

Abstract
The polyene macrolide antibiotic filipin is widely used as a probe for cholesterol in biological membranes. The filipin biosynthetic pathway of Streptomyces avermitilis contains two position-specific hydroxylases, C26-specific CYP105P1 and C1′-specific CYP105D6. In this study, we describe the three X-ray crystal structures of CYP105P1: the ligand-free wild-type (WT-free), 4-phenylimidazole-bound wild-type (WT-4PI), and ligand-free H72A mutant (H72A-free) forms. The BC loop region in the WT-free structure has a unique feature; the side chain of His72 within this region is ligated to the heme iron. On the other hand, this region is highly disordered and widely open in WT-4PI and H72A-free structures, respectively. Histidine ligation of wild-type CYP105P1 was not detectable in solution, and a type II spectral change was clearly observed when 4-phenylimidazole was titrated. The H72A mutant showed spectroscopic characteristics that were almost identical to those of the wild-type protein. In the H72A-free structure, there is a large pocket that is of the same size as the filipin molecule. The highly flexible feature of the BC loop region of CYP105P1 may be required to accept a large hydrophobic substrate.
Cytochrome P450 (P450 or CYP) enzymes are hemoproteins whose fifth axial heme iron ligand is a thiolate group and are found in wide variety of organisms (19, 30). The majority of P450s catalyze the monooxygenation (hydroxylation) of hydrophobic substrates (14). This reaction utilizes a dioxygen bound as the sixth iron ligand and various redox systems for the cleavage of the O-O bond (22, 24, 42). The substrate recognition sites (SRSs) of P450s are on the distal pocket of the heme, and their structural and conformational varieties are the basis of the diverse substrate specificity (10, 48). SRSs include two highly diverse regions: the BC loop (SRS-1) and the FG helices (SRS-2 and -3). They form an entrance to the pocket, and the BC loop often contains a short helix denoted as B′ helix.
The genus Streptomyces and related bacteria are distinguished by their ability to produce a broad array of biologically active secondary metabolites, including macrolide antibiotics. The first crystal structure of macrolide hydroxylase has been determined for P450eryF (CYP107A1) (6), which is involved in the biosynthesis of erythromycin. The crystal structures of two possible macrolide hydroxylases from Streptomyces coelicolor A3(2), CYP154C1 and CYP154A1, have also been reported (38, 39). The hydroxylase activity of these enzymes for at least one or two macrolide compounds has been confirmed in vitro. Recently, the crystal structure of P450 PikC (CYP107L1), which is responsible for the production of a number of macrolide antibiotics related to narbomycin and pikromycin, has been reported (47). Moreover, the crystal structures of macrolide monooxygenase P450epoK, which catalyzes epoxidation in the epothilone biosynthesis, have been determined in both the substrate-free and the substrate-bound forms (26).
In the genome sequence of an avermectin-producing industrial microorganism Streptomyces avermitilis (13), there are 33 CYP genes (17), whereas the genome of S. coelicolor A3(2) has only 18 CYP genes (18). Among them, the genes encoding CYP105P1 (PteC, SAV413) and CYP105D6 (PteD, SAV412) form the gene cluster for filipin biosynthesis with modular polyketide synthases (pteA1-pteA5), ferredoxin (fdxI, pteE), and putative zinc-binding dehydrogenase (pteB) genes (13). The 28-membered polyene macrolide antibiotic filipin (Fig. 1) is widely used as a probe for cholesterol in biological membranes (8, 52) and as a prominent diagnostic tool for type C Niemann-Pick disease (4, 5). The filipin complex is a mixture of four macrolides with minor differences in their hydroxylation pattern of the post-polyketide modification at C1′ and C26 positions (3, 34). Analysis of the mutants of the genes encoding CYP105P1 and CYP105D6 has revealed that these P450s catalyze hydroxylation at C26 and C1′ positions, respectively (H. Ikeda et al., unpublished data). The CYP105 family enzymes are frequently found in actinomycetes (28) and are closely related to P450nor (CYP55A1). P450nor, one of the most unusual P450 enzymes, catalyzes the reduction of nitric oxide to dinitrogen oxide in the fungal denitrification steps, utilizing NADH as the direct electron donor (27, 31). To date, the crystal structures of two CYP105 family enzymes, P450 MoxA and P450 SU-1 (CYP105A1), have been reported (49, 54). Although their biological roles remain unclear, the ability of these enzymes to hydroxylate a wide variety of compounds attracts considerable interest in various applications.
FIG. 1.

Structures of filipins (43a). Filipin I: R1 = H, R2 = H; filipin II: R1 = OH, R2 = H; filipin III: R1 = OH, R2 = OH. The molecular formula of filipin IV is the same as that of filipin III, and it might be a stereoisomer of filipin III.
In the present study, we conducted crystallographic, spectroscopic, and mutational analyses of CYP105P1 from S. avermitilis, and we present the structures in three different states. The ligand-free wild-type (WT-free) structure provides a unique state, whose histidine residue is ligated to the heme iron atom. Compared to the 4-phenylimidazole (4PI)-bound wild-type (WT-4PI) and the ligand-free H72A mutant (H72A-free) structures, it is suggested that the high flexibility of the BC loop of this enzyme is a key feature for incorporating the large hydrophobic substrate, filipin.
MATERIALS AND METHODS
Protein production and site-directed mutagenesis.
A gene encoding CYP105P1 was amplified by PCR with Easy-A high fidelity polymerase (Stratagene, La Jolla, CA) using the genomic DNA of S. avermitilis MA-4680 as a template and the primer pair 5′-CCC ATA TGC CCG AGC CCA CCG CCG ACG C-3′ and 5′-GCA CTA GTT CAG TGG TGG TGG TGC CAG GTG ACG GGC AGC TCG TGC ACG-3′ (sequences in boldface and with an underline represent the restriction endonuclease sites and His4 tags, respectively). The amplified fragment was digested with NdeI and SpeI and then inserted into the corresponding sites of pET-17b (Novagen, Madison, WI). Each expression plasmid was introduced into E. coli BL21 CodonPlus (DE3) (Stratagene), and the transformants were cultured in Luria-Bertani medium containing 100 mg of ampicillin and 20 mg of chloramphenicol/liter at 25°C for 20 h. After centrifugation, cells were suspended in 50 mM potassium phosphate (pH 7.5), 0.5 M NaCl, 10 mM imidazole, 0.1 mM dithiothreitol, and 10% (vol/vol) glycerol. Cell extracts were obtained by sonication and followed by centrifugation to remove cell debris. The protein was purified to homogeneity by sequential column chromatography involving HiTrap Chelating HP (5 ml), ResourceQ, and Hiload 16/60 Superdex 200 (GE Healthcare, Piscataway, NJ). The purified enzyme appeared as a single band corresponding to a molecular mass of 46 kDa on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (data not shown). The absorbance ratio of proteins purified in this manner was >2.0 at 420 nm compared to the ratio at 280 nm. The P450 content measured by CO difference spectroscopy (29) was also checked to verify purification quality. H72A and H69A/H72A mutants were prepared by using a QuikChange site-directed mutagenesis kit (Stratagene), and the sequence was confirmed by DNA sequencing. The primers 5′-GTG CCC CAC GCG TTG GCC ACC CAG GAC GGC GTC-3′ and 5′-GCG TCC AGG TGC CCG CCG CAC TGG CCA CCC AG-3′ and their complementary primers were used to introduce the H72A and H69A mutations.
Crystallography.
For crystallization, the protein was concentrated to more than 14 mg/ml in 10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, and 0.1 mM EDTA. Crystallization was performed using the sitting drop vapor diffusion method. Crystals of the WT-free form were grown at 4°C by mixing 1 μl of the protein solution (9 mg of protein/ml) and 1 μl of the reservoir solution consisting of 3.7 M sodium formate (pH 8.0). Crystals of the WT-4PI form were grown at 25°C by mixing 1 μl of the protein solution (10 mg of protein/ml and 2.5 mM 4-PI) and 1 μl of the reservoir solution, consisting of 3.8 M sodium formate (pH 8.0). Crystals of the H72A-free form were grown at 25°C by mixing 1 μl of the protein solution (10 mg of H72A mutant protein/ml) and 1 μl of the reservoir solution consisting of 4.2 M sodium formate (pH 8.0). X-ray diffraction data were collected by using a charged-coupled device camera at the BL-6A station at the Photon Factory and the NW12A station at the Photon Factory, AR, High Energy Accelerator Research Organization (KEK), Tsukuba, Japan. Without any cryoprotection, crystals were directly flash-cooled in a nitrogen stream at 100 K. Diffraction images were processed by using the HKL2000 program suite (32). The WT-free structure was solved by molecular replacement using MOLREP (50). The search model was prepared using the homology modeling server 3D-JIGSAW based on the structure of OxyB (2). The WT-4PI and H72A-free structures were determined by molecular replacement using the refined WT-free structure as the search model. The initial structural model building was conducted by using the program ARP/wARP (37). Manual model rebuilding, introduction of water molecules, and refinement were performed by using Coot (7) and Refmac5 (25). In the final refinement stage using Refmac5, bulk solvent correction, and TLS (i.e., parameterization of the translation, libration, and screw rotation displacements of pseudorigid bodies) refinement with the groups defined by the TLSMD server (33) was applied. The data collection and refinement statistics are shown in Table 1. Figures were prepared by using PyMol (version 0.99; DeLano Scientific LLC, Palo Alto, CA [http://www.pymol.org]). Due to PCR error, the WT-free and WT-4PI structures contained a functionally irrelevant mutation, M387I. The residue is located on the surface of protein, and it does not interact with other residues or symmetry-related molecules in the crystal packing. We have determined the wild-type ligand-free structure without the mutation at 2.1 Å resolution and have confirmed that it is almost identical with the WT-free (M387I) structure at the higher resolution presented here, except for the mutation site (data not shown). The spectroscopic feature of the M387I mutant was also identical to the wild-type protein under every condition we examined (data not shown). For all spectroscopic data shown here, the wild-type protein without the mutation was used.
TABLE 1.
Data collection and refinement statistics
| Data seta | WT-free | WT-4PI | H72A-free |
|---|---|---|---|
| Data collection statistics | |||
| Beamline | PF-AR NW12A | PF-AR NW12A | PF-BL6A |
| Wavelength (Å) | 1.000 | 1.000 | 1.000 |
| Space group | P43212 | P3121 | P43212 |
| Unit cell (Å) | a = b = 88.401; c = 193.885 | a = b = 143.725; c = 70.943 | a = b = 88.635; c = 191.473 |
| Resolution (Å)* | 50.00-1.95 (2.02-1.95) | 50.00-2.60 (2.69-2.60) | 50.00-2.40 (2.49-2.40) |
| No. of total reflections | 812,705 | 294,967 | 407,345 |
| No. of unique reflections | 57,002 | 26,300 | 30,777 |
| Completeness (%)* | 99.8 (100.0) | 100.0 (100.0) | 99.8 (100.0) |
| Redundancy* | 14.3 (14.1) | 11.2 (11.1) | 13.3 (12.7) |
| Mean I/σ(I)* | 65.3 (5.3) | 38.7 (4.3) | 48.3 (5.3) |
| Rmerge (%)* | 5.1 (40.9) | 6.9 (42.9) | 6.0 (47.2) |
| Refinement statistics | |||
| PDB code | 3E5J | 3E5K | 3E5L |
| Resolution range (Å) | 38.75-1.95 | 34.52-2.60 | 40.23-2.40 |
| No. of reflections | 53,921 | 24,928 | 29,066 |
| R factor/Rfree (%) | 19.0/21.1 | 17.6/23.6 | 18.4/23.5 |
| No. of TLS groups | 6 | 3 | 7 |
| Avg B-factor (Å2) | |||
| Protein | 29.5 | 39.6 | 40.2 |
| Heme | 30.8 | 35.9 | 38.3 |
| 4PI | 45.0 | ||
| Water | 53.5 | 50.3 | 54.2 |
| RMSD from ideal values | |||
| Bond length (Å) | 0.016 | 0.017 | 0.023 |
| Bond angle (°) | 1.604 | 3.549 | 2.186 |
| Ramachandran plot (%)b | |||
| Favored | 95.5 | 93.9 | 96.9 |
| Allowed | 2.9 | 4.5 | 2.1 |
| Outlier | 1.6 | 1.6 | 1.0 |
*, Values in parentheses are for the highest-resolution shell.
Determined by the RAMPAGE server (21).
Spectroscopy.
UV-visible absorption spectra were collected by using a JASCO-V550 spectrophotometer with a 1-cm path length quartz cuvette at room temperature (25°C). Absorption spectra of purified wild-type CYP105P1 in the ferric (resting), dithionite-reduced, and dithionite-reduced plus CO states are shown in Fig. S1 in the supplemental material. In order for the titrations to determine the binding of 4PI, 1 ml of assay buffer containing 50 mM potassium phosphate (pH 7.5), 0.1 mM dithiothreitol, 0.1 mM EDTA, and 10% (vol/vol) glycerol was used. The concentrations of the proteins were 5.5 μM (wild-type) and 4.8 μM (H72A), and 14 μl of stock solution (5 mM) and 9.2 μl of stock solution (20 mM) were added for the wild type and H72A mutant, respectively. Spectra were recorded after each addition of 4PI, and the original spectrum was subtracted from subsequent spectra to generate difference spectra. The dissociation constant (Kd) was calculated from the difference in absorbance (ΔA) at the peak (around 438 nm) from that at the trough (around 416 nm). Electron paramagnetic resonance (EPR) spectra were measured with JOEL spectrometer JES-FA100 equipped with an ES-CT470 liquid helium cryostat system, and the temperature was monitored with a model 9650 cryogenic temperature controller (Advanced Research Systems, Inc., Allentown, PA).
Protein structure accession number.
The coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession codes 3E5J, 3E5K, and 3E5L.
RESULTS
WT-free structure.
The WT-free structure of CYP105P1 was determined at 1.95-Å resolution and refined to an R factor of 19.0% (Rfree = 21.1%). The crystal contains one molecule in the asymmetric unit and exhibits a high Matthews coefficient (4.27 Å3/Da) and solvent content (71.2%). The CYP105P1 structure shows a typical P450 fold (Fig. 2A). The final model contains residues from Asp7 to His400, including the first residue of the four histidine tag residues, which is attached after the original C-terminal residue, Trp399. However, due to a disorder, a central part of the BC loop ranging from Thr79 to Pro82 was not included (Fig. 2A and 3A). A structural similarity search using DALI server (12) indicated that CYP105P1 shows high similarity to MoxA (PDB code 2Z36 chain A; Z score = 47.1, and root mean square deviation [RMSD] for 371 Cα atoms = 1.9 Å) (54), the ligand-free wide open form of P450 PikC (PDB code 2BVJ chain B; Z score = 45.6, and RMSD for 373 Cα atoms = 2.4 Å) (47), and the ligand-free form of P450nor (PDB code 1ROM; Z score = 44.4, and RMSD for 373 Cα atoms = 2.1 Å) (35). These three structures have relatively open FG helices compared to the similar P450 structures. P450 SU-1 has closed FG helices and shows relatively low structural similarity to CYP105P1 (PDB code 2ZBZ; Z score = 42.9, and RMSD for 371 Cα atoms = 2.6 Å) despite the close sequence similarity (49). CYP105P1 does not contain α helix in the BC loop region, whereas the MoxA, P450 SU-1, and P450nor structures have a B′ helix here (Fig. 3) (35). The BC loop region of the WT-free form of CYP105P1 takes a unique conformation, which is not similar to the partially disordered BC loop of ligand-free P450 PikC structures. The N-terminal part of the BC loop (Ala70 to His72) forms a short 310 helix, and the side chain imidazole group of His72 is ligated to the heme iron atom (Fig. 4A). The electron density map around this region was clearly observed (Fig. 4D). The local structure is stabilized by a hydrogen bond between the Nδ1 atoms of His72 and His69 (2.9 Å), as well as several other hydrogen bonds formed by water atoms. In this structure, five residues after the B helix, Phe63 to Val67, forms a β-strand (designated as β1-6 in Fig. 2A and 5).
FIG. 2.
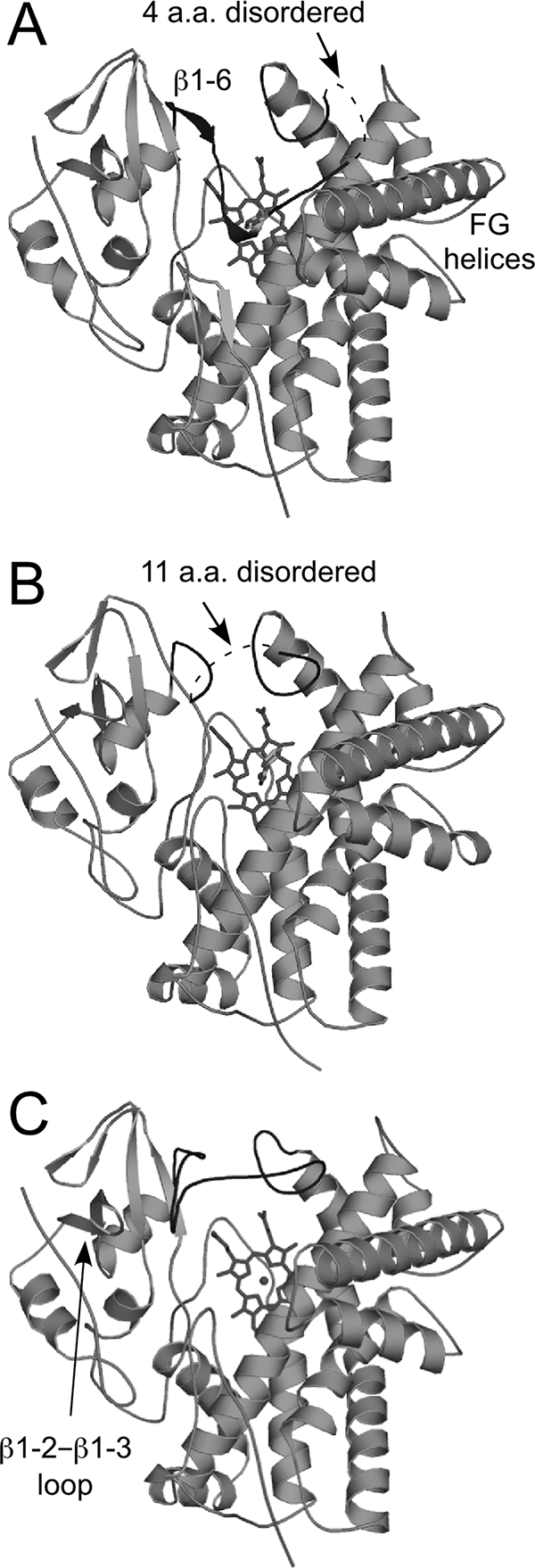
Overall structure of CYP105P1 in the WT-free (A), WT-4PI (B), and H72A-free (C) forms illustrated by ribbon diagrams. Heme and ligands are shown as stick models. The BC loop region is shown in dark gray.
FIG. 3.

(A) Stereographic superimposition of the three crystal structures of CYP105P1. A region from B helix to I helix is shown as ribbon models. FG helices and BC loop regions of WT-free, WT-4PI, and H72A-free structures are colored in blue, yellow, and orange, respectively. Two residues in the BC loop of H72A-free structure (Gly80 and Lys81 colored in magenta) exhibit relatively weak electron density. The side chains of His72 in WT-free and Ala72 in H72A-free structures are shown as stick models with carbon atoms green. (B) Comparison of H72A-free structure with two CYP105 structures, P450 SU-1 in complex with 1α,25-dihydroxyvitamin D3 (PDB code 2ZBZ) and P450 MoxA in complex with MES (PDB code 2Z36). FG helices, BC loop regions and bound ligands of CYP105P1, P450 SU-1, and P450 MoxA are colored orange, green, and magenta, respectively.
FIG. 4.

Structures near the heme distal site. Interactions of WT-free (A), WT-4PI (B), and H72A-free (C) forms, and the 2Fobs − Fcalc composite omit map of WT-free form contoured at 2.0 σ (D) and the Fobs − Fcalc omit electron density map of the 4PI molecule in the WT-4PI structures contoured at 3.0 σ (E) are shown.
FIG. 5.

Amino acid sequence alignment of CYP105 family members in the genome of S. avermitilis, MoxA, P450 SU-1, and P450nor in the BC loop and flanking regions. Secondary structures of the WT-free structure and the position of His72 residue of CYP105P1 are indicated above the sequence. Symbols below the alignment indicate the degree of conservation. The B′ helix region of structurally determined P450s, conserved FP/WP motif, and glycine residues in the BC loop region of CYP105P1 and CYP105D6 are highlighted by boxes, black/white inverse characters, and underlines, respectively.
WT-4PI structure.
The WT-4PI structure was determined at 2.6-Å resolution and refined to an R factor of 17.6% (Rfree = 23.6%) (Fig. 2B). The crystal is not isomorphous to the WT-free crystal, but it also exhibits a high Matthews coefficient (4.76 Å3/Da) and solvent content (74.2%), containing only one molecule in the asymmetric unit. The final model contains residues from Pro9 to His401, including two of the four histidine tag residues at the C terminus. However, the disordered region in the BC loop was longer than that of WT-free structure, ranging from Ala70 to Gln80. Therefore, the short 310 helix including His72 was not observed. Except for the BC loop region, WT-4PI and WT-free structures are essentially identical (RMSD for 335 Cα atoms = 0.75 Å). Flanking ends of the disordered region point toward outside of the molecule (Fig. 2B and 3A), and the β1-6 strand after the B helix is not formed in this structure. A Fobs − Fcalc omit map for the bound 4PI molecule is shown in Fig. 4E. The imidazole ring of 4PI occupies the same place of the side chain of His72 in WT-free structure (Fig. 4B). An imidazole nitrogen atom directly coordinates with the heme iron. The other imidazole nitrogen atom forms a hydrogen bond with Thr241 (2.9 Å). The phenyl ring of 4PI is surrounded by four hydrophobic residues: Leu88, Trp89, Ile236, and Phe286. These interactions are similar to those of other 4PI-bound P450 structures (11, 38, 40, 43, 53).
Spectroscopic properties of the wild type and H72A mutant.
A “type II” spectral change of P450 is a red shift of the Soret band, which is characterized by a difference spectrum with a peak at 425 to 435 nm and a trough at 390 to 413 nm (1, 16, 44). This change is caused by the replacement of the sixth ligand of the heme iron with a stronger field ligand, usually nitrogen-containing heteroaromatic compounds.
In order to investigate the effect of eliminating the side chain of His72 on spectral and structural features of CYP105P1, we constructed the H72A mutant. Figure 6 shows UV-visible absorption spectra of wild-type and H72A mutant and their titration results with 4PI. Unexpectedly, both samples exhibit almost identical results. Although they have slightly red-shifted Soret maxima in the low-spin resting state compared to those of standard P450 enzymes (∼418 nm), both samples illustrate typical type II spectral shift to 425 to 427 nm. The Kd values were estimated to be 12.5 ± 0.4 μM and 15.2 ± 1.0 μM for the wild type and H72A mutant, respectively. We have also constructed a double mutant, H69A/H72A, but it also exhibited spectral features essentially identical to those of the wild-type and H72A mutant (data not shown). The Soret maximum of H69A/H72A mutant in a ferric resting state without ligands was 421 nm. Figure 7 shows the EPR spectra of the wild-type and H72A mutant in the absence or presence of 4PI in the low spin region around g ∼ 2. These samples again exhibit virtually the same results (gz = 2.42, gy = 2.25, and gx = 1.91). There were no detectable signals in the high spin region around g ∼ 5.8 and 4.3. In the presence of 4PI, additional peaks appeared (g = 2.50 and 1.87). The g values are very similar to those of the 4PI complex of P450cam (CYP101A) (20). These results indicate that the His-ligated conformation of CYP105P1 is unlikely to be dominant in solution. We have also tried to detect the His-ligated state in solution by UV-visible absorption spectra measurements, changing the conditions to those similar to a crystallization setup; 0 to 4.8 M sodium formate (pH 8.0), 0 to 3.6 M sodium acetate (pH 8.0), or 0 to 3.0 M potassium chloride in 20 mM morpholinepropanesulfonic acid-NaOH (pH 7.4), but with a much lower protein concentration (∼3 μM). However, in every condition we tested, the Soret maximum did not shift from 419 to 422 nm (data not shown).
FIG. 6.

Type II binding spectral change of the wild-type (A) and H72A (B) mutant of CYP105P1 (ferric resting state) upon addition of increasing concentrations of 4PI. The inset shows difference spectra. See Materials and Methods for more details.
FIG. 7.

The X-band EPR spectra of the wild-type and H72A mutant of CYP105P1 in the absence or in the presence of 4PI. Instrument settings: microwave power, 1 mW; modulation amplitude, 0.2 mT; temperature, 20 K. Spectra were recorded for the wild-type (495 μM) and H72A mutant (280 μM) in 50 mM potassium phosphate (pH 7.5), 0.1 mM dithiothreitol, 0.1 mM EDTA, and 10% (vol/vol) glycerol, with or without 0.5 mM 4PI.
H72A-free structure.
The H72A-free structure was determined from a crystal isomorphous to the WT-free crystals at 2.4-Å resolution and refined to an R factor of 18.4% (Rfree = 23.5%) (Fig. 2C). The final model contains residues from Asp7 to His400. The H72A-free structure is essentially identical to WT-free (RMSD for 340 Cα atoms = 0.61 Å) and WT-4PI (RMSD for 350 Cα atoms = 0.62 Å) structures. The whole BC loop region was modeled in this structure. The BC loop region of the H72A-free form largely shifts upward, widely opening the entrance of the distal pocket (Fig. 3A). There is no significant secondary structure in this region. Interactions between the N-terminal part of the BC loop region (His69 to Gly76) and the β1-2-β1-3 loop (Thr40 to His43) stabilize the open conformation of BC loop (Fig. 2C). Ala72 residue is 17 Å away from the heme iron. Two residues in the middle part of the BC loop (Glu80 and Lys81, colored magenta in Fig. 3A) show relatively weak electron density and high B-factor. C-terminal region of the BC loop (Gly83 to Gly92) is in a conformation similar to those of WT-free and WT-4PI structures. In contrast to the BC loop region, the FG helices of the three structures are almost overlapping. The sixth ligand of heme iron is a water molecule and is stabilized by a hydrogen bond with the main-chain carbonyl oxygen of Asp237 (Fig. 4C). This observation is inconsistent with the spectroscopic results presented above; CYP105P1 is predominantly in a low spin state. Figure 3B shows structural comparison of the H72A-free structure with two CYP105 structures, P450 MoxA and P450 SU-1. As mentioned above, the FG helices of P450 SU-1 are relatively closed, and both of the P450 MoxA and P450 SU-1 structures contain the B′ helix.
DISCUSSION
Figure 8 shows molecular surface representations of the three CYP105P1 structures and two closely related P450s, MoxA and P450nor in a ligand-free state. The ligand-free structure of P450nor is in an open conformation, and heme is visible through a wide channel for NADH binding (Fig. 8F). MoxA is basically in an open conformation, although an MES (morpholineethanesulfonic acid) molecule is bound in the cavity (Fig. 8E). WT-free structure of CYP105P1 is also basically in an open conformation due to its open FG helices. However, the BC loop sinks onto the heme and completely covers the distal pocket (Fig. 8A). On the other hand, WT-4PI and H72A-free structures have an open BC loop, and heme is accessible from the top of the molecule (Fig. 8B and C). These facts indicate significant flexibility of BC loop of CYP105P1, unlike those of MoxA and P450nor. Among bacterial P450 enzymes similar to CYP105P1, OxyB (CYP165B3), P450 PikC, and P450 SU-1 have a partially disordered BC loop for three to seven amino acids in ligand-free forms (47, 49, 55). In contrast, there are a number of examples that the FG helices displace between ligand-free and ligand-bound forms, e.g., CYP119 from Sulfolobus solfataricus (36), P450nor (31), and CYP158A2 (56). When membrane-associated P450s incorporate lipophilic substrates, there is a distinct route from those of soluble P450s. In the case of mammalian CYP2B4, there is a wide channel orthogonal to that in soluble P450s, between the B and C helices and above the heme plane (41, 46). The opening of this channel is directly linked to extensive rearrangement of the BC loop and C helix, and substrates are thought to access the active site from phospholipid bilayers through this channel (57). Sterol 14α-demethylase (CYP51) from Mycobacterium tuberculosis also has a substrate-accessible channel at a similar position, which is open due to the disorder of the BC loop and C helix (41).
FIG. 8.

Molecular surface representations of CYP105P1 and related P450s. Top views of WT-free (A), WT-4PI (B), H72A-free (C), MoxA (E, PDB code 2Z36), and P450nor in ligand-free form (F, PDB code 1ROM) are shown. (D) Distal pocket of H72A-free structure. The FG helices and BC loop regions are colored in gray, and heme is shown as black spheres. Ligands (4PI and MES) are shown as white sticks.
To date, there are two examples of His-ligated P450 crystal structures. The first is the ligand-free structure of mammalian CYP2B4 (45). In the symmetric homodimer structure of CYP2B4 in a wide open conformation, FG helices of one molecule partially fill the cleft of another molecule. His226 residue, which is located at the tip of FG helices, forms an intermolecular coordinate bond to the axial sixth position of heme iron. This peculiar homodimeric state is also observed in solution by UV-visible spectroscopy with the Soret maximum at 424 nm. Another example is an engineered A264H mutant of P450 BM3 (9). Ala264 residue is located in the I helix and close to the heme iron, and mutation at this residue creates an unusual ligand set at the sixth position, e.g., glutamate (15), lysine, or histidine (9). The coordinated state is stable in solution and has been clearly characterized by spectroscopic methods, including UV-visible absorption spectra (Soret maximum at 427 nm), EPR (g = 2.50, 2.26, and 1.89) and magnetic circular dichroism.
Here we observed three different conformations of the BC loop of CYP105P1 in ligand-free and small inhibitor-bound crystal structures. There is no clear crystal packing interaction for the BC loop region in both the WT-free and H72A-free structures (see Fig. S1 in the supplemental material), but histidine ligation was not detectable in solution. Therefore, for an unknown reason, the His-ligated conformation may be stabilized only in crystals of the wild-type protein. These results strongly indicate that the BC loop of CYP105P1 is highly flexible in solution. When soluble P450s accept large macrolide substrates into their distal pocket, the entrance regions should have a certain conformational flexibility. Such a flexible feature of SRSs has been reported for another macrolide hydroxylase, P450 PikC (47). The crystal structure of P450 PikC in ligand-free form contains two molecules in an asymmetric unit, and both FG helices and BC loop regions are in significantly different conformations between these molecules. On substrate binding, a unique anchoring mechanism to trap the deoxysugar group of diverse substrates has been suggested. Figure 5 shows the sequence alignment around the BC loop regions of CYP105 families in the genome of S. avermitilis, as well as highly homologous P450s, whose structures have been determined. CYP105P1 lacks the conserved FP/WP motif. Moreover, CYP105P1 and CYP105D6 contain several glycine residues in this region. These sequence features may make the BC loop regions of the filipin hydroxylases flexible.
Interestingly, in the H72A-free structure, we found a large hydrophobic space under the FG helices sized about 10 × 20 Å (Fig. 8D). A nuclear magnetic resonance study has revealed that filipin III is a rod-shaped molecule 18 Å long, 7 to 8 Å wide, and 3 to 4 Å thick in solution (51). The large 28-membered ring is rigid, stabilized by both intramolecular hydrogen bonds of syn 1,3-polyols and a conjugated pentaene moiety, whereas the lateral aliphatic chain is highly flexible. The large pocket of the H72A-free structure appears to be able to accommodate the filipin molecule. However, our preliminary analysis indicated that the wild-type CYP105P1 did not exhibit any spectroscopic change when commercially available filipin complex (mixture of filipins I, II, III, and IV) was titrated (L.-H. Xu et al., unpublished data). The filipin complex contain only slight fraction of the substrate for CYP105P1, filipin I (ca. 4%) (3), and the major components (filipins II, III, and IV) may not bind to the heme pocket due to their hydroxyl group at C26 position. To understand the C26 position-specific hydroxylation mechanism of CYP105P1, knowledge of the complex structure with filipin would be indispensable.
Supplementary Material
Acknowledgments
We thank the staff of the Photon Factory for the X-ray data collection and S. Adachi, S. Y. Park, and A. Tomita for technical assistance.
This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (to H.S., no. 20248009).
Footnotes
Published ahead of print on 12 December 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Ahlstrom, M. M., and I. Zamora. 2008. Characterization of type II ligands in CYP2C9 and CYP3A4. J. Med. Chem. 511755-1763. [DOI] [PubMed] [Google Scholar]
- 2.Bates, P. A., L. A. Kelley, R. M. MacCallum, and M. J. Sternberg. 2001. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins, Suppl. 5 39-46. [DOI] [PubMed]
- 3.Bergy, M. E., and T. E. Eble. 1968. The filipin complex. Biochemistry 7653-659. [DOI] [PubMed] [Google Scholar]
- 4.Butler, J. D., J. Blanchette-Mackie, E. Goldin, R. R. O'Neill, G. Carstea, C. F. Roff, M. C. Patterson, S. Patel, M. E. Comly, A. Cooney, et al. 1992. Progesterone blocks cholesterol translocation from lysosomes. J. Biol. Chem. 26723797-23805. [PubMed] [Google Scholar]
- 5.Butler, J. D., M. E. Comly, H. S. Kruth, M. Vanier, M. Filling-Katz, J. Fink, N. Barton, H. Weintroub, J. M. Quirk, T. Tokoro, et al. 1987. Niemann-pick variant disorders: comparison of errors of cellular cholesterol homeostasis in group D and group C fibroblasts. Proc. Natl. Acad. Sci. USA 84556-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cupp-Vickery, J. R., and T. L. Poulos. 1995. Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat. Struct. Biol. 2144-153. [DOI] [PubMed] [Google Scholar]
- 7.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 602126-2132. [DOI] [PubMed] [Google Scholar]
- 8.Gimpl, G., and K. Gehrig-Burger. 2007. Cholesterol reporter molecules. Biosci. Rep. 27335-358. [DOI] [PubMed] [Google Scholar]
- 9.Girvan, H. M., H. E. Seward, H. S. Toogood, M. R. Cheesman, D. Leys, and A. W. Munro. 2007. Structural and spectroscopic characterization of P450 BM3 mutants with unprecedented P450 heme iron ligand sets. New heme ligation states influence conformational equilibria in P450 BM3. J. Biol. Chem. 282564-572. [DOI] [PubMed] [Google Scholar]
- 10.Gotoh, O. 1992. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 26783-90. [PubMed] [Google Scholar]
- 11.Ho, W. W., H. Li, C. R. Nishida, P. R. de Montellano, and T. L. Poulos. 2008. Crystal structure and properties of CYP231A2 from the thermoacidophilic archaeon Picrophilus torridus. Biochemistry 472071-2079. [DOI] [PubMed] [Google Scholar]
- 12.Holm, L., and C. Sander. 1995. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20478-480. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda, H., J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori, and S. Omura. 2003. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 21526-531. [DOI] [PubMed] [Google Scholar]
- 14.Isin, E. M., and F. P. Guengerich. 2007. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta 1770314-329. [DOI] [PubMed] [Google Scholar]
- 15.Joyce, M. G., H. M. Girvan, A. W. Munro, and D. Leys. 2004. A single mutation in cytochrome P450 BM3 induces the conformational rearrangement seen upon substrate binding in the wild-type enzyme. J. Biol. Chem. 27923287-23293. [DOI] [PubMed] [Google Scholar]
- 16.Kumaki, K., M. Sato, H. Kon, and D. W. Nebert. 1978. Correlation of type I, type II, and reverse type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J. Biol. Chem. 2531048-1058. [PubMed] [Google Scholar]
- 17.Lamb, D. C., H. Ikeda, D. R. Nelson, J. Ishikawa, T. Skaug, C. Jackson, S. Omura, M. R. Waterman, and S. L. Kelly. 2003. Cytochrome p450 complement (CYPome) of the avermectin-producer Streptomyces avermitilis and comparison to that of Streptomyces coelicolor A3(2). Biochem. Biophys. Res. Commun. 307610-619. [DOI] [PubMed] [Google Scholar]
- 18.Lamb, D. C., T. Skaug, H. L. Song, C. J. Jackson, L. M. Podust, M. R. Waterman, D. B. Kell, D. E. Kelly, and S. L. Kelly. 2002. The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem. 27724000-24005. [DOI] [PubMed] [Google Scholar]
- 19.Lamb, D. C., M. R. Waterman, S. L. Kelly, and F. P. Guengerich. 2007. Cytochromes P450 and drug discovery. Curr. Opin. Biotechnol. 18504-512. [DOI] [PubMed] [Google Scholar]
- 20.Lipscomb, J. D. 1980. Electron paramagnetic resonance detectable states of cytochrome P-450cam. Biochemistry 193590-3599. [DOI] [PubMed] [Google Scholar]
- 21.Lovell, S. C., I. W. Davis, W. B. Arendall III, P. I. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson, and D. C. Richardson. 2003. Structure validation by Cα geometry: φ, ψ, and Cβ deviation. Proteins 50437-450. [DOI] [PubMed] [Google Scholar]
- 22.McLean, K. J., M. Sabri, K. R. Marshall, R. J. Lawson, D. G. Lewis, D. Clift, P. R. Balding, A. J. Dunford, A. J. Warman, J. P. McVey, A. M. Quinn, M. J. Sutcliffe, N. S. Scrutton, and A. W. Munro. 2005. Biodiversity of cytochrome P450 redox systems. Biochem. Soc. Trans. 33796-801. [DOI] [PubMed] [Google Scholar]
- 23.Reference deleted.
- 24.Munro, A. W., H. M. Girvan, and K. J. McLean. 2007. Variations on a (t)heme: novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 24585-609. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53240-255. [DOI] [PubMed] [Google Scholar]
- 26.Nagano, S., H. Li, H. Shimizu, C. Nishida, H. Ogura, P. R. Ortiz de Montellano, and T. L. Poulos. 2003. Crystal structures of epothilone d-bound, epothilone B-bound, and substrate-free forms of cytochrome P450epoK. J. Biol. Chem. 27844886-44893. [DOI] [PubMed] [Google Scholar]
- 27.Nakahara, K., T. Tanimoto, K. Hatano, K. Usuda, and H. Shoun. 1993. Cytochrome P-450 55A1 (P-450dNIR) acts as nitric oxide reductase employing NADH as the direct electron donor. J. Biol. Chem. 2688350-8355. [PubMed] [Google Scholar]
- 28.O'Keefe, D. P., and P. A. Harder. 1991. Occurrence and biological function of cytochrome P450 monooxygenases in the actinomycetes. Mol. Microbiol. 52099-2105. [DOI] [PubMed] [Google Scholar]
- 29.Omura, T., and R. Sato. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 2392370-2378. [PubMed] [Google Scholar]
- 30.Ortiz de Montellano, P. R. 2005. Cytochrome P450: structure, mechanism, and biochemistry, 3rd ed. Kluwer Academic/Plenum Publishers, New York, NY.
- 31.Oshima, R., S. Fushinobu, F. Su, L. Zhang, N. Takaya, and H. Shoun. 2004. Structural evidence for direct hydride transfer from NADH to cytochrome P450nor. J. Mol. Biol. 342207-217. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276307-326. [DOI] [PubMed] [Google Scholar]
- 33.Painter, J., and E. A. Merritt. 2006. TLSMD web server for the generation of multi-group TLS models. J. Appl. Cryst. 39109-111. [Google Scholar]
- 34.Pandey, R. C., and K. L. Rinehart, Jr. 1970. Polyene antibiotics. V. Characterization of components of the filipin complex by mass spectrometry. J. Antibiot. 23414-417. [DOI] [PubMed] [Google Scholar]
- 35.Park, S. Y., H. Shimizu, S. Adachi, A. Nakagawa, I. Tanaka, K. Nakahara, H. Shoun, E. Obayashi, H. Nakamura, T. Iizuka, and Y. Shiro. 1997. Crystal structure of nitric oxide reductase from denitrifying fungus Fusarium oxysporum. Nat. Struct. Biol. 4827-832. [DOI] [PubMed] [Google Scholar]
- 36.Park, S. Y., K. Yamane, S. Adachi, Y. Shiro, K. E. Weiss, S. A. Maves, and S. G. Sligar. 2002. Thermophilic cytochrome P450 (CYP119) from Sulfolobus solfataricus: high resolution structure and functional properties. J. Inorg. Biochem. 91491-501. [DOI] [PubMed] [Google Scholar]
- 37.Perrakis, A., R. Morris, and V. S. Lamzin. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6458-463. [DOI] [PubMed] [Google Scholar]
- 38.Podust, L. M., H. Bach, Y. Kim, D. C. Lamb, M. Arase, D. H. Sherman, S. L. Kelly, and M. R. Waterman. 2004. Comparison of the 1.85 A structure of CYP154A1 from Streptomyces coelicolor A3(2) with the closely related CYP154C1 and CYPs from antibiotic biosynthetic pathways. Protein Sci. 13255-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Podust, L. M., Y. Kim, M. Arase, B. A. Neely, B. J. Beck, H. Bach, D. H. Sherman, D. C. Lamb, S. L. Kelly, and M. R. Waterman. 2003. The 1.92-Å structure of Streptomyces coelicolor A3(2) CYP154C1. A new monooxygenase that functionalizes macrolide ring systems. J. Biol. Chem. 27812214-12221. [DOI] [PubMed] [Google Scholar]
- 40.Podust, L. M., T. L. Poulos, and M. R. Waterman. 2001. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. USA 983068-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podust, L. M., L. V. Yermalitskaya, G. I. Lepesheva, V. N. Podust, E. A. Dalmasso, and M. R. Waterman. 2004. Estriol bound and ligand-free structures of sterol 14α-demethylase. Structure 121937-1945. [DOI] [PubMed] [Google Scholar]
- 42.Poulos, T. L. 2007. Structural biology of p450-oxy complexes. Drug Metab. Rev. 39557-566. [DOI] [PubMed] [Google Scholar]
- 43.Poulos, T. L., and A. J. Howard. 1987. Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry 268165-8174. [DOI] [PubMed] [Google Scholar]
- 43a.Richardson, T. I., and S. D. Rychnovsky. 1996. Filipin III: configuration assignment and confirmation by synthetic correlation. J. Org. Chem. 614219-4231. [DOI] [PubMed] [Google Scholar]
- 44.Schenkman, J. B., H. Remmer, and R. W. Estabrook. 1967. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 3113-123. [PubMed] [Google Scholar]
- 45.Scott, E. E., Y. A. He, M. R. Wester, M. A. White, C. C. Chin, J. R. Halpert, E. F. Johnson, and C. D. Stout. 2003. An open conformation of mammalian cytochrome P450 2B4 at 1.6-A resolution. Proc. Natl. Acad. Sci. USA 10013196-13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott, E. E., M. A. White, Y. A. He, E. F. Johnson, C. D. Stout, and J. R. Halpert. 2004. Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9-A resolution: insight into the range of P450 conformations and the coordination of redox partner binding. J. Biol. Chem. 27927294-27301. [DOI] [PubMed] [Google Scholar]
- 47.Sherman, D. H., S. Li, L. V. Yermalitskaya, Y. Kim, J. A. Smith, M. R. Waterman, and L. M. Podust. 2006. The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem. 28126289-26297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stout, C. D. 2004. Cytochrome p450 conformational diversity. Structure 121921-1922. [DOI] [PubMed] [Google Scholar]
- 49.Sugimoto, H., R. Shinkyo, K. Hayashi, S. Yoneda, M. Yamada, M. Kamakura, S. Ikushiro, Y. Shiro, and T. Sakaki. 2008. Crystal structure of CYP105A1 (P450SU-1) in complex with 1α,25-dihydroxyvitamin D3. Biochemistry 474017-4027. [DOI] [PubMed] [Google Scholar]
- 50.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 301022-1025. [Google Scholar]
- 51.Volpon, L., and J. Lancelin. 2000. Solution NMR structures of the polyene macrolide antibiotic filipin III. FEBS Lett. 478137-140. [DOI] [PubMed] [Google Scholar]
- 52.Wachtler, V., and M. K. Balasubramanian. 2006. Yeast lipid rafts? An emerging view. Trends Cell Biol. 161-4. [DOI] [PubMed] [Google Scholar]
- 53.Yano, J. K., L. S. Koo, D. J. Schuller, H. Li, P. R. Ortiz de Montellano, and T. L. Poulos. 2000. Crystal structure of a thermophilic cytochrome P450 from the archaeon Sulfolobus solfataricus. J. Biol. Chem. 27531086-31092. [DOI] [PubMed] [Google Scholar]
- 54.Yasutake, Y., N. Imoto, Y. Fujii, T. Fujii, A. Arisawa, and T. Tamura. 2007. Crystal structure of cytochrome P450 MoxA from Nonomuraea recticatena (CYP105). Biochem. Biophys. Res. Commun. 361876-882. [DOI] [PubMed] [Google Scholar]
- 55.Zerbe, K., O. Pylypenko, F. Vitali, W. Zhang, S. Rouset, M. Heck, J. W. Vrijbloed, D. Bischoff, B. Bister, R. D. Sussmuth, S. Pelzer, W. Wohlleben, J. A. Robinson, and I. Schlichting. 2002. Crystal structure of OxyB, a cytochrome P450 implicated in an oxidative phenol coupling reaction during vancomycin biosynthesis. J. Biol. Chem. 27747476-47485. [DOI] [PubMed] [Google Scholar]
- 56.Zhao, B., F. P. Guengerich, A. Bellamine, D. C. Lamb, M. Izumikawa, L. Lei, L. M. Podust, M. Sundaramoorthy, J. A. Kalaitzis, L. M. Reddy, S. L. Kelly, B. S. Moore, D. Stec, M. Voehler, J. R. Falck, T. Shimada, and M. R. Waterman. 2005. Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem. 28011599-11607. [DOI] [PubMed] [Google Scholar]
- 57.Zhao, Y., M. A. White, B. K. Muralidhara, L. Sun, J. R. Halpert, and C. D. Stout. 2006. Structure of microsomal cytochrome P450 2B4 complexed with the antifungal drug bifonazole: insight into P450 conformational plasticity and membrane interaction. J. Biol. Chem. 2815973-5981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.