

Abstract
Mutations within the polyamine biosynthetic pathway of Leishmania donovani, the etiological agent of visceral leishmaniasis, confer polyamine auxotrophy to the insect vector or promastigote form of the parasite. However, whether the infectious or amastigote form of the parasite requires an intact polyamine pathway has remained an open question. To address this issue, conditionally lethal Δodc mutants lacking ornithine decarboxylase (ODC), the rate-limiting enzyme in polyamine biosynthesis, were created by double targeted gene replacement within a virulent strain of L. donovani. ODC-deficient promastigotes and axenic amastigotes were auxotrophic for polyamines and capable of robust growth only when exogenous putrescine was supplied in the culture medium, confirming that polyamine biosynthesis is an essential nutritional pathway for L. donovani promastigotes. To assess whether the Δodc lesion also affected the ability of amastigotes to sustain a robust infection, macrophage and mouse infectivity experiments were performed. Parasite loads in murine macrophages infected with each of two independent Δodc knockout lines were decreased ∼80% compared to their wild-type counterpart. Furthermore, α-difluoromethylornithine, a suicide inhibitor of ODC, inhibited growth of wild-type L. donovani amastigotes and effectively cured macrophages of parasites, thereby preventing host cell destruction. Strikingly, however, parasitemias of both Δodc null mutants were reduced by 6 and 3 orders of magnitude, respectively, in livers and spleens of BALB/c mice. The compromised infectivity phenotypes of the Δodc knockouts in both macrophages and mice were rescued by episomal complementation of the genetic lesion. These genetic and pharmacological studies strongly implicate ODC as an essential cellular determinant that is necessary for the viability and growth of both L. donovani promastigotes and amastigotes and intimate that pharmacological inhibition of ODC is a promising therapeutic paradigm for the treatment of visceral and perhaps other forms of leishmaniasis.
Leishmania is a digenetic protozoan parasite that causes a spectrum of pathologies in humans that range in severity from self-healing cutaneous lesions to visceral leishmaniasis, the latter being an invariably fatal disease in the absence of drug treatment. The extracellular, flagellated promastigote stage resides in the insect vector, sand flies of the Phlebotominae subfamily, while the intracellular amastigote form inhabits the phagolysosome of macrophages and other reticuloendothelial cells within the mammalian host. There is no effective vaccine for leishmaniasis, and therefore, chemotherapy is the only means available to combat the disease. Unfortunately, the current arsenal of antileishmanial drugs is far from ideal, principally due to toxicity for the host, for which a lack of target specificity is the chief culprit, and to the acquisition of drug resistance (23, 38). Thus, the identification and validation of new drug targets, particularly for treating visceral leishmaniasis, are imperative.
One pathway that has been clinically validated as an antiparasitic drug target is that for polyamine biosynthesis. The polyamines, putrescine, spermidine, and spermine, are ubiquitous organic cations that play critical roles in a variety of key cellular processes, including growth, differentiation, and macromolecular synthesis (5, 29, 30, 52). d,l-α-Difluoromethylornithine (DFMO), a suicide inhibitor of ornithine decarboxylase (ODC), the enzyme that catalyzes the rate-limiting step in the polyamine biosynthetic pathway (37), has shown remarkable therapeutic efficacy in treating African sleeping sickness caused by Trypanosoma brucei gambiense (2, 14, 20, 51, 55), a protozoan parasite phylogenetically related to Leishmania. DFMO is also effective at killing other genera of protozoan parasites (1, 10, 24, 45), including Leishmania promastigotes (32, 34, 39, 45, 50), and studies have shown that DFMO can also inhibit short-term Leishmania donovani infections in mice (27, 34) and hamsters (40). Moreover, inhibitors of S-adenosylmethionine decarboxylase (ADOMETDC), the enzyme that provides the aminopropyl group for spermidine synthesis, are also effective antitrypanosomal agents (3, 8, 9, 15, 18, 54).
The polyamine biosynthetic pathway of Leishmania consists of four enzymes: arginase (ARG), ODC, ADOMETDC, and spermidine synthase (SPDSYN). ARG, the first enzyme of this pathway, catalyzes the conversion of arginine to ornithine. Subsequently, ornithine is decarboxylated by ODC to form putrescine, which is then converted to spermidine through the concerted actions of ADOMETDC and SPDSYN. Unlike mammalian cells, however, Leishmania parasites do not synthesize or make use of spermine (4, 31).
The genes encoding the leishmanial ARG, ODC, ADOMETDC, and SPDSYN proteins have all been cloned, and a battery of conditionally lethal null mutants of Leishmania mexicana (Δarg mutant) (49) and L. donovani (Δodc, Δspdsyn, and Δadometdc mutants) (31, 47, 48) have been constructed by targeted gene disruption. Characterization of these knockouts demonstrated that the Δarg L. mexicana promastigotes can survive only in the presence of added ornithine, putrescine, or spermidine (49) whereas Δodc L. donovani promastigotes require putrescine or spermidine supplementation (31) and Δadometdc and Δspdsyn L. donovani promastigotes can proliferate only if spermidine is supplied in the culture medium (47, 48). Thus, an intact polyamine biosynthetic pathway is essential for the viability and growth of Leishmania promastigotes.
Despite the plethora of biochemical and genetic studies of polyamine biosynthesis in promastigotes, little is known about polyamine synthesis in amastigotes. The intracellular milieu in which amastigotes replicate is presumably rich in polyamines, and Basselin et al. (6) have reported that axenic amastigotes of L. mexicana are capable of transporting both putrescine and spermidine. Recently it was established that Δarg L. mexicana, a cutaneous strain of Leishmania, is only partially compromised in its ability to infect murine macrophages and mice (22). The finding that Δarg L. mexicana parasites are able to establish an infection, albeit less effectively than wild-type parasites, implies that L. mexicana amastigotes do not need an intact polyamine biosynthetic pathway to survive in vivo because they can effectively scavenge sufficient amounts of ornithine and/or putrescine/spermidine from the phagolysosome of the skin macrophages in which the cutaneous infection takes place.
To determine whether the amastigote stage of L. donovani, a visceralizing and much more serious pathogen that resides primarily in the liver and spleen, requires an intact polyamine pathway or can survive exclusively on polyamines acquired from the host environment, we have evaluated the capacities of two Δodc L. donovani strains to infect murine macrophages and mice. Here we report that parasite loads in macrophages infected with Δodc parasites are significantly reduced from those in macrophages infected with the wild-type L. donovani counterpart, and parasite burdens in infected livers and spleens of mice infected with the knockout parasites are profoundly compromised. These studies demonstrate that L. donovani amastigotes require ODC activity to sustain a robust infection in mice and are not capable of scavenging polyamines from the host milieu in amounts sufficient to sustain an infection. These findings indicate that targeting ODC is a rational therapeutic paradigm for the treatment of leishmaniasis.
MATERIALS AND METHODS
Parasites.
All genetically manipulated parasites were derived from the wild-type LdBob strain of L. donovani (26), which was originally obtained from Stephen Beverley (Washington University, St. Louis, MO). LdBob promastigotes and axenic amastigotes were routinely cultivated at 26°C, pH 7.4, and 37°C, pH 5.5, respectively, in the culture media previously reported (26). All parasite strains were cycled back and forth between the promastigote and axenic amastigote forms several times prior to infectivity experiments with macrophages or mice. Wild-type parasites were routinely cultured in medium with no drug supplementations, while the Δodc mutants were propagated in 50 μg/ml hygromycin, 20 μg/ml G418, and 200 μM putrescine or just 200 μM putrescine alone. The “add-back” strains, designated the Δodc1[pXG-BSD-ODC] and Δodc2[pXG-BSD-ODC] strains (see below), which harbor a multicopy episomal plasmid containing a full-length ODC gene, were routinely propagated in the same medium as wild-type parasites.
For growth experiments in putrescine and/or DFMO, wild-type LdBob promastigotes or axenic amastigotes were seeded at 1.0 × 105 parasites/ml in their respective growth media, and putrescine and/or DFMO were added as appropriate. Promastigotes were enumerated daily and axenic amastigotes after 4 days by using a hemacytometer.
For limiting dilution assays to quantify parasite loads in mice, serial fourfold dilutions of liver and spleen cell lysates were cultivated in 96-well plates in promastigote growth medium to which 10% fetal bovine serum and 200 μM putrescine were added. Growth in individual wells was monitored after 2 weeks by visual inspection.
Transfections.
The LdBob Δodc knockouts were constructed using the same drug resistance cassettes, pX63-HYG-Δodc and pX63-NEO-Δodc, employed in the derivation of the same mutants in the avirulent 1S Sudanese strain of L. donovani (31). These targeting vectors encompassed either the hygromycin or neomycin phosphotransferase markers bounded by the 5′ and 3′ flanking regions of ODC, and each was linearized with HindIII-BglII and gel purified prior to transfection as described previously (31). Wild-type LdBob promastigotes were first transfected with linearized pX63-HYG-Δodc according to standard procedures (31) and plated on semisolid agar plates containing 50 μg/ml hygromycin. The correct integration events in the ODC/odc heterozygotes were verified by Southern blotting, and then one ODC/odc clone was subjected to a second round of transfection using the excised targeting construct from pX63-NEO-Δodc and plated on semisolid medium containing 50 μg/ml hygromycin, 20 μg/ml G418, and 200 μM putrescine. The presence of hygromycin on the selective plates employed during the second round of transfection was to ensure selection of clones in which only the wild-type allele was displaced from the ODC/odc heterozygote. Colonies were picked and expanded and Southern blot analysis employed to confirm that homologous recombination had correctly supplanted the wild-type ODC allele in the ODC/odc heterozygote. Two independent but isogenic Δodc clones, the Δodc1 and Δodc2 clones, were chosen for further analysis, and these knockout lines were routinely maintained in growth medium with 200 μM putrescine but without hygromycin or G418.
The Δodc null mutation was then functionally complemented in both the Δodc1 and Δodc2 lines by transfection with a chimeric vector containing the full-length L. donovani ODC open reading frame ligated into the pXG-BSD leishmanial expression plasmid (25). These lines were selected in 20 μg/ml blasticidin in the absence of added polyamine and were designated the Δodc1[pXG-BSD-ODC] and Δodc2[pXG-BSD-ODC] lines, respectively. The Δodc1[pXG-BSD-ODC] and Δodc2[pXG-BSD-ODC] “add-backs” were employed as positive controls in virtually all of the experiments in this investigation.
Southern and Western blot analyses.
Genomic DNA from wild-type and Δodc parasites was prepared for Southern blot analysis using the DNAeasy kit (Qiagen Inc., Valencia, CA) according to the manufacturer's protocol. DNA was digested with SalI and probed with a 1.0-kb fragment of the ODC coding region using highly stringent hybridization conditions.
Parasite lysates from exponentially growing cell populations were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (35) and blotted onto either nitrocellulose or Nytran membranes (Schleicher and Schuell, Keene, NH), and Western blot analysis was performed according to standardized procedures (53). The membranes were probed with polyclonal antibodies raised to the L. donovani ODC, ADOMETDC, and SPDSYN (31, 46-48), with commercially available anti-α-tubulin mouse monoclonal antibody (DM1A) (Calbiochem., La Jolla, CA), or with antisera to the L. donovani A2 protein (59), which was generously furnished to the Ullman laboratory by Greg Matlashewski (McGill University, Montreal, CA).
Infection of peritoneal macrophages.
Macrophages were harvested 5 days after intraperitoneal injection of 1.2 ml of 3% Brewer thioglycolate into BALB/c mice. Peritoneal macrophages (2.0 × 105) were then affixed to four-well Lab-TekII chamber slides (Nalge Nunc International Corp., Rochester, NY) in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 4 mM l-glutamine, 1.5 g/liter sodium bicarbonate, and 4.5 g/liter glucose and subsequently infected with a 10-fold excess of stationary-phase parasites. After 12 h, residual extracellular promastigotes were removed by gently washing the adherent macrophages three times with phosphate-buffered saline. Cells were then rinsed and growth medium changed on a daily basis. After 72 h, intracellular amastigotes were identified after staining of parasite-infected macrophages by using the Diff-Quik kit and viewed on a Zeiss Axiovert 200 M microscope using a ×60 oil immersion lens. Stained parasites and cells were photographed with an AxioCam MR camera, followed by parasite and macrophage enumeration. For studies involving DFMO, peritoneal macrophages were processed as described above except that after the initial 12 h of infection, the growth medium was replaced with medium containing 50 μM DFMO and the macrophages and parasites were allowed to incubate for an additional 72 h before staining and photographing.
Infectivity studies with mice.
Groups of four female BALB/c mice (Charles River Laboratories, Wilmington, MA) were each inoculated by tail vein injection with 5 × 106 of either wild-type, Δodc1, Δodc1[pXG-BSD-ODC], Δodc2, or Δodc2[pXG-BSD-ODC] stationary-phase promastigotes. Prior to the mouse inoculations, each of the five strains employed in this experiment was also passaged once through mice for 12 days in order to eliminate parasites that had become attenuated in response to prolonged culture. Livers and spleens were harvested 4 weeks afterward as described previously (13). Single-cell suspensions from mouse organs were prepared by passage through a 70-μm cell strainer (BD Falcon), and parasitemias were then determined in 96-well microtiter plates using a standardized limiting-dilution assay (13).
RESULTS
Expression of polyamine biosynthetic enzymes in L. donovani promastigotes and axenic amastigotes.
L. donovani promastigotes are polyamine prototrophs and express the entire complement of polyamine biosynthetic enzymes (31, 47-49). However, little is known about polyamine biosynthesis in amastigotes. As a prelude to examining the consequences of a genetic loss of polyamine biosynthesis for the ability of amastigotes to propagate within animals, the expression levels of polyamine biosynthetic proteins in promastigotes and axenic amastigotes were compared. Western blots of fractionated L. donovani lysates probed with monospecific polyclonal antisera raised to ODC, ADOMETDC, and SPDSYN revealed that all three enzymes are expressed in axenic amastigotes at levels equivalent to those observed in promastigotes (Fig. 1). These lysates were also probed with antibodies to A2, an amastigote-specific antigen (59), in order to establish authenticity of the axenic amastigote culture, and with a commercial monoclonal antibody to tubulin, a protein that is not expressed in a stage-specific fashion, as the loading control. The robust and comparable expression of ODC, ADOMETDC, and SPDSYN in both promastigotes and axenic amastigotes demonstrated that amastigotes express the requisite enzymatic machinery to convert ornithine into putrescine and spermidine.
FIG. 1.

Expression of polyamine biosynthetic pathway proteins in L. donovani promastigotes and amastigotes. Lysates of wild-type L. donovani promastigotes (Pro) and axenic amastigotes (Am) were prepared from exponentially growing parasites and equal amounts of protein from both extracts fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After blotting onto nitrocellulose filters was done, the blots were probed with antibodies to ODC (A), ADOMETDC (B), SPDSYN (C), A2, an amastigote-specific protein (59) (D), or tubulin (E). The sources of these antibodies have been reported (46, 59). Molecular mass markers are indicated to the left of each blot.
Molecular characterization of LdBob Δodc lines.
Although we have previously reported the phenotypic characterization of Δodc L. donovani promastigotes, this analysis was undertaken with an avirulent L. donovani strain (31). Thus, the Δodc lesion was recreated in LdBob, a virulent L. donovani strain that is capable of establishing visceralizing infections in mice (19, 26). Southern blot analysis of genomic DNA prepared from wild-type and mutant parasites clearly demonstrated the allelic rearrangements that triggered the loss of ODC coding sequences in the Δodc1 and Δodc2 clones (Fig. 2A). Ethidium bromide staining indicated equal loading of chromosomal DNA on all three lanes of the Southern blot (Fig. 2B). Loss of the wild-type ODC locus in the two null mutants was corroborated by Western blot analysis of lysates from wild-type, Δodc1, and Δodc2 parasites (Fig. 2C). Polyclonal antibodies recognized an ∼85-kDa protein, the size of ODC predicted from translation of the ODC open reading frame (28), in wild-type, Δodc1[pXG-BSD-ODC], and Δodc2[pXG-BSD-ODC] cell lysates, and this band was absent in extracts prepared from both Δodc1 and Δodc2 parasites. Simultaneous probing of the immunoblot with antibodies to tubulin ensured that comparable amounts of parasite extract were loaded onto each lane of the gel.
FIG. 2.

Molecular characterization of the ODC locus and ODC expression in the Δodc knockouts. Genomic DNA from wild-type, Δodc1, and Δodc2 parasites and protein lysates from wild-type, Δodc1, Δodc2, Δodc1[pXG-BSD-ODC], and Δodc2[pXG-BSD-ODC] parasites were obtained by standard protocols. (A) 2 μg of genomic DNA from wild-type, Δodc1, or Δodc2 parasites was digested with SalI and hybridized to a 1.0-kb fragment of the ODC coding region that was prepared by PCR. Molecular size markers are indicated to the left. (B) Ethidium bromide-stained gel that was employed in the Southern blot in panel A. (C) Cell extracts prepared from 1.0 × 107 wild-type, Δodc1, Δodc2, Δodc1[pXG-BSD-ODC] (Δodc1[pODC]), or Δodc[pXG-BSD-ODC] (Δodc2[pODC]) promastigotes were fractionated by sodium dodecyl sulfate electrophoresis and the blots probed with polyclonal antibodies against L. donovani ODC and a commercial monoclonal antibody that recognizes tubulin. The tubulin antibody was employed to verify equivalent loading of protein on all lanes.
Nutritional phenotypes of LdBob Δodc parasites.
As anticipated, Δodc1 and Δodc2 promastigotes exhibited polyamine auxotrophy and, unlike wild-type parasites, could not proliferate in polyamine-deficient medium (Fig. 3). This polyamine auxotrophy could be circumvented genetically by complementation with a covering ODC plasmid or pharmacologically by supplying either putrescine (Fig. 3) or spermidine (data not shown) in the culture medium. The growth phenotypes of axenic amastigote forms of the two Δodc L. donovani lines mirrored that of the promastigotes. The Δodc1 and Δodc2 axenic amastigotes were auxotrophic for polyamines, and the polyamine auxotrophy could be circumvented by supplementation of the culture medium with putrescine. Wild-type, Δodc1[pXG-BSD-ODC], and Δodc2[pXG-BSD-ODC] axenic amastigotes, as expected, grew robustly in polyamine-deficient medium (Fig. 3).
FIG. 3.

Growth phenotypes of Δodc promastigotes and axenic amastigotes. Wild-type, Δodc1, Δodc1[pXG-BSD-ODC] (Δodc1[pODC]), Δodc2, and Δodc2[pXG-BSD-ODC] (Δodc2[pODC]) promastigotes (A) and axenic amastigotes (B) were each incubated in their respective growth media in the absence or presence of 200 μM putrescine. Parasites were enumerated after 5 days by using a hemocytometer. Promastigotes were maintained at pH 7.4 and 28°C, while axenic amastigotes were cultured at pH 5.5 and 37°C.
Survival of Δodc parasites in peritoneal macrophages.
To examine the role of parasite ODC on intracellular amastigote survival and proliferation, the abilities of wild-type, Δodc1, Δodc1[pXG-BSD-ODC], Δodc2, and Δodc2[pXG-BSD-ODC] parasites to sustain an infection in peritoneal macrophages were assessed. These experiments demonstrated that parasite burdens of macrophages infected with the two Δodc mutants after 3 days were ∼20% of that of wild-type parasites, and this reduction in parasitemia was not observed with the episomally complemented Δodc1[pXG-BSD-ODC] and Δodc2[pXG-BSD-ODC] transfectants (Fig. 4). After a 16-h incubation period, however, parasite numbers in macrophages were equivalent for all strains (∼7 amastigotes/macrophage), indicating that the Δodc lesion did not affect parasite attachment, internalization, or transformation to the amastigote forms (data not shown).
FIG. 4.
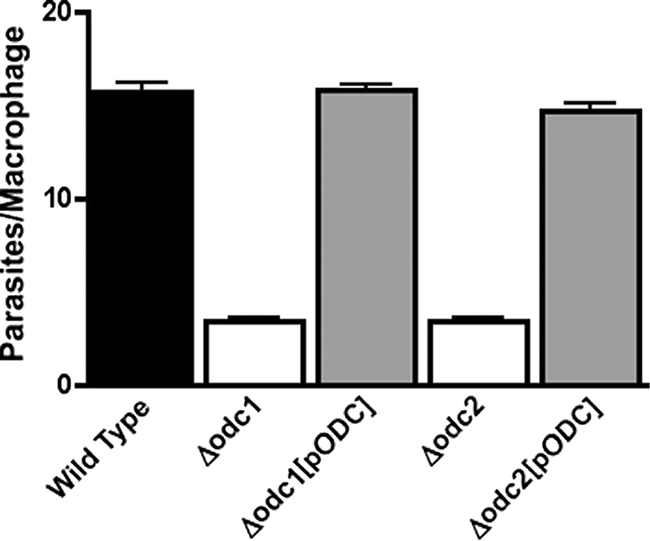
Survival of Δodc L. donovani in peritoneal murine macrophages. Peritoneal macrophages were infected with either wild-type, Δodc1, Δodc1[pXG-BSD-ODC] (Δodc1[pODC]), Δodc2, or Δodc2[pXG-BSD-ODC] (Δodc2[pODC]) stationary-phase promastigotes at a parasite/macrophage ratio of 10:1 as described in Materials and Methods. Cells were stained after 72 h and macrophages and amastigotes enumerated visually. The results are the averages and ranges of two experiments that were each performed in triplicate (n = 6).
To test whether DFMO, the suicide inhibitor of ODC (37), could pharmacologically simulate the effects of ODC deficiency on amastigote viability, DFMO was evaluated as an antiparasitic agent against both axenic and tissue culture amastigotes. DFMO was toxic toward axenic amastigotes of wild-type L. donovani, with a 50% effective concentration of ∼40 μM (Fig. 5A), a value similar to that reported for promastigotes (28, 32). DFMO toxicity was abrogated in the presence of 200 μM putrescine, since axenic amastigotes were completely refractory to 500 μM DFMO. DFMO was not tested against either of the two Δodc knockout lines, because these strains do not replicate efficiently in macrophages in the absence of putrescine (Fig. 4) and lack the pharmacologic target of DFMO, or the “add-back” lines, which are refractory to DFMO because of ODC overproduction (46).
FIG. 5.

Effects of DFMO on L. donovani axenic and tissue culture amastigotes. (A) Wild-type axenic amastigotes were seeded at 5 × 104/100 μl in various concentrations of DFMO in the absence (▪) or presence (▴) of 200 μM putrescine. Parasites were counted after 96 h by using a hemacytometer and plotted as a percentage of control growth in the absence of DFMO as a function of DFMO concentration. (B) Culture of peritoneal macrophages 96 h after initial exposure to wild-type L. donovani at a ratio of 10 promastigotes per macrophage. (C) Peritoneal macrophages infected with parasites as for panel B but treated with 50 μM DFMO. (D) Uninfected macrophages treated with 50 μM DFMO. Cells were stained and examined under the microscope after 96 h as described in Materials and Methods. These experiments were repeated one additional time with equivalent results.
In order to demonstrate DFMO efficacy on intracellular amastigotes, peritoneal macrophages infected with wild-type L. donovani were exposed to DFMO. While the parasite infection obliterated the untreated macrophage culture after 96 h, 50 μM DFMO protected the macrophage culture from destruction, and no parasites were observed by visual inspection (Fig. 5B and C) after 96 h. A control experiment demonstrated that 50 μM DMFO by itself had no obvious deleterious effect on the primary macrophages that had not been exposed to parasites (Fig. 5D).
In vivo infections with Δodc L. donovani.
To determine the effects of a genetic deficiency of parasite ODC on virulence in an animal model, parasite burdens in both liver and spleen tissue were assessed for BALB/c mice injected with either wild-type, Δodc1, Δodc1[pXG-BSD-ODC], Δodc2, or Δodc2[pXG-BSD-ODC] cells (Fig. 6). The abilities of both the Δodc1 and Δodc2 parasites to infect mice were severely diminished, and the virulence defect was rescued by episomal complementation (Fig. 6). The average liver and spleen parasite loads of mice infected with wild-type parasites was ∼2 × 108 parasites/g and 2 × 105 parasites/g, respectively (Fig. 6A and B). It should be noted that the absolute numbers of wild-type parasites in liver and spleen on a per-gram basis differed by 3 orders of magnitude, a discrepancy typically observed for L. donovani infections (11, 21, 33, 41). In contrast, parasitemias of mice infected with Δodc1 or Δodc2 parasites 4 weeks postinfection were ∼102 parasites/g liver and ∼2 × 102 to 3 × 102 parasites/g spleen, respectively. Parasitemias in livers and spleens of mice infected with the Δodc1[pXG-BSD-ODC] “add-back” parasites were restored essentially to wild-type levels, whereas the parasite burdens in livers and spleens of mice infected with the Δodc2[pXG-BSD-ODC] complemented line were also significantly higher than those with the uncomplemented parental knockout parasites but did not reach wild-type levels (Fig. 6A and B). These data were also corroborated in a separate experiment in which parasite burdens were ascertained by enumeration of amastigotes in Giemsa-stained liver slices. Strikingly, no parasites were detected in all six of the mice infected with the Δodc1 parasites in this mouse experiment (data not shown).
FIG. 6.
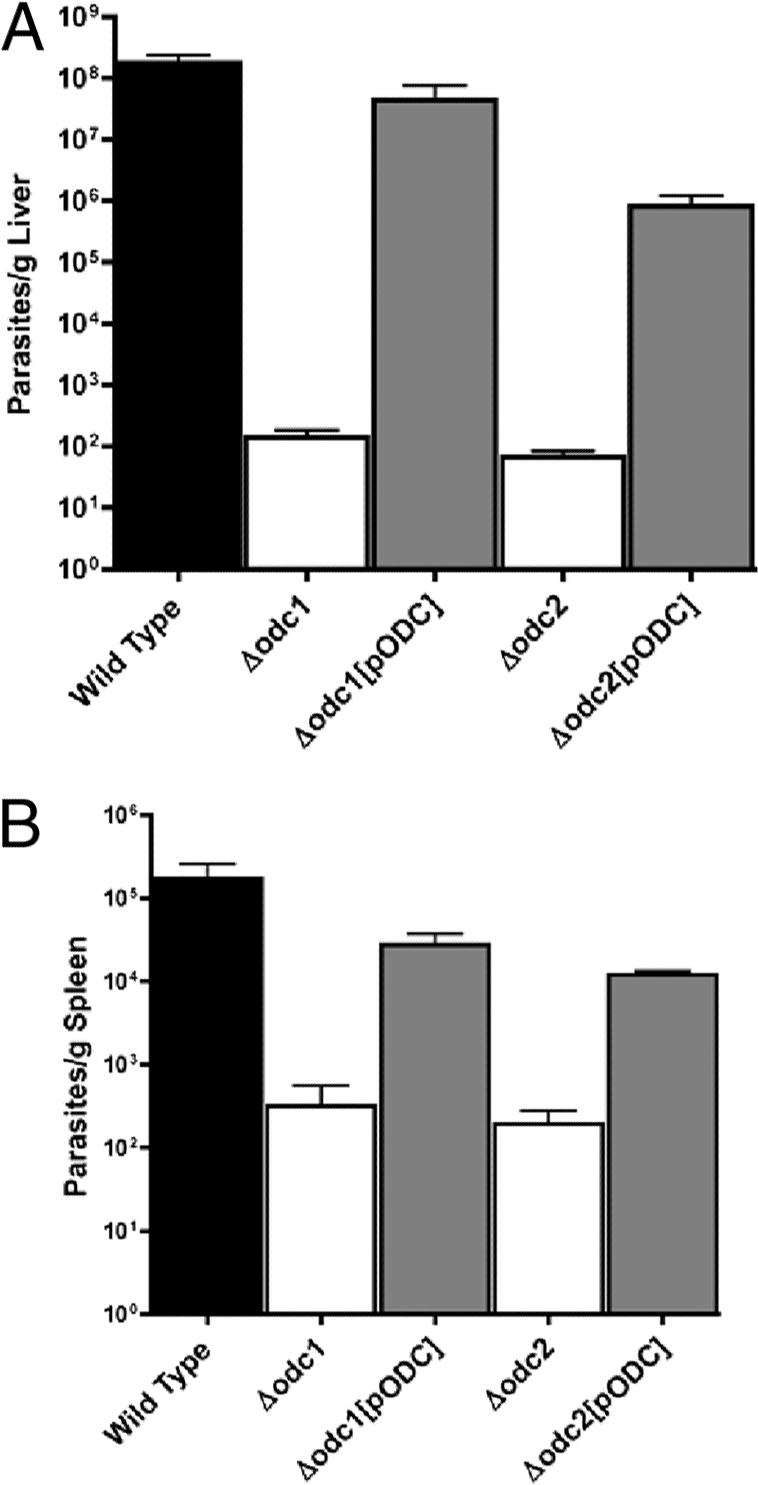
Parasite burdens in livers and spleens of mice infected with wild-type, Δodc, and “add-back” parasites. (A and B) Five separate groups of BALB/c mice were infected with either wild-type, Δodc1, Δodc1[pXG-BSD-ODC] (Δodc1[pODC]), Δodc2, or Δodc2[pXG-BSD-ODC] (Δodc2[pODC]) stationary-phase promastigotes as described in Materials and Methods. Mice were sacrificed after 4 weeks and parasite loads in liver (A) or spleen (B) preparations determined by limiting dilution.
DISCUSSION
Previous studies have established ODC as a valid therapeutic target in Leishmania promastigotes (32, 34, 39, 45, 50), but the role of ODC has not been definitively established for amastigotes, the human infectious form of the parasite. The demonstration that two independent Δodc lines were severely compromised in their abilities to establish infections in BALB/c mice provides powerful genetic evidence that ODC plays an essential housekeeping function in the amastigote stage of the parasite. The average liver and spleen parasite loads of mice infected with Δodc1 or Δodc2 parasites, as assessed by limiting dilution, were 6 and 3 orders of magnitude lower, respectively, than those of wild-type parasites (Fig. 6).
That the extremely attenuated phenotype of the Δodc1 or Δodc2 parasites can be ascribed to the allelic replacements at the ODC locus and not to secondary mutations was genetically confirmed by episomal rescue of the genetic lesion. Two separate “add-back” lines exhibited dramatically increased capacities, compared to the knockout parental parasites, to infect mouse organs at levels close to those obtained with wild-type parasites (Fig. 6 and data not shown). Whether the small disparities in parasite loads that were obtained between the two lines harboring ODC-containing episomes and wild-type parasites could be ascribed to plasmid stability, levels of ODC expression, or simply experimental variation cannot be known. Because a loss of extrachromosomal DNAs, such as plasmids, occurs in Leishmania in the absence of selective pressure (7, 12, 16, 56), the rate of plasmid elimination and the level of ODC expression in rodent experiments are impossible to control.
Although no parasites were detected in the liver samples of any of the six mice infected with the Δodc1 parasites when they were analyzed by visual inspection (data not shown), a separate experiment showed that each liver and spleen of mice injected with either Δodc1 or Δodc2 promastigotes continued to harbor several hundred persistent parasites 4 weeks postinoculation (Fig. 6A and B). Whether these residual parasites persisted by accessing residual pools of exogenous polyamines available in the phagolysosome or simply endured polyamine starvation conditions for the 4-week experiment and were in the process of disappearing cannot be easily addressed using the mouse system, since BALB/c mice naturally clear L. donovani infections from the liver but small numbers of parasites can persist in the spleen over the lifetime of the animal (11, 21, 58).
The strikingly compromised virulence phenotype of Δodc L. donovani in comparison to that of wild-type parasites was surprising given that previous experiments by our group have demonstrated that Δarg L. mexicana promastigotes that are also auxotrophic for polyamines are still capable of sustaining a cutaneous infection in mice, even though the lesions develop more slowly than in mice infected with wild-type or Δarg[pARG] “add-back” parasites (22). The remarkable discrepancy in the abilities of Δodc L. donovani and Δarg L. mexicana to establish infections in mice could be ascribed to species (L. mexicana versus L. donovani) or host milieu (cutaneous versus visceral) variation or to the specific nature of the genetic lesion within the polyamine pathway (Δarg versus Δodc). Because Keithly and Fairlamb found that DFMO is effective against the visceral strains, L. donovani and L. chagasi, but not against the cutaneous species, L. major and L. mexicana (34), these preliminary data insinuate that the difference in infectivity may be due to species or host milieu differences. From a metabolic perspective, ARG deficiency in Leishmania can be rescued by either ornithine or putrescine (49), whereas a Δodc lesion can be circumvented only by exogenous putrescine (31). It is certainly possible that variations exist in the ornithine and putrescine pools within the phagolysosomes of macrophages in the skin (Langerhans cells, skin dendritic cells, and histiocytes) or liver (Kupffer cells, parenchymal cells, and nonresident macrophages) and spleen (splenic macrophages), in which L. mexicana or L. donovani resides, respectively, and could account for the virulence disparity observed between Δarg L. mexicana and Δodc L. donovani. Differential ornithine and putrescine uptake capacities of L. mexicana and L. donovani could also contribute to virulence differences between the two knockout lines.
The enormous diminution in parasite burdens conferred by loss of ODC is mirrored by the infectivity differences observed between wild-type and Δodc parasites in murine peritoneal macrophages. Parasite loads in macrophages infected with the Δodc1 or Δodc2 null mutants were ∼80% reduced from those obtained in macrophages exposed to wild-type L. donovani (Fig. 4). The infectivity deficit with each knockout was eliminated by complementation, since parasite loads in macrophages 3 days post-infection with either wild-type or Δodc[pXG-BSD-ODC] parasites were equivalent (Fig. 4). Although the differences in the parasite loads observed between wild-type and Δodc parasites in macrophages is much smaller than those observed in mice, the macrophage and mouse experiments are performed using quite disparate experimental paradigms. In particular, polyamine starvation conferred by the Δodc mutation is likely to require more time than the duration of the time-limited (72-h) macrophage experiment to fully manifest itself as a growth impediment. Furthermore, macrophages, unlike mice, do not clear infections by wild-type parasites and eventually die (Fig. 5B), and therefore, in vitro infections are limited by experimental constraints.
Although ODC is a validated target for African trypanosomiasis, it has been generally alleged that ODC is not a target for leishmaniasis, presumably due to the existence of salvageable polyamines in the host phagolysosome to which the parasites would have access. DFMO toxicity toward promastigotes of several Leishmania species is well documented (32, 34, 39, 45). Furthermore, ODC is the sole target for DFMO action in L. donovani, since DFMO toxicity is completely circumvented by pharmacological supplementation of the growth medium with putrescine (32) or by overexpression of ODC from an episome (46). However, several review articles have asserted that the amastigote stage of the parasite is generally refractory to DFMO (17, 36). Yet DFMO has shown promise against experimental Leishmania infections in mice and hamsters, although in none of these experimental infections did DFMO achieve a cure. However, these rodent studies were preliminary and the time courses of drug administration limited (27, 34, 40). Our investigations established that a Δodc lesion radically impaired the ability of L. donovani to sustain an infection in BALB/c mice and revealed that genetic or pharmacological impairment of ODC significantly lowered parasite loads in macrophages in vitro. These findings imply, contrary to previous speculations (17, 36), that putrescine is either not present in the phagolysosome or at least not found in sufficient amounts to sustain a leishmanial infection when de novo polyamine production is obliterated. Whether genetic lesions in other polyamine biosynthetic enzymes, ADOMETDC and SPDSYN, also negatively impact the ability of L. donovani to colonize macrophages and establish infections in rodents remains to be examined. This will be tested by reconstruction of the Δadometdc and Δspdsyn lesions that have already been generated in the avirulent DI700 L. donovani strain (47, 48) with the wild-type LdBob virulence background.
It is important to note that although these studies bolster ODC as a potential drug target for treating leishmaniasis, DFMO itself, the suicide inhibitor of ODC, is an unlikely “lead” candidate drug. DFMO is expensive to synthesize, binds only weakly to parasite ODC proteins (Ki values for the T. brucei and L. donovani ODCs are 130 μM and 170 μM, respectively) (43), is needed in hectogram amounts to be effective against T. brucei infections, requires prolonged and multiple intravenous administrations in a hospital setting, and exhibits poor pharmacokinetics (20, 42, 44, 57). Nevertheless, our findings implicated ODC as a valid drug target for treating visceral leishmaniasis and will hopefully spur the development of other pharmacological inhibitors of ODC in Leishmania.
Acknowledgments
This work was supported in part by Public Health Service Grant AI 41622 from the National Institute of Allergy and Infectious Diseases.
We thank Nicola S. Carter for her critical reading of the manuscript prior to submission. We thank Yani Chen and Bayan Sudan for their technical expertise with some of the mouse experiments.
We have no financial conflicts of interest in this work.
Editor: W. A. Petri, Jr.
Footnotes
Published ahead of print on 8 December 2008.
REFERENCES
- 1.Assaraf, Y. G., J. Golenser, D. T. Spira, G. Messer, and U. Bachrach. 1987. Cytostatic effect of DL-alpha-difluoromethylornithine against Plasmodium falciparum and its reversal by diamines and spermidine. Parasitol. Res. 73313-318. [DOI] [PubMed] [Google Scholar]
- 2.Bacchi, C. J., and P. P. McCann. 1987. Parasitic protozoa and polyamines, p. 317-344. In P. P. McCann, A. E. Pegg, and A. Sjoerdsma (ed.), Inhibition of polyamine metabolism: biological significance and basis for new therapies. Academic Press, Orlando, FL.
- 3.Bacchi, C. J., H. C. Nathan, N. Yarlett, B. Goldberg, P. P. McCann, A. J. Bitonti, and A. Sjoerdsma. 1992. Cure of murine Trypanosoma brucei rhodesiense infections with an S-adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 362736-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacchi, C. J., and N. Yarlett. 1995. Polyamine metabolism. In J. J. Marr and M. Muller (ed.), Biochemistry and molecular biology of parasites, p. 119-131. Academic Press Ltd., London, United Kingdom.
- 5.Bachrach, U. 2005. Naturally occurring polyamines: interaction with macromolecules. Curr. Protein Pept. Sci. 6559-566. [DOI] [PubMed] [Google Scholar]
- 6.Basselin, M., G. H. Coombs, and M. P. Barrett. 2000. Putrescine and spermidine transport in Leishmania. Mol. Biochem. Parasitol. 10937-46. [DOI] [PubMed] [Google Scholar]
- 7.Beverley, S. M. 1991. Gene amplification in Leishmania. Annu. Rev. Microbiol. 45417-444. [DOI] [PubMed] [Google Scholar]
- 8.Bitonti, A. J., T. L. Byers, T. L. Bush, P. J. Casara, C. J. Bacchi, A. B. Clarkson, Jr., P. P. McCann, and A. Sjoerdsma. 1990. Cure of Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense infections in mice with an irreversible inhibitor of S-adenosylmethionine decarboxylase. Antimicrob. Agents Chemother. 341485-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bitonti, A. J., J. A. Dumont, and P. P. McCann. 1986. Characterization of Trypanosoma brucei brucei S-adenosyl-L-methionine decarboxylase and its inhibition by Berenil, pentamidine and methylglyoxal bis(guanylhydrazone). Biochem. J. 237685-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bitonti, A. J., P. P. McCann, and A. Sjoerdsma. 1987. Plasmodium falciparum and Plasmodium berghei: effects of ornithine decarboxylase inhibitors on erythrocytic schizogony. Exp. Parasitol. 64237-243. [DOI] [PubMed] [Google Scholar]
- 11.Blackwell, J. M. 1996. Genetic susceptibility to leishmanial infections: studies in mice and man. Parasitology 112 (Suppl.)S67-S74. [PubMed] [Google Scholar]
- 12.Boucher, N., F. McNicoll, M. Laverdiere, A. Rochette, M. N. Chou, and B. Papadopoulou. 2004. The ribosomal RNA gene promoter and adjacent cis-acting DNA sequences govern plasmid DNA partitioning and stable inheritance in the parasitic protozoan Leishmania. Nucleic Acids Res. 322925-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buffet, P. A., A. Sulahian, Y. J. Garin, N. Nassar, and F. Derouin. 1995. Culture microtitration: a sensitive method for quantifying Leishmania infantum in tissues of infected mice. Antimicrob. Agents Chemother. 392167-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burri, C., and R. Brun. 2003. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 90(Supp. 1)S49-S52. [DOI] [PubMed] [Google Scholar]
- 15.Chang, K. P., R. F. Steiger, C. Dave, and Y. C. Cheng. 1978. Effects of methylglyoxal bis(ganylhydrazone) on trypanosomatid flagellates: inhibition of growth and nucleoside incorporation in Trypanosoma brucei. J. Protozool. 25145-149. [DOI] [PubMed] [Google Scholar]
- 16.Clayton, C. E. 1999. Genetic manipulation of kinetoplastida. Parasitol. Today 15372-378. [DOI] [PubMed] [Google Scholar]
- 17.Cohen, S. S. 1998. A guide to the polyamines. Oxford University Press, Oxford, United Kingdom.
- 18.Danzin, C., P. Marchal, and P. Casara. 1990. Irreversible inhibition of rat S-adenosylmethionine decarboxylase by 5′-([(Z)-4-amino-2-butenyl]methylamino)-5′-deoxyadenosine. Biochem. Pharmacol. 401499-1503. [DOI] [PubMed] [Google Scholar]
- 19.Debrabant, A., M. B. Joshi, P. F. Pimenta, and D. M. Dwyer. 2004. Generation of Leishmania donovani axenic amastigotes: their growth and biological characteristics. Int. J. Parasitol. 34205-217. [DOI] [PubMed] [Google Scholar]
- 20.Docampo, R., and S. N. Moreno. 2003. Current chemotherapy of human African trypanosomiasis. Parasitol. Res. 90(Supp. 1)S10-S13. [DOI] [PubMed] [Google Scholar]
- 21.Garg, R., and A. Dube. 2006. Animal models for vaccine studies for visceral leishmaniasis. Indian J. Med. Res. 123439-454. [PubMed] [Google Scholar]
- 22.Gaur, U., S. C. Roberts, R. P. Dalvi, I. Corraliza, B. Ullman, and M. E. Wilson. 2007. An effect of parasite-encoded arginase on the outcome of murine cutaneous leishmaniasis. J. Immunol. 1798446-8453. [DOI] [PubMed] [Google Scholar]
- 23.Geary, T. G., S. A. Edgar, and J. D. Jensen. 1986. Drug resistance in protozoa, p. 209-236. In W. C. Campbell and R. S. Rew (ed.), Chemotherapy of parasitic diseases. Plenum Press, New York, NY.
- 24.Gillin, F. D., D. S. Reiner, and P. P. McCann. 1984. Inhibition of growth of Giardia lamblia by difluoromethylornithine, a specific inhibitor of polyamine biosynthesis. J. Protozool. 31161-163. [DOI] [PubMed] [Google Scholar]
- 25.Goyard, S., and S. M. Beverley. 2000. Blasticidin resistance: a new independent marker for stable transfection of Leishmania. Mol. Biochem. Parasitol. 108249-252. [DOI] [PubMed] [Google Scholar]
- 26.Goyard, S., H. Segawa, J. Gordon, M. Showalter, R. Duncan, S. J. Turco, and S. M. Beverley. 2003. An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol. Biochem. Parasitol. 13031-42. [DOI] [PubMed] [Google Scholar]
- 27.Gradoni, L., M. A. Iorio, M. Gramiccia, and S. Orsini. 1989. In vivo effect of eflornithine (DFMO) and some related compounds on Leishmania infantum preliminary communication. Farmaco 441157-1166. [PubMed] [Google Scholar]
- 28.Hanson, S., J. Adelman, and B. Ullman. 1992. Amplification and molecular cloning of the ornithine decarboxylase gene of Leishmania donovani. J. Biol. Chem. 2672350-2359. [PubMed] [Google Scholar]
- 29.Janne, J., L. Alhonen, T. A. Keinanen, M. Pietila, A. Uimari, E. Pirinen, M. T. Hyvonen, and A. Jarvinen. 2005. Animal disease models generated by genetic engineering of polyamine metabolism. J. Cell Mol. Med. 9865-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janne, J., L. Alhonen, M. Pietila, and T. A. Keinanen. 2004. Genetic approaches to the cellular functions of polyamines in mammals. Eur. J. Biochem. 271877-894. [DOI] [PubMed] [Google Scholar]
- 31.Jiang, Y., S. C. Roberts, A. Jardim, N. S. Carter, S. Shih, M. Ariyanayagam, A. H. Fairlamb, and B. Ullman. 1999. Ornithine decarboxylase gene deletion mutants of Leishmania donovani. J. Biol. Chem. 2743781-3788. [DOI] [PubMed] [Google Scholar]
- 32.Kaur, K., K. Emmett, P. P. McCann, A. Sjoerdsma, and B. Ullman. 1986. Effects of DL-alpha-difluoromethylornithine on Leishmania donovani promastigotes. J. Protozool. 33518-521. [DOI] [PubMed] [Google Scholar]
- 33.Kaye, P. M., P. Gorak, M. Murphy, and S. Ross. 1995. Strategies for immune intervention in visceral leishmaniasis. Ann. Trop. Med. Parasitol. 89(Suppl. 1)75-81. [DOI] [PubMed] [Google Scholar]
- 34.Keithly, J. S., and A. H. Fairlamb. 1987. Inhibition of Leishmania species by alpha-difluoromethylornithine, p. 749-756. In D. T. Hart (ed.), Leishmaniasis: the current status and new strategies for control, vol. 163. Plenum Press, New York, NY. [Google Scholar]
- 35.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 36.McCann, P., C. J. Bacchi, W. L. Hanson, G. D. Cain, H. C. Nathan, S. H. Hutner, and A. Sjoerdsma. 1981. Effect on parasitic protozoa of alpha-difluoromethylornithine, an inhibitor of ornithine decarboxylase. Adv. Polyamine Res. 397-110. [Google Scholar]
- 37.Metcalf, B. W., P. Bey, C. Danzin, M. J. Jung, P. Casara, and J. P. Vevert. 1978. Catalytic irreversible inhibition of mammalian ornithine decarboxylase (E.C. 4.1.1.17) by substrate and product analogues. J. Am. Chem. Soc. 1002551-2553. [Google Scholar]
- 38.Mishra, J., A. Saxena, and S. Singh. 2007. Chemotherapy of leishmaniasis: past, present and future. Curr. Med. Chem. 141153-1169. [DOI] [PubMed] [Google Scholar]
- 39.Mukhopadhyay, R., P. Kapoor, and R. Madhubala. 1996. Characterization of alpha-difluoromethylornithine resistant Leishmania donovani and its susceptibility to other inhibitors of the polyamine biosynthetic pathway. Pharmacol. Res. 3443-46. [DOI] [PubMed] [Google Scholar]
- 40.Mukhopadhyay, R., and R. Madhubala. 1993. Effect of a bis(benzyl)polyamine analogue, and DL-alpha-difluoromethylornithine on parasite suppression and cellular polyamine levels in golden hamster during Leishmania donovani infection. Pharmacol. Res. 28359-365. [DOI] [PubMed] [Google Scholar]
- 41.Mullen, A. B., A. J. Baillie, and K. C. Carter. 1998. Visceral leishmaniasis in the BALB/c mouse: a comparison of the efficacy of a nonionic surfactant formulation of sodium stibogluconate with those of three proprietary formulations of amphotericin B. Antimicrob. Agents Chemother. 422722-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Na-Bangchang, K., F. Doua, J. Konsil, W. Hanpitakpong, B. Kamanikom, and F. Kuzoe. 2004. The pharmacokinetics of eflornithine (alpha-difluoromethylornithine) in patients with late-stage T.b. gambiense sleeping sickness. Eur. J. Clin. Pharmacol. 60269-278. [DOI] [PubMed] [Google Scholar]
- 43.Osterman, A., N. V. Grishin, L. N. Kinch, and M. A. Phillips. 1994. Formation of functional cross-species heterodimers of ornithine decarboxylase. Biochemistry 3313662-13667. [DOI] [PubMed] [Google Scholar]
- 44.Pepin, J., and F. Milord. 1994. The treatment of human African trypanosomiasis. Adv. Parasitol. 331-47. [DOI] [PubMed] [Google Scholar]
- 45.Reguera, R. M., R. B. Fouce, J. C. Cubria, M. L. Bujidos, and D. Ordonez. 1995. Fluorinated analogues of L-ornithine are powerful inhibitors of ornithine decarboxylase and cell growth of Leishmania infantum promastigotes. Life Sci. 56223-230. [DOI] [PubMed] [Google Scholar]
- 46.Roberts, S. C., Y. Jiang, J. Gasteier, B. Frydman, L. J. Marton, O. Heby, and B. Ullman. 2007. Leishmania donovani polyamine biosynthetic enzyme overproducers as tools to investigate the mode of action of cytotoxic polyamine analogs. Antimicrob. Agents Chemother. 51438-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts, S. C., Y. Jiang, A. Jardim, N. S. Carter, O. Heby, and B. Ullman. 2001. Genetic analysis of spermidine synthase from Leishmania donovani. Mol. Biochem. Parasitol. 115217-226. [DOI] [PubMed] [Google Scholar]
- 48.Roberts, S. C., J. Scott, J. E. Gasteier, Y. Jiang, B. Brooks, A. Jardim, N. S. Carter, O. Heby, and B. Ullman. 2002. S-adenosylmethionine decarboxylase from Leishmania donovani. Molecular, genetic, and biochemical characterization of null mutants and overproducers. J. Biol. Chem. 2775902-5909. [DOI] [PubMed] [Google Scholar]
- 49.Roberts, S. C., M. J. Tancer, M. R. Polinsky, K. M. Gibson, O. Heby, and B. Ullman. 2004. Arginase plays a pivotal role in polyamine precursor metabolism in Leishmania. Characterization of gene deletion mutants. J. Biol. Chem. 27923668-23678. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez, C. P., J. Mucci, N. S. Gonzalez, A. Ochoa, M. M. Zakin, and I. D. Algranati. 1997. Alpha-difluoromethylornithine-resistant cell lines obtained after one-step selection of Leishmania mexicana promastigote cultures. Biochem. J. 324847-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schechter, P. J., J. L. R. Barlow, and A. Sjoerdsma. 1987. Clinical aspects of inhibition of ornithine decarboxylase with emphasis on therapeutic trials of eflornithine (DFMO) in cancer and protozoan diseases, p. 345-364. In P. P. McCann, A. E. Pegg, and A. Sjoerdsma (ed.), Inhibition of polyamine metabolism: biological significance and basis for new therapies. Academic Press, Orlando, FL.
- 52.Tabor, C. W., and H. Tabor. 1984. Polyamines. Annu. Rev. Biochem. 53749-790. [DOI] [PubMed] [Google Scholar]
- 53.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 764350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ulrich, P., R. W. Grady, and A. Cerami. 1982. The trypanocidal activity of various aromatic bisguanylhydrazones in vivo. Drug Dev. Res. 2219-228. [Google Scholar]
- 55.Van Nieuwenhove, S., P. J. Schechter, J. Declercq, G. Bone, J. Burke, and A. Sjoerdsma. 1985. Treatment of gambiense sleeping sickness in the Sudan with oral DFMO (DL-alpha-difluoromethylornithine), an inhibitor of ornithine decarboxylase; first field trial. Trans. R. Soc. Trop. Med. Hyg. 79692-698. [DOI] [PubMed] [Google Scholar]
- 56.Vergnes, B., D. Sereno, J. Tavares, A. Cordeiro-da-Silva, L. Vanhille, N. Madjidian-Sereno, D. Depoix, A. Monte-Alegre, and A. Ouaissi. 2005. Targeted disruption of cytosolic SIR2 deacetylase discloses its essential role in Leishmania survival and proliferation. Gene 36385-96. [DOI] [PubMed] [Google Scholar]
- 57.Wang, C. C. 1995. Molecular mechanisms and therapeutic approaches to the treatment of African trypanosomiasis. Annu. Rev. Pharmacol. Toxicol. 3593-127. [DOI] [PubMed] [Google Scholar]
- 58.Wilson, M. E., S. M. Jeronimo, and R. D. Pearson. 2005. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb. Pathog. 38147-160. [DOI] [PubMed] [Google Scholar]
- 59.Zhang, W. W., H. Charest, E. Ghedin, and G. Matlashewski. 1996. Identification and overexpression of the A2 amastigote-specific protein in Leishmania donovani. Mol. Biochem. Parasitol. 7879-90. [DOI] [PubMed] [Google Scholar]