into the μI–II primed side cleft than previously observed. These structures also show the movement of a flexible gating loop out of the binding pocket in the presence of a ligand moiety stretching into the S-primed subsites. More importantly, the structure of μI–II bound to 1 demonstrates a novel non-covalent interaction between μI–II and the inhibitor, where the aromatic adenine moiety of 1 stacks against a conserved tryptophan in the catalytic cleft of μI–II and forms a hydrogen bond to the side chain of Glu300. These interactions may explain why 1 inhibits μI–II activity more potently than the nearly identical compound 2, which possesses a non-aromatic primed side substituent. The information gained from these structures should provide valuable insights for developing more specific inhibitors that can simultaneously target both the unprimed and primed subsites of calpains.
Figure 1.

Chemical structures of 1 and 2, the two α-ketoamide calpain inhibitors used in this study. Both compounds contain a P1 α-aminobutanoic acid and P2 leucine residue, but the primed side extension of 1 ends with adenine ring, while that of 2 is terminated by a piperazine ring. The structure of the commercially available calpain inhibitor AK-295 is also shown for comparison.
Results
Evaluating the inhibitory potency of 1 and 2 on μI–II and m-calpain
Upon calcium-activation, the rate of cleavage of the FRET substrate by m-calpain (Figure 2A) was approximately four times higher than that catalyzed by μI–II (Figure 2B). In the absence of either inhibitor, the autolytic inactivation of m-calpain caused the fluorescence intensity to plateau after just a few minutes of reaction. However, this autolytic inactivation was not observed with μI–II. Upon addition of either inhibitor, the increase in fluorescence was immediately attenuated, albeit more noticeably with m-calpain. For both m-calpain and μI–II, 1 caused a more distinct attenuation of fluorescence when compared to 2, indicating that the former is a more potent calpain inhibitor.
Figure 2.

Inhibitory effect of 1 and 2 on m-calpain (A) and μI–II (B). Duplicate assays were performed by adding 1.3 μM (EDANS)-EPLFAERK-(DABCYL) (1), 125 nM calpain (2) and 4 mM CaCl2 (3), then averaged to yield the plots shown. Autolytic inactivation of m-calpain, but not μI–II, was observed when no inhibitor was added (X). Immediately following addition of each inhibitor (4), the increase in fluorescence intensity was attenuated. 1(Y) caused a more distinct attenuation of fluorescence in both the m-calpain and μI–II assays, indicating that it is a more potent inhibitor when compared to 2 (Z).
X-ray crystal structures of the complexes of μI–II with 1 and 2
Owing to its stability and resistance to autolytic inactivation,8 μI–II was used to form calpain-inhibitor complexes with 1 and 2. A summary of crystallographic data collection and refinement statistics is presented in Table 1. Within the catalytic cleft of μI–II, the electron density of each inhibitor was clearly observed in the inner S-unprimed and S-primed subsites (Figure 3A and B). Electron density was weakest around the P3 phenyl ring of both inhibitors, suggesting that this group was more flexible and formed limited interactions with the S3-unprimed region. Nevertheless, when compared to the previously solved structures of μI–II bound to leupeptin or the α-ketoamide inhibitor 3,15 the position of the P3 phenyl ring in 1 and 2 was consistent with that of the P3 diethylene glycol in 3 and the P3 leucyl group in leupeptin; all four P3 groups overlap closely along the surface of domain I (Figure 4A).
Table 1.
Summary from Data Collection and Structure Refinement
| μI–II-1 complex | μI–II-2 complex | |
|---|---|---|
| Crystal Parameters | ||
| Space group | P212121 | P212121 |
| a (Å) | 40.43 | 40.57 |
| b (Å) | 70.24 | 70.57 |
| c (Å) | 110.41 | 110.41 |
| Data Collection Statistics | ||
| Resolution limit (Å) | 1.80–59.23 (1.80–1.90) | 1.60–35.29 (1.60–1.69) |
| No. of measured reflections | 196538 (28320) | 229854 (15208) |
| No. of unique reflections | 29796 (4224) | 40801 (4932) |
| Completeness (%) | 99.2 (98.4) | 96.0 (82.6) |
| I/σI | 35.1 (15.1) | 17.9 (1.6) |
| Rmerge (%) | 4.0 (12.2) | 7.8 (60.2) |
| Refinement Statistics | ||
| Resolution range (Å) | 1.80–27.22 (1.80–1.847) | 1.60–35.29 (1.60–1.642) |
| Size of free reflection set (%) | 5 (5) | 5 (5) |
| Rwork | 15.6 (16.9) | 18.9 (33.8) |
| Rfree | 19.6 (24.1) | 22.2 (34.7) |
| No. of atoms (no H) protein/solvent/Ca2+/Cl−/glycerol | 2644/305/2/3/12 | 2663/268/2/3/12 |
| Bond angle rmsd (deg) | 1.47 | 1.273 |
| Bond length rmsd (Å) | 0.016 | 0.012 |
| Average B-factor (Å2) | 14.3 | 15.8 |
| B-factor from Wilson plot (Å2) | 13.0 | 17.5 |
Figure 3.

Crystal structures of 1 and 2 bound to the active site of μI–II. The electron density of 1 (calculated with coefficients 2mFobs – DFcalc, φcalc and contoured at 1σ) and the inhibitor’s polar contacts with μI–II are shown in panels A & C, respectively, while those for 2 are shown in panels B & D. The carve feature of PyMOL33 was used to limit the display of electron density to a distance of 2 Å from the inhibitors. Bonds are colored by atom type: carbon=gray (μI–II), carbon = green (1), carbon = cyan (2), nitrogen = blue, oxygen = red, sulfur = orange. The peptide subsites are labelled below panel C.
Figure 4.
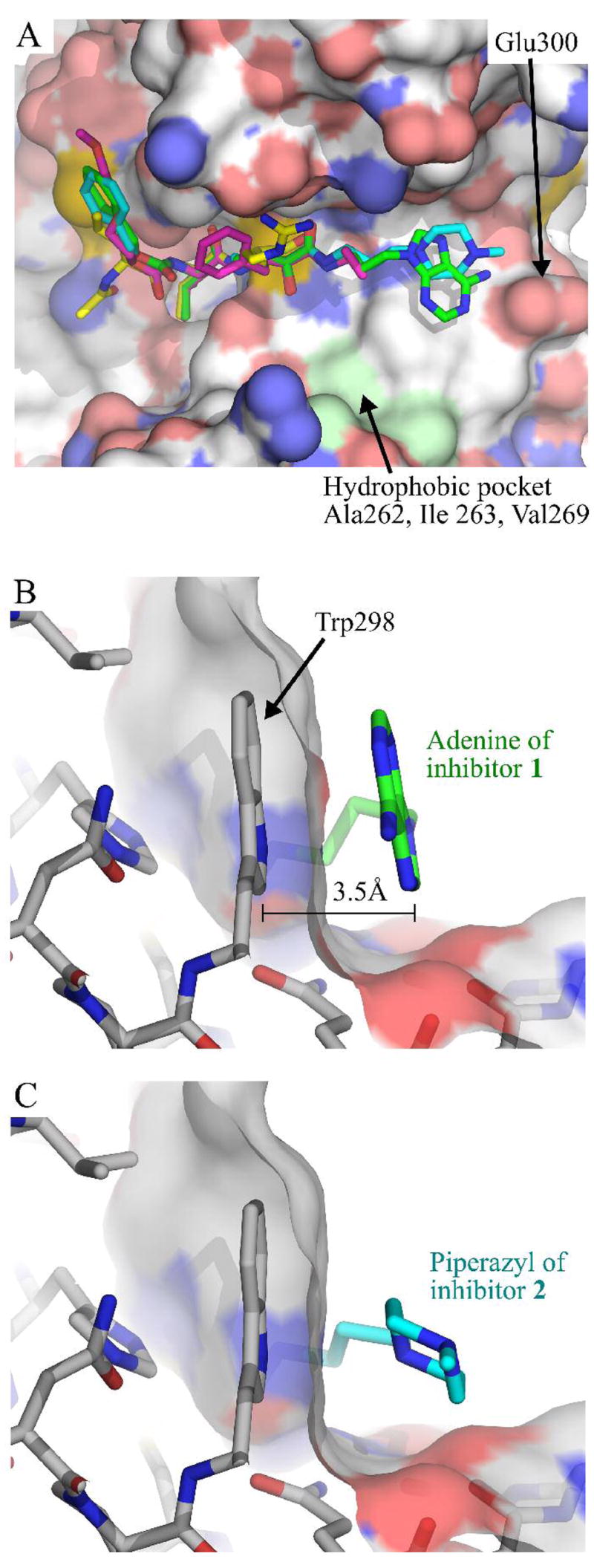
(A) Structural overlap of calpain inhibitors, achieved by aligning the polypeptide chain of each structure. Inhibitors are shown as sticks and accompanied by a surface representation of μI–II from the μI–II-1 complex. Atoms are colored as in Figure 3. The overlap of 1, 2, 3 and leupeptin shows a high degree of structural alignment from the P3 to the P1 residue. The hydrophobic pocket formed by Ala262, Ile263 and Val269 is highlighted in light green. (B & C). Orientation of terminal primed side ring groups of 1 and 2 relative to Trp298 in μI–II. A transparent surface representation of μI–II is also shown. (B) Primed side aromatic stacking interaction between the adenine moiety of 1 and W298. (C) This stacking interaction is absent for the non-aromatic piperazyl group of 2. Atoms are colored as for Figure 3.
Both inhibitors 1 and 2 are stabilized within the active site through interactions that are conserved in the structures of μI–II bound to leupeptin or 3. On the unprimed side, hydrogen bonds are formed between the inhibitor’s peptide backbone atoms (amide nitrogen and carbonyl oxygen of the P2 residue and amide nitrogen of P1 residue) and Gly208 and Gly271 in the catalytic cleft (Figure 3C and D). At the α-ketoamide warhead of 1, 2 and 3, the newly formed hydroxyl group, which arises from nucleophilic attack by the active site thiol, forms a hydrogen bond with the imidazole ring of His272, while the carbonyl oxygen of the carboxyamide provides further stabilization via two polar contacts with Gln109 and Cys115.
As would be expected from the highly conserved hydrogen bonding interactions, a high degree of structural alignment was observed at the P2 leucine of each of the inhibitors 1, 2, 3 and leupeptin (Figure 4A). In fact, the backbone and side chains adopt nearly identical conformations. This strong overlap continued into the backbone of the P1 residue and its respective side chain β- and γ-carbons. In addition to the conserved hydrogen bonds, another contributing factor to the high degree of structural alignment is the narrowness of the binding pocket around the P1 residue. As described by Cuerrier et al.,15 this narrow pocket imposes restrictions on the ability of the P1 residue to rotate around the Cα-Cβ bond.
While the structures of leupeptin and 3 are terminated at, or shortly after the warhead, 1 and 2 possess much longer primed side extensions. Interestingly, the greatest degree of structural divergence between these two inhibitors is concentrated within this region. Structural alignment deteriorates substantially in the 3-carbon chain that lies on the C-terminal side of the warhead and is ultimately transmitted to the terminal ring groups (Figure 4A). It appears that the primed-side 3-carbon chain in 1 may adopt a different conformation from that of 2 in order to facilitate an aromatic stacking interaction between the adenine moiety of 1 and the side chain of Trp298. As shown in Figure 4B, it stacks in a coplanar conformation with respect to tryptophan’s indole group, leaving a gap of only 3.5 Å between the two and allowing the formation of a hydrogen bond to Glu300. Neither this coplanar stacking nor any polar contacts were observed between the non-aromatic piperazyl ring of 2 and the primed side pocket (Figure 4C). The absence of this stacking interaction and a hydrogen bond to Glu300 therefore likely accounts for the weaker electron density around the piperazyl ring when compared to that of the adenine moiety (Figure 3A and B).
For both inhibitor-bound structures, although the electron density of μI–II was strong in most areas, the density was observed to be considerably weaker between residues 251–261, which comprise a known flexible loop region. Fitting these residues to the available density resulted in a loop that adopted an “open” conformation, with Glu261 displaced away from the S1′ subsite. This “open” conformation closely resembles the one reported in the structure of 3 bound to μI–II, but contrasts with the “closed” conformation observed in the leupeptin-bound structure (Figure 5).
Figure 5.

Overlay of four μI–II structures, showing the two possible conformations of the gating loop (residues 256–261). Glu261 in μI–II and the bound inhibitor 1 are shown as sticks. The primed side extensions of 1 and 2 cause Glu261 in the gating loop to be displaced, resulting in an “open” conformation. This “open” conformation closely resembles that of the structure of μI–II bound to 3, where a P1′-cyclopropyl group causes Glu261 to be displaced. When bound to leupeptin, which lacks a primed side extension, the gating loop is in a “closed” position, with Glu261 blocking the S1’ subsite. The polypeptides are colored as follows: green = μI–II-1 complex, cyan = μI–II-2 complex, magenta = μI–II-3 complex, yellow = μI–II-leupeptin complex.
Discussion
As noted in previously solved μI–II structures, residues 251–261 in domain II comprise a flexible loop that possesses weak electron density and is considered to act as a gate for the catalytic cleft.11,15,20, When μI–II is bound to an inhibitor that extends only into the unprimed side, such as leupeptin, the flexible “gating” loop adopts the “closed” conformation, with Glu261 obstructing the S1′ subsite (Figure 5). However, in the presence of an inhibitor such as 1 or 2, which possesses a primed side group that extends into the S2′-subsite, the loop is oriented away from the active site in a much more “open” manner. This “open” conformation was first observed in the structure of μI–II bound to 3, where the inhibitor’s P1′ cyclopropyl group was thought to displace Glu261 from the S1′ site.15 Although the purpose of the “open” and “closed” loop conformations has yet to be determined, it is possible that they could be signaling different catalytic stages, possibly via interactions with domains (e.g. domain III) that are absent in the isolated proteolytic core. The open loop conformation for instance, might be signaling the initial substrate-binding event where the polypeptide chain would stretch across the cleft. However, once catalysis has yielded the acyl-enzyme intermediate, which no longer possesses a primed side extension, the loop may close to signify the near completion of catalysis.
In addition to providing insight about the flexible gating loop, the crystal structures produced from this study also revealed the importance of the newly discovered aromatic stacking interaction between the primed-side adenine moiety of 1 and the indole ring of Trp298. This class of non-covalent interactions, commonly referred to as π-π stacking, has been widely documented in protein-ligand complexes, especially when the ligand is comprised of nucleic acids.21–26 One study in particular examined the stacking interactions between adenine and aromatic residues in the active site of DNA glycosylases.27 Since these enzymes remove damaged bases during the first step of base excision repair28 and have aromatic residues lining their active sites, it was thought that π-π stacking was responsible for substrate recognition and stabilization. By examining the available crystal structures of DNA glycosylases and performing computational analyses of the aromatic interactions, it was determined that out of the four aromatic amino acids (His, Phe, Tyr and Trp), the strongest stacking with adenine occurred in the presence of tryptophan, an apparent consequence of its side chain’s large surface area and dipole moment.27 Furthermore, the optimal perpendicular separation between tryptophan and adenine was determined to range from 3.5 Å to 3.6 Å, which matches the distance observed in the complex of μI–II with 1.
Based on the energetic consequences of π-π stacking and considering that compounds 1 and 2 differ only with respect to their terminal primed side ring groups, the aromatic interaction therefore may explain the greater inhibitory potency of 1 over 2 observed in the fluorimetry assays (Figure 2). Through this stacking, the terminal primed side end of 1 is held more rigidly in place and likely works in conjunction with the unprimed side polar contacts to properly orient the α-keto carbonyl relative to the active site thiol (Figure 3). In contrast, the absence of this aromatic interaction in the structure of μI–II with inhibitor 2 causes the piperazyl ring to exhibit greater flexibility, which could be transmitted towards the warhead group, thereby impairing the α-keto carbonyl from achieving the necessary orientation required for potent inhibition.
Incidentally, the residue that facilitates the π-π stacking interaction, Trp298, is also involved in the later stage of active site assembly, where its side chain undergoes a radical change in position.6 Upon binding of the second and final Ca2+ ion to the μ-calpain protease core, the indole ring of Trp298 moves from its initial wedge-like position between the catalytic domains into the orientation observed in the inhibitor-bound structures. In this position, it helps shield two of the three catalytic triad residues (His272 and Asn296) from solvent exposure and thus facilitates catalysis. Therefore, the π-π stacking is done by Trp298 in its catalytically active conformation, suggesting that inhibitor 1 binds preferentially to the Ca2+-bound, active form of calpain.
Although the π-π stacking interaction on the primed side cleft is promising from the perspective of structure-guided drug design, it alone is expected to be insufficient to specifically target calpains since other cysteine proteases also possess a tryptophan residue that resides in a similar position to that in the catalytic conformation of calpain.6. As shown in Figure 6, when the structures of the leupeptin-bound papain, human liver cathepsin B and E-64-bound cathepsin K are aligned with that of the μI–II-1 complex, the analogous tryptophan residue in these other cysteine proteases could still interact with the adenine ring of 1, albeit perhaps less optimally when compared to Trp298. What the other papain-like cysteine proteases lack, though, is a residue equivalent to Glu300, so they would be unable to form a hydrogen bond to the amino group of the adenyl moiety. Along these lines, the stacked adenine ring could still be used as a scaffold for other functional groups, which could form contacts with structural features that are unique to calpain’s primed side cleft. For instance, the shallow hydrophobic pocket formed by Ala262, Ile263 and Val269 (Figure 4A) is not observed in the primed side cleft of papain, cathepsin B or cathepsin K. Therefore, addition of an aliphatic extension to the adenine ring could facilitate interactions with the hydrophobic pocket. In addition, the replacement of the primary amino group with a positively charged substituent on the adenine moiety of 1 could enhance the electrostatic contacts with the Glu300 side chain. Modification of compound 1 to exploit these calpain-exclusive interactions holds promise for the development of new potent and selective inhibitors.
Figure 6.

Overlay of Trp298 in the μI–II-1 complex (μI–II in white, 1 in green) with the corresponding tryptophan in papain (magenta), cathepsin B (yellow) and cathepsin K (cyan). This overlay was achieved by first aligning three catalytic site residues (cysteine, histidine, tryptophan) of papain, cathepsin B and cathepsin K with those of the μI–II-1 structure. Inhibitor 1 and the overlaid cysteine, histidine and tryptophan residues are shown as sticks. The relatively similar alignments between the tryptophans suggests that aromatic stacking, by itself, will be insufficient to specifically target an inhibitor into the calpain active site. The PDB entries for the cysteine proteases are as follows: papain bound to leupeptin (1POP), human liver cathepsin B (1HUC) and cathepsin K bound to E-64 (1ATK). Nitrogens are colored blue, while oxygens are red.
In conclusion, we have demonstrated that interactions with calpain’s S-primed subsites represent an important aspect of inhibitor design that should not be ignored. As was highlighted in the two crystal structures, the aromatic stacking interaction and hydrogen bond between the primed side adenine ring of 1 and Trp298 and Glu300, respectively, of μI–II, provided the inhibitor with a decisive potency advantage over an equivalent inhibitor lacking a terminal aromatic group. Being the first non-covalent interactions reported on the primed side of a calpain-inhibitor complex, the two high-resolution structures described in this study should contribute greatly to the field of structure-guided drug design and in particular, to the development of potent and specific inhibitors of calpains.
Experimental Procedures
Materials
The rat μI–II construct is composed of Gly27-Asp356 from rat μ-calpain and contains an N-terminal Met and C-terminal Leu-Glu-(His)6 tag. The full-length rat m-calpain, comprised of the 80 and 21 kDa subunits, contains the C-terminal Lys-Leu-Ala-Ala-Leu-Glu-(His)6 tag. Both constructs were recombinantly expressed in E. coli and purified as previously described.8,29. The calpain substrate (EDANS)-EPLFAERK-(DABCYL) was synthesized at the Alberta Peptide Institute (Edmonton, Alberta, Canada) and in the Peptide Synthesis Laboratory of the Protein Function Discovery Facility at Queen’s University (Kingston, Ontario, Canada). All other reagents were obtained from common sources.
Synthesis of the α-ketoamide compounds
3-(Benzyloxycarbonyl-L-leucylamino)-N-(3-(6-amino-9H-purin-9-yl)propyl)-2-oxopentanamide (1, Z-Leu-Abu-CONH-(CH2)3-adenin-9-yl)
A mixture of adenine (4.05 g, 30 mM), 1-bromo-3-chloropropane (21.3 g, 13.4 mM), and potassium carbonate (10.4 g, 75 mM) in DMF (200 mL) was stirred at room temperature under argon for 4 days, then filtrated and evaporated to dryness. The crude product was washed with water and dried. Recrystallization from ethanol gave 9-(3-chloropropyl)adenine in 59% yield.
The 9-(3-chloropropyl)adenine (1.9 g, 9 mM) and sodium azide (1.75 g, 27 mM) in DMF was stirred at 80°C for 24 h, cooled to room temperature and filtered. The solid was washed with CH2Cl2. The solvent was removed from the combined filtrates and the residue was taken up in water with sonication. The aqueous layer was extracted with CH2Cl2 (3 × 60 mL). After removing solvent the crude product was recrystallized from ethanol to give 9-(3-azidopropyl)adenine as a white crystalline solid in 81% yield.
The 9-(3-azidopropyl)adenine(0.5 g, 2.3 mM) and 5 % palladium on carbon (0.5 g) in methanol was reacted with hydrogen gas at room temperature for 22 h. The catalyst was removed by filtration, and the solvent removed to give 9-(3-aminopropyl)adenine as a white solid in 76% yield. 1H NMR (DMSO-d6): 1.80 (m, 2H, CH2), 2.45 (m, 2H, CH2), 3.35 (s, 2H, NH2), 4.20 (m, 2H, CH2), 7.20 (s, 2H, NH2), 8.10 (s, 2H, CH). MS (ED+): 193.0.
The ketoamide product Z-Leu-Abu-CONH-(CH2)3-adenin-9-yl was obtained from 9-(3-aminopropyl)adenine and the ketoacid Z-Leu-Abu-COOH30 using the EDC/HOBt coupling method, purified by column chromatography on silica gel with 85:15 CH2Cl2:MeOH as the eluant, then recrystallized from CH3COOEt/hexane to give a white solid (27% yield). 1H NMR (CDCl3): 0.91 (m, 9H, CH3 of Val and Abu), 1.60–1.80 (m, 5H, CH2 and CH of Leu and Abu), 2.00 (m, 2H, CH2), 3.20 (2H, CH2), 4.24 (m, 3H, CH2 and α-H), 5.11 (s, 2H, Z), 5.20 (m, 1H, α-H), 6.20 (s, 1H, NH), 6.80 (b, 1H, NH), 7.20–7.40 (m, 6H, Ph and NH), 7.85 (d, 1H, CH of adenine), 8.36 (d, 1H, CH of adenine).
3-(Benzyloxycarbonyl-L-leucylamino)-N-(3-(4-methylpiperazin-1-yl)propyl)-2-oxopentanamide (2, Z-Leu-Abu-CONH-(CH2)3-(4-methylpiperazin-1-yl)
A solution of N-(3-bromopropyl)phthalimide (8.04 g, 30 mM) in xylene (60 mL) was added dropwise to a solution of 1-methylpiperazine (6.61 g, 66 mM) in xylene (90 mL) at 70 °C. After the addition was complete, the mixture was heated under reflux for 20 h. A precipitate was removed by filtration and the filtrate was concentrated. The crude product was purified by silica gel chromatography with 9:1 CH2Cl2:MeOH to give the product N-(3-(4-methylpiperazin-1-yl)propyl)phthalimide as an oil in 72% yield.
A solution of N-(3-(4-methylpiperazin-1-yl)propyl)phthalimide (6.2 g, 21.6 mM) and hydrazine monohydrate (1.13 g, 26 mM) in ethanol (60 mL) and methanol (60 mL) was refluxed for 4 h. After cooling to room temperature, concentrated HCl (2.4 mL) was added and the mixture heated under reflux for another 1 h. After removing the solvent, water (100 mL) was added, the mixture stirred and insoluble material removed by filtration. Solid K2CO3 (1.2 eq) and CH2Cl2 (100 mL) was added to the aqueous layer, the mixture stirred, and filtered. The organic layer was washed with water (3 × 20 mL). The combined aqueous layers were washed with Et2O. Water was removed from the organic layers, they were then dried and evaporated to give 1-(3-aminopropyl)-4-methylpiperazine as a oil in 39% yield. 1H NMR (CDCl3): 1.55 (m, 2H, CH2), 2.20 (s, 3H, CH3), 2.34 (t, 2H, CH2), 2.67 (t, 2H, CH2). MS (ES+): 157.9.
The ketoamide Z-Leu-Abu-CONH-(CH2)3-(4-methylpiperazin-1-yl) was synthesized from 1-(3-aminopropyl)-4-methylpiperazine and Z-Leu-Abu-COOH30 using the EDC/HOBt coupling method, and purified twice by column chromatography on silica gel using 80:20 CH2Cl2:MeOH and 85:15 CH2Cl2:MeOH as the eluant to give a yellow semi-solid in 16% yield. 1H NMR (CDCl3): 0.91 (m, 9H, CH3 of Val and Abu), 1.60–1.80 (m, 5H, CH2 and CH), 2.00 (m, 2H, CH2), 2.44 (s, 3H, CH3 of piperazine), 2.50–2.65 (m, 8H, CH2 of piperazine), 3.30 (m, 2H, CH2), 4.20 (m, 3H, CH2 and α-H), 5.10 (s, 2H, Z), 5.15 (m, 1H, α-H), 6.70 (b, 1H, NH), 7.20–7.30 (m, 6H, Ph and NH), 8.60 (b, 1H, NH).
Fluorimetric analysis of μI–II and m-calpain inhibition by 1 and 2
Enzymatic activity was monitored in real-time using a 1.5 mL quartz cuvette and a Perkin-Elmer LS50-B Luminescence Spectrophotometer. Duplicate assays were performed with 1.3 μM (EDANS)-EPLFAERK-(DABCYL) (12) and 125 nM μI–II or m-calpain in a buffer solution containing 50 mM HEPES-HCl (pH 7.7) and 10 mM DTT. The reaction was initiated at the 420 s time mark using 4 mM CaCl2 and monitored at excitation and emission wavelengths of 335 nm and 500 nm, respectively. After a waiting period of 100 s, the inhibitor solution (either 1 or 2 dissolved in DMSO) was added at a concentration of 1 μM (for the m-calpain assays) or 50 μM (for the μI–II assays). Control assays were performed by adding an equivalent volume of DMSO after the 100 s delay.
Determination of μI–II-1 and μI–II-2 crystal structures
A solution of 325 μM μI–II in 10 mM HEPES-HCl (pH 7.7) and 10 mM DTT was first incubated with 2 mM 1 or 2 at room temperature for 30 min. This solution was then mixed with an equal volume of a precipitant solution to cover conditions that expanded around 1.5–2.0 M NaCl, 10 mM CaCl2, and 0.1 M MES (pH 5.75–6.5). Using the hanging drop vapor diffusion method, crystals were obtained and subsequently soaked in a solution of the mother liquor supplemented with 20% glycerol, prior to data collection. Crystallographic data was collected at beamline X6A at the National Synchrotron Light Source, Brookhaven National Laboratory. The processing of data was performed using Mosflm31 and Scala32 from the CCP4 program suite.33 The inhibitor-bound structure was solved using the Ca2+-bound μI–II (PDB entry 1KXR) as the model for molecular replacement in MOLREP.34 The molecular topology descriptions of the inhibitors 1 and 2 were generated with the Dundee PRODRG2 server.35 Model refinement and building were performed using REFMAC536 and Coot,37 respectively. The structures shown in Figures 3 through 6 were prepared with PyMOL.38
Supplementary Material
Analytical data (high resolution mass determination and HPLC tracings) for target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors would like to thank Sherry Gauthier for technical assistance and Marc Allaire from beamline X6A at the National Synchrotron Light Source, Brookhaven National Laboratory, for assisting with the crystallographic data collection. We thank Christina Hampton and Dr. Facundo Fernandez in the School of Chemistry and Biochemistry at Georgia Tech for the HPLC/MS data on the calpain inhibitors.
This work was funded by a grant from the Canadian Institute for Health Research to P.L.D. and National Institutes of Health (R21 NS053801) to J.C.P. J.Q. is supported by a studentship from the Government of Canada’s Natural Sciences and Engineering Research Council. P.L.D. holds a Canada Research Chair in Protein Engineering.
Footnotes
PDB: coordinates and structure factors for the complexes of the protease core of μ-calpain with 1 and 2 have been deposited with the protein data bank (2R9C and 2R9F, respectively).
References
- 1.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 2.Carragher NO. Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr Pharm Des. 2006;12:615–638. doi: 10.2174/138161206775474314. [DOI] [PubMed] [Google Scholar]
- 3.Wang KK, Yuen PW. Development and therapeutic potential of calpain inhibitors. Adv Pharmacol. 1997;37:117–152. doi: 10.1016/s1054-3589(08)60949-7. [DOI] [PubMed] [Google Scholar]
- 4.Sorimachi H, Suzuki K. The structure of calpain. J Biochem (Tokyo) 2001;129:653–664. doi: 10.1093/oxfordjournals.jbchem.a002903. [DOI] [PubMed] [Google Scholar]
- 5.Hosfield CM, Elce JS, Davies PL, Jia Z. Crystal structure of calpain reveals the structural basis for Ca2+-dependent protease activity and a novel mode of enzyme activation. EMBO J. 1999;18:6880–6889. doi: 10.1093/emboj/18.24.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moldoveanu T, Jia Z, Davies PL. Calpain activation by cooperative Ca2+ binding at two non-EF-hand sites. J Biol Chem. 2004;279:6106–6114. doi: 10.1074/jbc.M310460200. [DOI] [PubMed] [Google Scholar]
- 7.Pal GP, Elce JS, Jia Z. Dissociation and aggregation of calpain in the presence of calcium. J Biol Chem. 2001;276:47233–47238. doi: 10.1074/jbc.M105149200. [DOI] [PubMed] [Google Scholar]
- 8.Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca2+ switch aligns the active site of calpain. Cell. 2002;108:649–660. doi: 10.1016/s0092-8674(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 9.Neffe AT, Abell AD. Developments in the design and synthesis of calpain inhibitors. Curr Opin Drug Discov Devel. 2005;8:684–700. [PubMed] [Google Scholar]
- 10.Davis TL, Walker JR, Finerty PJ, Jr, Mackenzie F, Newman EM, Dhe-Paganon S. The crystal structures of human calpains 1 and 9 imply diverse mechanisms of action and auto-inhibition. J Mol Biol. 2007;366:216–229. doi: 10.1016/j.jmb.2006.11.037. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Hanzlik RP, Weaver RF, Schonbrunn E. Molecular mode of action of a covalently inhibiting peptidomimetic on the human calpain protease core. Biochemistry. 2006;45:701–708. doi: 10.1021/bi052077b. [DOI] [PubMed] [Google Scholar]
- 12.Donkor IO. A survey of calpain inhibitors. Curr Med Chem. 2000;7:1171–1188. doi: 10.2174/0929867003374129. [DOI] [PubMed] [Google Scholar]
- 13.Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 14.Cuerrier D, Moldoveanu T, Davies PL. Determination of peptide substrate specificity for mu-calpain by a peptide library-based approach: the importance of primed side interactions. J Biol Chem. 2005;280:40632–40641. doi: 10.1074/jbc.M506870200. [DOI] [PubMed] [Google Scholar]
- 15.Cuerrier D, Moldoveanu T, Inoue J, Davies PL, Campbell RL. Calpain inhibition by alpha-ketoamide and cyclic hemiacetal inhibitors revealed by X-ray crystallography. Biochemistry. 2006;45:7446–7452. doi: 10.1021/bi060425j. [DOI] [PubMed] [Google Scholar]
- 16.Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem. 2006;281:12824–12832. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Patil GS, Golubski ZE, Hori H, Tehrani K, Foreman JE, Eveleth DD, Bartus RT, Powers JC. Peptide alpha-keto ester, alpha-keto amide, and alpha-keto acid inhibitors of calpains and other cysteine proteases. J Med Chem. 1993;36:3472–3480. doi: 10.1021/jm00074a031. [DOI] [PubMed] [Google Scholar]
- 18.Chatterjee S, Dunn D, Tao M, Wells G, Gu ZQ, Bihovsky R, Ator MA, Siman R, Mallamo JP. P2-achiral P′-extended alpha-ketoamide inhibitors of calpain I. Bioorg Med Chem Lett. 1999;9:2371–2374. doi: 10.1016/s0960-894x(99)00392-3. [DOI] [PubMed] [Google Scholar]
- 19.Donkor IO, Han J, Zheng X. Design and synthesis molecular modeling studies and calpain inhibitory activity of novel alpha-ketoamides incorporating polar residues at the P1′-position. J Med Chem. 2004;47:72–79. doi: 10.1021/jm0301336. [DOI] [PubMed] [Google Scholar]
- 20.Moldoveanu T, Campbell RL, Cuerrier D, Davies PL. Crystal structures of calpain-E64 and -leupeptin inhibitor complexes reveal mobile loops gating the active site. J Mol Biol. 2004;343:1313–1326. doi: 10.1016/j.jmb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 22.Valegard K, Murray JB, Stonehouse NJ, van den Worm S, Stockley PG, Liljas L. The three-dimensional structures of two complexes between recombinant MS2 capsids and RNA operator fragments reveal sequence-specific protein-RNA interactions. J Mol Biol. 1997;270:724–738. doi: 10.1006/jmbi.1997.1144. [DOI] [PubMed] [Google Scholar]
- 23.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 24.Price SR, Evans PR, Nagal K. Crystal structure of the spliceosomal U2B″-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature. 1998;394:645–650. doi: 10.1038/29234. [DOI] [PubMed] [Google Scholar]
- 25.Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- 26.Nolan SJ, Shiels JC, Tuite JB, Cecere KL, Baranger AM. Recognition of an essential adenine at a protein-RNA interface: comparison of the contributions of hydrogen bonds and a stacking arrangement. J Am Chem Soc. 1999;121:8951–8952. [Google Scholar]
- 27.Rutledge LR, Campbell-Verduyn LS, Hunter KC, Wetmore SD. Characterization of nucleobase-amino acid stacking interactions utilized by a DNA repair enzyme. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:19652–19663. doi: 10.1021/jp061939v. [DOI] [PubMed] [Google Scholar]
- 28.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moldoveanu T, Hosfield CM, Jia Z, Elce JS, Davies PL. Ca2+-induced structural changes in rat m-calpain revealed by partial proteolysis. Biochim Biophys Acta. 2001;1545:245–254. doi: 10.1016/s0167-4838(00)00286-7. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Ortega-Vilain AC, Patil GS, Chu DL, Foreman JE, Eveleth DD, Powers JC. Novel peptidyl α-keto amide inhibitors of calpains and other cysteine proteases. J Med Chem. 1996;39:4089–4098. doi: 10.1021/jm950541c. [DOI] [PubMed] [Google Scholar]
- 31.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 32.Evans PR. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 33.COLLABORATIVE COMPUTATIONAL PROJECT NUMBER 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Vagin AT, Teplyakov A. An automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 35.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. DeLano Scientific; Palo Alto, CA, USA: 2003. The PyMOL Molecular Graphics System. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analytical data (high resolution mass determination and HPLC tracings) for target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.