

Abstract
Chlamydia pneumoniae infection has been associated with chronic obstructive airway disease (COPD), asthma, and atherosclerosis. Inflammation and airway remodeling in asthma and COPD result in subepithelial fibrosis that is characterized by the deposition of interstitial collagens and fibronectin. The progression of atherosclerosis is also accompanied by an increased production of interstitial collagens in the intima. As shown by reverse transcription-PCR and immunoblotting, infection of human fibroblasts and smooth muscle cells by C. pneumoniae TW-183 downregulated the expression of type I and III collagen and fibronectin, whereas the level of type IV collagen remained unchanged. Conditioned medium from infected fibroblasts as well as epithelial WISH cells also reduced the expression of interstitial collagens and fibronectin in uninfected cells. In experiments using blocking antibodies, beta interferon was found to contribute to the inhibitory effects of conditioned medium collected from infected fibroblasts. In contrast, downregulation of matrix protein expression by conditioned medium from epithelial cells was caused by interleukin-1α, which was not secreted from fibroblasts following chlamydial infection. C. pneumoniae-mediated inhibition of collagen and fibronectin expression was diminished following transfection of fibroblasts with specific small interfering RNA targeting the transcription factor CCAAT/enhancer-binding protein β. The downregulation of interstitial collagens and fibronectin by the Chlamydia-induced host cell cytokine response may modulate tissue remodeling processes in airway diseases. In atherosclerosis the inhibition of collagen synthesis by C. pneumoniae infection may promote plaque vulnerability, thereby increasing the risk of plaque rupture.
The association of Chlamydia pneumoniae, an intracellular bacterial pathogen, with chronic obstructive airway disease (COPD), asthma, and atherosclerosis was initially established by studies that described the presence of increased antibody titers in patients compared to healthy control persons (2, 14, 41). The discussion on a pathogenic role of C. pneumoniae in these diseases remains controversial. Differentiating a previous contact with the pathogen from an active infection by serological methods is frequently difficult (19). Although the presence of chlamydiae in the airways of asthmatics and COPD patients as well as in atherosclerotic plaques could be demonstrated by PCR, it has to be considered that PCR methods for the detection of C. pneumoniae are laboratory specific and that different PCR-based clinical studies produce contradictory results (4, 18, 43, 50). In some cases chlamydiae could be cultivated from respiratory specimens of patients with asthma and COPD as well as from atherosclerotic plaques (18, 52).
Mouse models have shown that C. pneumoniae infection may promote bronchial hyperresponsiveness, a central characteristic of asthma and COPD, and the formation and progression of atherosclerotic plaques; however, the precise mechanisms by which the pathogen may contribute to disease pathology are not fully understood (5, 8). Chlamydiae can obviously evade defense mechanisms of the host immune system by the inhibition of host cell apoptosis, suppression of antigen presentation pathways, and the ability to convert into persistent forms during their intracellular replication cycle (33). Doubtless, cases of recurrent C. pneumoniae-caused pharyngitis and pneumonia despite appropriate antibiotic therapy are due to the persistence of the pathogen, and chronic inflammation induced by cycles of persistence and productive infection might also explain the association with chronic airway diseases and atherosclerosis (25).
C. pneumoniae may contribute to COPD, asthma, and atherosclerosis by the induction and perpetuation of chronic inflammation via activation of a T cell-dependent immune response and induction of cytokine release from infected host cells. Chronic inflammation in COPD and asthma leads to remodeling processes associated with airway wall thickening, impaired lung function, and abnormal contraction to bronchospastic stimuli (10). In asthmatic patients airway remodeling is characterized by the formation of mucus plaques, hyperplasia of myofibroblasts and smooth muscle cells (SMC), and subepithelial fibrosis (3). Airway remodeling is also a key feature of COPD. The airways of the patients display epithelial cell metaplasia, hyperplasia of myofibroblasts and SMC, and supepithelial fibrosis, which is mostly less prominent than in chronic asthma (3). Subepithelial fibrosis primarily occurs at the lamina reticularis in which fibronectin and collagens of types I, III, and V that are mainly produced by fibroblasts accumulate (10). The activation of fibroblasts to upregulate matrix protein synthesis has been linked to cytokines secreted by inflammatory as well as structural cells (10).
Because C. pneumoniae infection stimulates the production of matrix metalloproteinases (MMPs) the pathogen may promote increased matrix protein turnover in the airways during respiratory infection (35, 40). It can also be hypothesized that C. pneumoniae may contribute to airway remodeling in COPD and asthma by modulating the synthesis of interstitial collagens and fibronectin. The accumulation of these matrix compounds is also a characteristic of the progression of atherosclerosis. Early fatty streak lesions that result from the deposition of macrophages and low-density lipoprotein cholesterol in the intima beneath the endothelial layer develop into fibrous atherosclerotic plaques that are characterized by a central lipid-rich core and a fibrous cap consisting of SMC, fibroblasts, and interstitial collagens (38). The hypothesis that C. pneumoniae modulates matrix protein synthesis by mesenchymal cells might provide a pathogenic link between the chlamydial infection, obstructive airway diseases, and atherosclerosis. Therefore, it was of interest to investigate the effect of C. pneumoniae on the expression of collagens and fibronectin in infected epithelial cells, fibroblasts, and SMC. To clarify the role of induced cytokines in the regulation of collagen and fibronectin gene expression, fibroblasts were stimulated with conditioned medium collected from Chlamydia-infected cells.
MATERIALS AND METHODS
Cell cultures and C. pneumoniae infection.
Epithelial WISH cells (ATCC CCL-25) and human dermal fibroblasts (C-12300; PromoCell, Heidelberg, Germany) were grown in minimal essential medium (OptiMEM; Gibco, Invitrogen, Karlsruhe, Germany) with 10% fetal calf serum (FCS) (PromoCell). Human aortic SMC (C-12533; PromoCell) were subcultured in SMC growth medium 2 (PromoCell).
C. pneumoniae strain TW-183 (obtained from the Institute of Ophthalmology, London, United Kingdom) was propagated in buffalo green monkey (BGM) cells as described previously (36). Infectivity titers of chlamydial stocks were quantified by titrating the number of inclusion-forming units per milliliter in BGM cells. Mycoplasma contaminations were excluded using a MycoDtect DNA array (Greiner Bio-One, Frickenhausen, Germany). For infection experiments, subcultures of WISH cells, fibroblasts, and SMC were grown in 35-mm-diameter culture wells (six-well plates). The cells were inoculated with C. pneumoniae at multiplicities of infection (MOIs) of 1, 5, and 10. For mock-infected cultures, diluted harvests of uninfected BGM cells were added. After centrifugation at 4,000 × g at 37°C for 45 min, the inoculum was decanted, and the cells were further incubated with OptiMEM containing 2% FCS. In some experiments, as indicated in Fig. 3, cell monolayers were inoculated with chlamydiae that were inactivated by UV light (15 W at a distance of 30 cm for 15 min) or heat (75°C for 10 min). Chlamydial protein synthesis was inhibited with chloramphenicol (100 μg/ml; Sigma-Aldrich, Hamburg, Germany).
FIG. 3.

Effects of heat and UV inactivation of chlamydiae and of chloramphenicol treatment on collagen and fibronectin mRNA levels in fibroblasts. (A) Cells were infected at an MOI of 5 or centrifuged with an equivalent dose of UV or heat-inactivated chlamydiae. The number of inactivated bacteria was calculated based on the number of inclusion-forming units of the corresponding chlamydial stock before treatment. (B) Mock-infected and infected cells were treated with 100 μg of chloramphenicol per ml. RT-PCR was conducted on total RNA isolated at 24 h after stimulation. FN, fibronectin; Col α1(I), type I collagen α1 chain; Col α1(III), type III collagen α1 chain.
RT-PCR.
Total RNA was prepared using an RNeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcriptase PCR (RT-PCR) was carried out as described previously (35). The sequences of specific primers used in this work are given in Table 1. PCR products were electrophoresed on 1% agarose gels and visualized with SYBR green staining. The volumes (optical density × mm2) of the band images were quantitated with Multi-Analyst PC software (Bio-Rad, Munich, Germany) and normalized against the pyruvate dehydrogenase (PDH) signal from the same sample.
TABLE 1.
Sequences of primers used for RT-PCR
| Primer specificity | Primer paira | Product size (bp) | Reference |
|---|---|---|---|
| Type I collagen α1 | 5′-CCCACCAATCACCTGCGTACAGA-3′ | 209 | 27 |
| 5′-TTCTTGGTCGGTGGGTGACTCTGA-3′ | |||
| Type III collagen α1 | 5′-GAGATGTCTGGAAGCCAGAACCAT-3′ | 207 | 27 |
| 5′-GATCTCCCTTGGGGCCTTGAGGT-3′ | |||
| Type IV collagen α1 | 5′-GCTCACCAGGACCAGTGGGT-3′ | 310 | 31 |
| 5′-TCACCTTTAGGTCCTGGCTG-3′ | |||
| Fibronectin | 5′-CCGTGGGCAACTCTGTC-3′ | 438 | 15 |
| 5′-TGCGGCAGTTGTCACAG-3′ | |||
| COX-2 | 5′-CGAGGTGTATGTATGAGTGTG-3′ | 540 | 45 |
| 5′-TCTAGCCAGAGTTTCACCGTA-3′ | |||
| MMP-1 | 5′-ATGCGCACAAATCCCTTCTACC-3′ | 247 | 51 |
| 5′-TTTCCTCAGAAAGAGCAGCATCG-3′ | |||
| IDO | 5′-ACAGACCACAAGTCACAGCG-3′ | 662 | 26 |
| 5′-AACTGAGCAGCATGTCCTCC-3′ | |||
| IFN-β | 5′-GATTCATCTAGCACTGGCTGG-3′ | 186 | 23 |
| 5′-CTTCAGGTAATGCAGAATCC-3′ | |||
| C/EBPβ | 5′-ACAGCGACGAGTACAAGATCC-3′ | 195 | 17 |
| 5′-GCAGCTGCTTGAACAAGTTCC-3′ | |||
| IL-8 | 5′-CTTGGCAGCCTTCCTGATTT-3′ | 263 | 54 |
| 5′-CAGCCCTCTTCAAAAACTTC-3′ | |||
| PDH | 5′-GGTATGGATGAGGACCTGGA-3′ | 105 | 37 |
| 5′-CTTCCACAGCCCTCGACTAA-3′ |
Sense and antisense, respectively.
Western blotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of cell lysates and Western blot assays were performed according to the protocol of a previous study (36). To separate collagen and fibronectin proteins, 5% polyacrylamide gels were used. Blots were incubated with 1:1,000 dilutions of rabbit polyclonal antibodies to human collagen type I (Rockland/Biomol, Hamburg, Germany), type III (Rockland/Biomol), type IV (Rockland/Biomol), fibronectin (H-300; Santa Cruz Biotechnology, Heidelberg, Germany), or CCAAT/enhancer-binding protein β (C/EBPβ) (C-19; Santa Cruz Biotechnology). As a reference band, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected with a rabbit polyclonal antibody (FL-335; Santa Cruz Biotechnology, Heidelberg, Germany). Alkaline phosphatase-conjugated goat anti-rabbit immunoglobulin G (Dianova, Hamburg, Germany) was used as secondary antibody at a dilution of 1:2,000. The bands were visualized with 5-bromo-4-chloro-3-indolylphosphate toluidine salt-p-nitroblue tetrazolium chloride (Sigma Fast; Sigma-Aldrich, Hamburg, Germany).
Confocal microscopy.
Fibroblasts grown on 11-mm-diameter glass coverslips were infected with C. pneumoniae at an MOI of 5. At 48 h after infection, coverslips were removed from the culture tubes, washed with phosphate-buffered saline, and fixed with methanol. For immunostaining, the cells were incubated with rabbit polyclonal antibodies to collagens (types I, III, and IV) (Rockland/Biomol) or fibronectin (Santa Cruz Biotechnology) at a dilution of 1:50 overnight at 4°C. The secondary antibody, a horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (Dako), was incubated at a dilution of 1:200 for 2 h at room temperature, and specific binding was detected with (E)-N-ethoxyethyl-4-[2-(4-hydroxyphenyl)ethenyl]chinolinium bromide, a fluorescent substrate of peroxidase (22). Staining was analyzed using a confocal laser scanning microscope (TCS SP5; Leica, Heidelberg, Germany). The fluorescence marker was excited with the Ar laser at an excitation wavelength of 488 nm and recorded using a band-pass filter (500 to 550 nm) in the green channel.
Stimulation of fibroblasts with conditioned medium and neutralization of cytokine activities.
Conditioned medium from mock-infected and infected fibroblasts and WISH cells was collected at 48 h after infection, clarified by centrifugation at 20,000 × g at 4°C for 30 min, and inoculated onto uninfected fibroblasts (3 ml per 35-mm-diameter well). For the neutralization of cytokine activities, goat polyclonal antibodies to human basic fibroblast growth factor ([bFGF] AF-00-NA), beta interferon ([IFN-β] AF-00-NA), or interleukin-1α ([IL-1α] AF-200-NA) (all from R&D Systems, Wiesbaden, Germany) were added to conditioned medium at concentrations of 0.3 and 1 μg/ml. Medium without and with antibodies was incubated at 37°C for 1 h before its inoculation onto fibroblast monolayers. In some experiments, infected fibroblasts were treated with NS-398 (N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide) (Qbiogene-Alexis, Grünberg, Germany).
ELISAs.
Cytokine and prostaglandin E2 (PGE2) levels in culture supernatants were measured by enzyme-linked immunosorbent assays (ELISAs) purchased from PromoCell (IL-1α), R&D Systems (IL-1β and PGE2), and Biosource (IFN-β; Fujirebio Inc., Ratingen, Germany). Assays were performed according to the supplier's protocols.
IFN-β and IL-1α stimulation of fibroblasts.
Fibroblasts grown in six-well plates were stimulated with recombinant IFN-β (PromoCell) or IL-1α (PromoCell) at different concentrations. Cytokines were diluted in OptiMEM with 2% FCS.
RNA interference.
HP Validated small interfering RNA (siRNA) targeting C/EBPβ (SI02777292) and nonsilencing All Stars Negative Control siRNA were purchased from Qiagen. Fibroblasts were transfected using an RNAi Human Starter Kit (Qiagen) according to the manufacturer's instructions. Transfected cells were incubated for 48 h before infection with C. pneumoniae. The efficiency of the transfection method was confirmed by using AlexaFluor 488-labeled All Stars Negative Control siRNA (data not shown).
Statistical analysis.
Statistical comparisons were made using a Student's t test. P values of ≤0.05 were considered to be statistically significant.
RESULTS
Expression of collagens and fibronectin in C. pneumoniae-infected cells.
In the first experiments we evaluated the expression of collagen (α1 chains) and fibronectin mRNAs in epithelial cells (WISH), fibroblasts, and SMC by RT-PCR analysis. Figure 1 shows that the infection of fibroblasts with C. pneumoniae resulted in decreased mRNA levels of interstitial collagens I and III, whereas the expression of collagen type IV was not altered. In infected fibroblasts smaller amounts of fibronectin mRNA were found than in mock-infected cells. The effects of C. pneumoniae infection on SMC were identical to those observed in fibroblast cultures (Fig. 1). Epithelial cells expressed only type IV collagen and fibronectin, and the infection did not affect transcription of these genes (Fig. 1).
FIG. 1.

Expression of type I collagen α1 chain [Col α1(I)], type III collagen α1 chain [Col α1(III)], type IV collagen α1 chain [Col α1(IV)], and fibronectin (FN) genes in WISH cells, fibroblasts, and SMC infected with C. pneumoniae TW-183 at various MOIs. Total RNA was extracted at 24 h after infection. mRNA levels were determined by RT-PCR. Detection of PDH served as control. RNA isolated from fibroblasts was used as a positive control in WISH cell assays. N.C., not calculated.
Further experiments on collagen protein synthesis were performed with fibroblasts. Immunoblot assays of cell lysates revealed that C. pneumoniae clearly reduced the amounts of type I and III collagen as well as fibronectin proteins when fibroblasts were infected at an MOI of 5 or 10 (Fig. 2A). These doses resulted in 4 and 8% of inclusion-containing cells in the cultures, respectively (n = 4). Type I collagen was detected as a double band of approximately 80 kDa and 70 kDa. Type III collagen and fibronectin showed single bands of approximately 95 kDa and >200 kDa, respectively (Fig. 2A). Chlamydia-exposed cells showed smaller amounts of these matrix proteins at 48 and 72 h after infection than mock-infected cells (Fig. 2B). Collagen IV appeared as a 95-kDa single band in mock-infected fibroblasts (Fig. 2A and B). Upon infection a double band of collagen IV was induced at 48 h and 72 h after infection, but the total amount of the protein did not seem to be significantly changed (Fig. 2A and B). In immunoblotting assays of SMC, similar results were obtained in comparison to fibroblasts (data not shown). Immunofluorescence staining of fibroblasts confirmed the downregulation of interstitial collagen and fibronectin protein synthesis in response to chlamydial infection (Fig. 2C). The fluorescence intensity of type IV collagen showed no visible change in infected cells (Fig. 2C).
FIG. 2.

Collagen and fibronectin protein synthesis in fibroblasts infected with C. pneumoniae. (A) Matrix protein levels in cells infected with various doses of chlamydiae. Cell lysates were prepared at 48 h after infection. (B) Time course of collagen and fibronectin (FN) protein synthesis. Fibroblasts were infected at an MOI of 5. GAPDH was stained as a reference band. (C) Indirect immunofluorescence staining of collagens and fibronectin in mock-infected and C. pneumoniae-infected fibroblasts. Cells were infected at an MOI of 5 and stained at 48 h after infection. Fibroblasts show a less intense staining for type I and III collagens (Col I and Col III, respectively) and fibronectin but not for type IV collagen (Col IV) following infection. Bar, 50 μm.
The downregulation of collagen types I and III as well as of fibronectin expression in fibroblasts required infectious chlamydiae because cells exposed to UV and heat-inactivated bacteria produced mRNA levels similar to the levels in mock-infected cells (Fig. 3A). Moreover, the inhibition of bacterial protein synthesis with chloramphenicol prevented the decrease in matrix protein gene expression (Fig. 3B).
Identification of C. pneumoniae-induced cytokines mediating collagen and fibronectin expression.
Downregulation of collagen and fibronectin expression may be caused by the direct effects of chlamydiae on the activation of host cell signaling pathways or via soluble mediators that are released from infected cells and act in a paracrine manner. To evaluate the involvement of soluble mediators, conditioned medium collected from mock-infected and infected cell cultures was examined for the ability to modulate matrix protein expression in fibroblasts. Conditioned medium harvested from Chlamydia-infected fibroblasts at 48 h after infection caused a significant reduction in type I and III collagen as well as fibronectin mRNA levels in fibroblasts in a similar manner compared to direct infection of the cells (Fig. 4). Collagen type IV mRNA expression remained unchanged. Interestingly, the incubation of fibroblasts with conditioned medium from infected epithelial cells also results in a decrease in interstitial collagen and fibronectin gene expression (Fig. 4). PGE2 represented a potential mediator responsible for the observed effects (32). However, infected fibroblasts that were treated with the cyclooxygenase 2 (COX-2) inhibitor NS-398 showed no differences in collagen and fibronectin mRNA levels compared to infected cells incubated in medium alone (data not shown). The efficacy of NS-398 treatment was confirmed by determination of PGE2 in culture supernatants. At 48 h after infection, the PGE2 level increased 9.9-fold (from 192 ± 84 pg/ml in mock-infected cultures to 1,897 ± 30 pg/ml in infected cultures; n = 3). In infected cultures treated with NS-398 the production of PGE2 was downregulated to 122 ± 38 pg/ml.
FIG. 4.

Potential of conditioned medium harvested from C. pneumoniae-infected cells to downregulate the mRNA levels of interstitial collagens and fibronectin in fibroblasts. Uninfected fibroblasts were treated with conditioned medium from fibroblasts and WISH cells that were infected with increasing doses of chlamydiae. Total RNA was prepared at 24 h after incubation of the cells with conditioned medium. mRNA levels were analyzed by RT-PCR. Relative mRNA expression was calculated from the normalization of the collagen and fibronectin signals against the PDH signal from the same sample. *, P ≤ 0.05, compared with values for mock-infected controls (Student's t test, n = 4). Collagen chains are indicated in the form Col α1(I) for type I collagen α1 chain, e.g.
Having established that PGE2 is not involved in C. pneumoniae-induced downregulation of matrix protein gene expression, we used conditioned medium from infected fibroblasts in incubations with specific antibodies against cytokines and then added it to uninfected cells. These experiments focused on bFGF and IFN-β because both mediators have been reported to suppress interstitial collagen expression and are known to be produced by Chlamydia-infected cells (20, 34, 36, 46). Antibodies to bFGF had no effect on the capacity of the conditioned medium to inhibit matrix protein synthesis (Fig. 5A). As a positive control of bFGF neutralization, mRNA levels of MMP-1 were analyzed. Treatment with bFGF antibodies abolished the ability of the conditioned medium to induce MMP-1 expression (Fig. 5A). When conditioned medium was treated with IFN-β neutralizing antibody at a concentration of 1 μg/ml, the downregulation of type I and III collagen and fibronectin expression was abolished or reduced, indicating that IFN-β represents a Chlamydia-induced mediator that modulates matrix protein synthesis (Fig. 5B). As a positive control of IFN-β neutralization, the expression of indoleamine 2,3-dioxygenase (IDO), an IFN-β-inducible gene, was examined. IDO gene transcription was induced in fibroblasts by conditioned medium from infected cells and downregulated when IFN-β antibody was added (Fig. 5B).
FIG. 5.
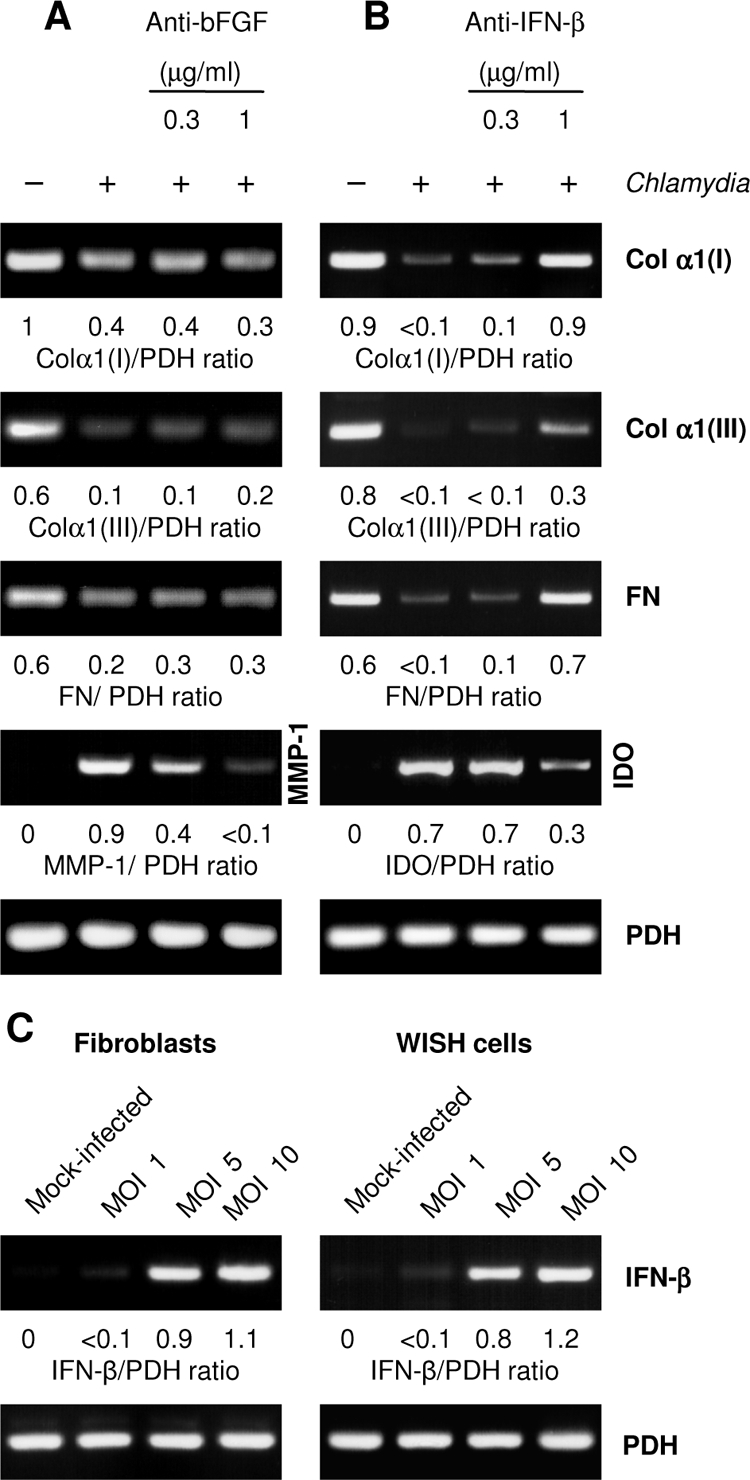
Neutralization of bFGF and IFN-β in conditioned medium from C. pneumoniae-infected fibroblasts. Conditioned medium from infected fibroblasts was incubated with neutralizing antibodies against bFGF (A) or IFN-β (B) before being added to uninfected fibroblasts. Total RNA was extracted from the cells 24 h later. mRNA levels of type I collagen α1 [Col α1(I)], type III collagen α1 [Col α1(III)], and fibronectin (FN) were determined by RT-PCR. MMP-1 (A) and IDO (B) mRNA levels were analyzed as controls of the neutralization of bFGF and IFN-β, respectively. (C) Determination of IFN-β mRNA levels in infected fibroblasts and WISH cells.
RT-PCR analysis confirmed that in fibroblasts IFN-β was expressed in response to C. pneumoniae infection (Fig. 5C). As measured by ELISA, culture supernatants of infected fibroblasts contained 22 ± 11 IU/ml of IFN-β at 48 h after infection (n = 3).
IFN-β mRNA was also found in epithelial WISH cells following infection (Fig. 5C). However, the supernatants did not contain measurable amounts of IFN-β. Moreover, inhibitory effects of the conditioned medium from WISH cells on the expression of type I and III collagen and of fibronectin were not affected after incubation with IFN-β neutralizing antibody (Fig. 6A). This led us to suppose that another cytokine must be involved. Surprisingly, IFN-β antibody treatment suppressed the slight induction of IDO in fibroblasts exposed to conditioned medium from WISH cells, indicating the presence of minimal levels of IFN-β below the detection limit of the ELISA of 2 IU per ml (Fig. 6A).
FIG. 6.

Neutralization of IFN-β and IL-1α in conditioned medium from C. pneumoniae-infected WISH cells. Conditioned medium was incubated with neutralizing antibodies against IFN-β (A) or IL-1α (B) and then used to study the effect on the expression of type I collagen α1 [Col α1(I)], type III collagen α1 [Col α1(III)], and fibronectin (FN) in fibroblasts. RT-PCR was conducted on total RNA isolated from the cells at 24 h after incubation. IDO (A) and COX-2 (B) mRNA levels were analyzed as controls of the neutralization of IFN-β and IL-1α, respectively. (C) Release of IL-1α from C. pneumoniae-infected WISH cells in comparison to cells following mock infection. IL-1α was measured in culture supernatants by ELISA. *, P ≤ 0.05 compared with values for mock-infected cells (Student's t test, n = 4).
In further experiments the role of IL-1α was evaluated because IL-1 has also been reported to possess the ability to inhibit collagen synthesis (9). Neutralizing antibody to IL-1α abolished the inhibitory effect of conditioned medium from WISH cells on the expression of interstitial collagens and fibronectin when added at concentrations of 0.3 and 1 μg/ml (Fig. 6B). Analysis of COX-2 mRNA levels was used as a positive control of the neutralization of IL-1α activity (Fig. 6B).
Increased production of IL-1α by WISH cells following C. pneumoniae infection was confirmed by ELISA (Fig. 6C). In culture supernatants of infected fibroblasts, no IL-1α could be detected. Furthermore, neither WISH cells nor fibroblasts produced the IL-1 isoform β.
To verify that IFN-β and IL-1α act as Chlamydia-induced factors downregulating interstitial collagen and fibronectin expression, uninfected fibroblasts were stimulated with recombinant cytokines. Both IFN-β and IL-1α caused a decrease in the levels of type I and III collagen and of fibronectin mRNAs (Fig. 7).
FIG. 7.
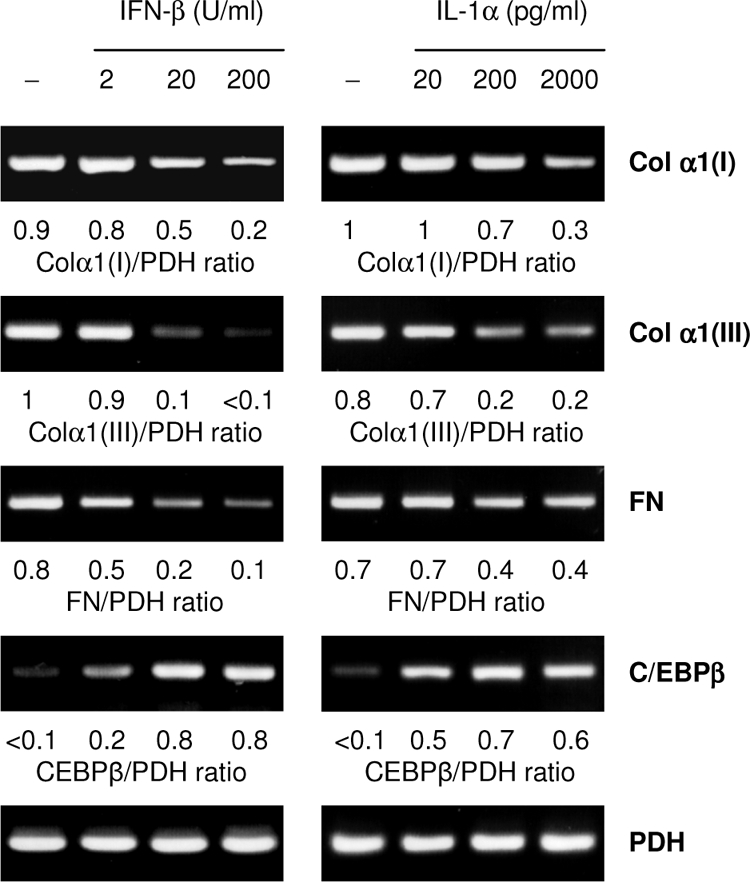
Effects of exogenous IFN-β and IL-1α on the expression of type I collagen α1 [Col α1(I)], type III collagen α1 [Col α1(III)], fibronectin (FN), and C/EBPβ mRNA in fibroblasts. The cells were incubated with increasing amounts of cytokines for 24 h before RT-PCR analysis was performed.
Role of C/EBPβ in diminished expression of interstitial collagens and fibronectin.
C/EBPβ has been identified as a transcription factor involved in the suppression of gene expression of type I collagen α1 and α2 chains (12, 13). Fibroblasts expressed enhanced levels of C/EBPβ mRNA when they were stimulated with recombinant IFN-β or IL-1α at increasing concentrations (Fig. 7). Correspondingly, C/EBPβ mRNA levels were increased in fibroblasts that were directly infected with C. pneumoniae or incubated with conditioned medium from infected cells (data not shown). Western blot analysis demonstrated that a specific 45-kDa band representing the full-length C/EBPβ form was increased upon infection (Fig. 8A).
FIG. 8.

Effect of siRNA targeting C/EBPβ on reduced expression of type I collagen α1 [Col α1(I)], type III collagen α1 [Col α1(III)], and fibronectin (FN) in C. pneumoniae-infected fibroblasts. (A) Western blot analysis of C/EBPβ protein in fibroblasts following infection. Cell lysates were prepared at 48 h after infection. GAPDH was stained as a reference band. (B) Cells were transfected with C/EBPβ-specific and control nonsilencing siRNA, incubated for 48 h, and then infected with C. pneumoniae. Downregulation of C/EBPβ mRNA levels in siRNA-transfected fibroblasts was assessed by RT-PCR. Analysis of IL-8 mRNA levels served as a positive control for C/EBPβ-mediated gene expression. Total RNA was extracted from the cells at 24 h after infection.
To examine the role of C/EBPβ in C. pneumoniae-mediated inhibition of collagen and fibronectin expression, fibroblasts were transfected with a specific siRNA before infection. Nonsilencing siRNA served as a negative control. Only the specific siRNA reduced the increase in C/EBPβ mRNA levels in response to chlamydial infection (Fig. 8B). In transfected fibroblasts the amounts of C/EBPβ mRNA were smaller and the mRNA levels of type I and III collagen and fibronectin were higher than in cells transfected with a control siRNA (Fig. 8B). Because C/EBPβ has been identified as a transcription factor involved in Chlamydia-induced IL-8 production, downregulation of IL-8 mRNA levels in response to C/EBPβ siRNA transfection was determined as a positive control (Fig. 8B) (7).
DISCUSSION
C. pneumoniae may play a pathogenic role in obstructive airway diseases. Fibrosis is an important feature of structural changes in the airways of asthma and COPD patients. The communication between epithelial cells and fibroblasts is essentially involved in remodeling processes characterized by an increased deposition of interstitial collagens and fibronectin (48). The results of this study demonstrate that the capacity of fibroblasts to express type I and III collagen and fibronectin is reduced in response to C. pneumoniae infection as well as following stimulation with conditioned medium from Chlamydia-infected epithelial cells. These findings indicate that infected host cells mainly release cytokines that exert inhibitory effects on matrix protein synthesis.
Collagen proteins are secreted as procollagens; they are cleaved, and the mature chains aggregate to form fibrils. For the analysis of collagen gene expression, we used primers targeting the genes of α1 chains of type I, III, and IV collagens. Collagen type I is a heterotrimer of two α1 and one α2 protein chains organized in a helical structure. The type I collagen α1 and α2 genes are coordinately regulated at the transcriptional level (11). Type III collagen consists of fibrils of three identical α1 protein chains, and type IV collagen is a non-fibrillar network-forming protein of two α1 chains and one α2 chain. Decreased expression of type I and III collagen α1 mRNAs in C. pneumoniae-infected fibroblast cultures was accompanied by reduced amounts of collagen proteins, as shown by Western blotting and immunofluorescence microscopy. The protein bands detected by the antibodies used in the Western blot assays also represent the α1 chains (according to the manufacturer). Although chlamydial infection did not affect the mRNA level of type IV collagen in fibroblasts, in Western blot assays the separation of the corresponding protein into a double band was observed, which might be explained by altered posttranslational modifications in infected cultures.
Downregulation of interstitial collagen and fibronectin expression by C. pneumoniae infection was mediated by host cell-derived cytokines that act in a paracrine manner. In the conditioned medium from infected fibroblasts, IFN-β was identified as a factor involved in this process. It is well known that IFN-γ can inhibit the expression of interstitial collagens, but there are few reports describing similar effects for IFN-β (12, 46). Transcription of the IFN-β gene was induced in fibroblasts as well as in WISH cells. However, only the supernatants of fibroblast cultures contained measurable amounts of IFN-β, and it can be assumed that WISH cells produced IFN-β only at a very low level. IL-1α could be identified as the WISH cell-derived factor that decreases interstitial collagen and fibronectin expression in fibroblasts. The role of IL-1 in the regulation of collagen synthesis is not completely understood, but the IL-1 isoform β has been described to inhibit type I α1 collagen gene expression in human fibroblasts (9). The proximal promoter sequence of type I α1 and α2 genes contains elements that are bound by ubiquitous transcription factors, including C/EBPβ (CCAAT/enhancer-binding protein β), a member of the CEBP family of basic leucine zipper DNA-binding proteins recognizing the CCAAT site. C/EBPβ can act as a negative regulator of interstitial collagen genes (11, 29). The fibronectin promoter also contains a CCAAT site (1). We could confirm that both IFN-β and IL-1α upregulate the production of C/EBPβ. Ghosh et al. reported that C/EBPβ binding to its cognate cis-acting recognition sites underlies the suppression of type I collagen gene expression in fibroblasts in response to IFN-γ and the naturally occurring truncated p20 isoform of C/EBPβ (liver-enriched transcription inhibitory protein) can abrogate the effects of IFN-γ (12). In C. pneumoniae-infected fibroblasts an increase in the amount of the C/EBPβ p45 full-length form was detected. Using siRNA targeting the C/EBPβ gene, we could demonstrate that the transcription factor is involved in the decreased expression of type I and III collagen and fibronectin in response to C. pneumoniae infection. It can be proposed that the Chlamydia-induced cytokines IFN-β and IL-1α negatively regulate matrix protein gene transcription through upregulation of C/EBPβ.
Mouse models have demonstrated that IFN-β and IL-1α contribute to the pulmonary inflammatory response to C. pneumoniae infection (28, 39). The findings of this study suggest that these cytokines impair the capacity of fibroblasts to synthesize interstitial collagens and fibronectin. It is unclear whether fibroblasts represent important target cells for C. pneumoniae in the lung. Recently, in a pig model of respiratory Chlamydia suis infection, this cell type was found to be infected, suggesting that fibroblasts can function as host cells for chlamydiae in the respiratory tract (30). In COPD patients, type II alveolar epithelial cells, in addition to macrophages, were identified as target cells for C. pneumoniae during long-lasting infection (40). The results of our experiments suggest that epithelial cells are an important source of IL-1α, which may decrease matrix protein synthesis by fibroblasts in a paracrine manner. Because asthma as well as COPD is accompanied by the subepithelial accumulation of interstitial collagens and fibronectin, it can be proposed that the host cell cytokine response to C. pneumoniae does not promote subepithelial fibrosis as a characteristic of tissue remodeling and airway obstruction (10). On the other hand, the induction of MMPs and cytokines inhibiting collagen production may increase matrix turnover, thereby allowing the spread of chlamydiae into the lung interstitium via infected macrophages and promoting the recruitment of inflammatory cells to the site of infection. Macrophages respond to C. pneumoniae infection with the release of tumor necrosis factor alpha, a cytokine which also functions as a negative regulator of collagen gene expression (11, 16).
Transforming growth factor β is considered to be an essential fibrogenic cytokine that contributes to the pathogenesis of obstructive airway diseases (10, 49). Because very little is known about the induction of transforming growth factor β during respiratory chlamydial infection, further studies are needed to identify mechanisms of the specific immune response that may cause an upregulation of fibrogenic mediators (53).
COPD encompasses two main phenotypes: chronic bronchitis and emphysema, which is characterized by the enlargement of peripheral air spaces as a consequence of degenerative collagen lesions and destructive processes that occur in the alveolar walls (3). It has been hypothesized that Chlamydia psittaci and C. pneumoniae infections may play a role in the development of emphysema (6, 47). Chlamydiae stimulate the production of MMPs upon host cell infection and, in turn, induce a cytokine response that reduces fibroblast collagen expression (35, 40). Both effects may lead to degenerative airway wall lesions.
Inhibitory effects of C. pneumoniae on the synthesis of interstitial collagens and fibronectin may be important for its potential role in atherosclerosis. Interstitial collagens of types I and III are the major matrix components of the fibrous cap of fibroatheromatous lesions, and the integrity of the collagenous skeleton determines the stability of the plaque (24). It is known that a thin fibrous cap with small amounts of collagen increases the plaque vulnerability and the risk of rupture with subsequent thrombosis, events that are mainly responsible for acute ischemic syndromes (24). Collagens and fibronectin in atherosclerotic plaques are mainly produced by SMC, a cell type that has been identified as a target cell of C. pneumoniae in the arterial wall (44). In this study we could observe that not only fibroblasts but also SMC express lower levels of interstitial collagens and fibronectin following infection. Reduced matrix protein synthesis in Chlamydia-infected atherosclerotic lesions may promote the development of a more vulnerable plaque phenotype (42).
In conclusion, this report demonstrates that C. pneumoniae activates epithelial cells, fibroblasts, and SMC to produce cytokines that downregulate the expression of interstitial collagens and fibronectin. In vivo these host cell responses may modulate tissue remodeling processes and contribute to a pathogenic role of C. pneumoniae in chronic airway diseases and atherosclerosis.
Acknowledgments
This work was supported by grants B307-04004 from the Interdisciplinary Centre for Clinical Research of Jena and CAPNETZ 01KL0104 from the Bundesministerium für Bildung und Forschung, Germany.
We are grateful to Katrin Prager for excellent technical assistance.
Editor: J. L. Flynn
Footnotes
Published ahead of print on 1 December 2008.
REFERENCES
- 1.Alonso, C. R., J. George, C. G. Pesce, D. M. Bissell, and A. R. Kornblihtt. 2002. Fibronectin transcription in liver cells: promoter occupation and function in sinusoidal endothelial cells and hepatocytes. Biochem. Biophys. Res. Commun. 2951077-1084. [DOI] [PubMed] [Google Scholar]
- 2.Beaty, C. D., J. T. Grayston, S. P. Wang, C.-C. Kuo, C. S. Reto, and T. R. Martin. 1991. Chlamydia pneumoniae, strain TWAR, infection in patients with chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1441408-1410. [DOI] [PubMed] [Google Scholar]
- 3.Bergeron, C., and L.-P. Boulet. 2006. Structural changes in airway diseases: characteristics, mechanisms, consequences, and pharmacologic modulation. Chest 1291068-1087. [DOI] [PubMed] [Google Scholar]
- 4.Biscione, G. L., J. Corne, A. J. Chauhan, and S. L. Johnston. 2004. Increased frequency of detection of Chlamydophila pneumoniae in asthma. Eur. Respir. J. 24745-749. [DOI] [PubMed] [Google Scholar]
- 5.Blasi, F., S. Aliberti, L. Allegra, G. Piatti, P. Tarsia, J. M. Ossewarde, V. Verweij, F. P. Nijkamp, and G. Folkerts. 2007. Chlamydophila pneumoniae induces a sustained airway hyperresponsiveness and inflammation in mice. Respir. Res. 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandén, E., J. Gnarpe, G. Hillerdal, L. Orre, C. M. Sköld, M. Löfdahl, H. Koyi, and G. Tornling. 2007. Detection of Chlamydia pneumoniae on cytospin preparations from bronchoalveolar lavage in COPD patients and in lung tissue from advanced emphysema. Int. J. Chron. Obstruct. Pulmon. Dis. 2643-650. [PMC free article] [PubMed] [Google Scholar]
- 7.Buchholz, K. R., and R. S. Stephens. 2006. Activation of the host cell proinflammatory interleukin-8 response by Chlamydia trachomatis. Cell. Microbiol. 81768-1779. [DOI] [PubMed] [Google Scholar]
- 8.de Kruif, M. D., E. C. M. van Gorp, T. T. Keller, J. M. Ossewarde, and H. ten Kate. 2005. Chlamydia pneumoniae infections in mouse models: relevance for atherosclerosis research. Cardiovasc. Res. 65317-327. [DOI] [PubMed] [Google Scholar]
- 9.Diaz, A., E. Munoz, R. Johnston, J. H. Korn, and S. A. Jimenez. 1993. Regulation of human fibroblast alpha 1(I) procollagen gene expression by tumor necrosis factor-α, interleukin-1β and prostaglandin W2. J. Biol. Chem. 26810364-10371. [PubMed] [Google Scholar]
- 10.Elias, J. A., Z. Zhu, G. Chupp, and R. J. Homer. 1999. Airway remodeling in asthma. J. Clin. Investig. 1041001-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh, A. K. 2002. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp. Biol. Med. 227301-314. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh, A. K., S. Bhattacharyya, Y. Mori, and J. Varga. 2006. Inhibition of collagen gene expression by interferon-γ: novel role of the CCAAT/enhancer-binding protein β (C/EBPβ). J. Cell. Physiol. 207251-260. [DOI] [PubMed] [Google Scholar]
- 13.Greenwel, P., S. Tanaka, D. Penkov, W. Zhang, M. Olive, J. Moll, C. Vinson, M. di Liberto, and F. Ramirez. 2000. Tumor necrosis factor alpha inhibits type I collagen synthesis through repressive CCAAT/enhancer-binding protein. Mol. Cell. Biol. 20912-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn, D. L., R. W. Dodge, and R. Golubjatnikov. 1991. Association of Chlamydia pneumoniae (strain TWAR) infection with wheezing, asthmatic bronchitis, and adult-onset asthma. JAMA 266225-230. [PubMed] [Google Scholar]
- 15.Han, S., J. D. Ritzenthaler, H. N. Rivera, and J. Roman. 2005. Peroxisome proliferator-activated receptor-gamma ligands suppress fibronectin gene expression in human lung carcinoma cells: involvement of both CRE and Sp1. Am. J. Physiol. Lung Cell. Mol. Physiol. 289L419-L428. [DOI] [PubMed] [Google Scholar]
- 16.Heinemann, M., M. Susa, U. Simnacher, R. Marre, and A. Essig. 1996. Growth of Chlamydia pneumoniae induces cytokine production and expression of CD14 in a human monocytic cell line. Infect. Immun. 644872-4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homma, J., R. Yamanaka, N. Yajima, N. Tsuchiya, N. Genkai, M. Sano, and R. Tanaka. 2006. Increased expression of CCAAT/enhancer binding protein beta correlates with prognosis in glioma patients. Oncol. Rep. 15595-601. [PubMed] [Google Scholar]
- 18.Ieven, M. M., and V. Y. Hoymans. 2005. Involvement of Chlamydia pneumoniae in atherosclerosis: more evidence for lack of evidence. J. Clin. Microbiol. 4319-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnston, S. L., and R. J. Martin. 2005. Chlamydophila pneumoniae and Mycoplasma pneumoniae: a role in asthma pathogenesis? Am. J. Respir. Crit. Care Med. 1721078-1089. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy, S. H., H. Qin, L. Lin, and E. M. Tan. 1995. Basic fibroblast growth factor regulates type I collagen and collagenase production in human smooth muscle cells. Am. J. Pathol. 146764-771. [PMC free article] [PubMed] [Google Scholar]
- 21.Reference deleted.
- 22.Krieg, R., A. Eitner, W. Günther, and K.-J. Halbhuber. 2007. Optimization of heterocyclic 4-hydroxystyryl derivatives for histological localization of endogenous and immunobound peroxidase activity. Biotech. Histochem. 82235-262. [DOI] [PubMed] [Google Scholar]
- 23.Li, X. L., M. Boyanapalli, X. Weihua, D. V. Kalvakolanu, and B. Hassel. 1998. Induction of interferon synthesis and activation of interferon-stimulated genes by liposomal transfection reagents. J. Interferon Cytokine Res. 18947-952. [DOI] [PubMed] [Google Scholar]
- 24.Libby, P. 1995. Molecular bases of the acute coronary syndrome. Circulation 912844-2850. [DOI] [PubMed] [Google Scholar]
- 25.Miyashita, N., H. Fukano, H. Hara, K. Yoshida, Y. Niki, and T. Matsushima. 2002. Recurrent pneumonia due to persistent Chlamydia pneumoniae infection. Intern. Med. 4130-33. [DOI] [PubMed] [Google Scholar]
- 26.Nagineni, C. N., K. Pardhasaradhi, M. C. Martins, B. Detrick, and J. J. Hooks. 1996. Mechanisms of interferon-induced inhibition of Toxoplasma gondii replication in human retinal pigment epithelial cells. Infect. Immun. 644188-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nusgens, B. V., P. Humbert, A. Rougier, A. C. Colige, M. Haftek, C. A. Lambert, A. Richard, P. Creidi, and C. M. Lapière. 2001. Topically applied vitamin C enhances the mRNA level of collagens I and III, their processing enzymes and tissue inhibitor of matrix metalloproteinase 1 in the human dermis. J. Investig. Dermatol. 116853-859. [DOI] [PubMed] [Google Scholar]
- 28.Penttilä, T., A. Haveri, A. Tammiruusu, J. M. Vuola, R. Lahesmaa, and M. Puolakkainen. 2008. Chlamydia pneumoniae infection in IL-10 knock-out mice: accelerated clearance but severe pulmonary inflammatory response. Microb. Pathogen. 4525-29. [DOI] [PubMed] [Google Scholar]
- 29.Ramirez, F., S. Tanaka, and G. Bou-Gharios. 2006. Transcriptional regulation of the human α2(I) collagen gene (COL1A2), an informative model system to study fibrotic diseases. Matrix Biol. 25365-372. [DOI] [PubMed] [Google Scholar]
- 30.Reinhold, P., N. Kirschvink, D. Theegarten, and A. Berndt. 2008. An experimentally induced Chlamydia suis infection in pigs results in severe lung function disorders and pulmonary inflammation. Vet. Res. 3935. [DOI] [PubMed] [Google Scholar]
- 31.Rinaldi, N., M. Willhauck, D. Weis, B. Brado, P. Kern, M. Lukoschek, M. Schwarz-Eywill, and T. F. E. Barth. 2001. Loss of collagen type IV in rheumatoid synovia and cytokine effect on the collagen type-IV gene expression in fibroblast-like synoviocytes from rheumatoid arthritis. Virchows Arch. 439675-682. [DOI] [PubMed] [Google Scholar]
- 32.Riquet, F. B., W. F. Lai, J. R. Birkhead, L. F. Suen, G. Karsenty, and M. B. Goldring. 2000. Suppression of type I collagen gene expression by prostaglandins in fibroblasts is mediated at the transcriptional level. Mol. Med. 6705-719. [PMC free article] [PubMed] [Google Scholar]
- 33.Roan, N. R., and M. N. Starnbach. 2008. Immune-mediated control of Chlamydia infection. Cell. Microbiol. 109-19. [DOI] [PubMed] [Google Scholar]
- 34.Rödel, J., A. Groh, H. Vogelsang, M. Lehmann, M. Hartmann, and E. Straube. 1998. Beta interferon is produced by Chlamydia trachomatis-infected fibroblast-like synoviocytes and inhibits gamma interferon-induced HLA-DR expression. Infect. Immun. 664491-4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rödel, J., D. Prochnau, K. Prager, E. Pentcheva, M. Hartmann, and E. Straube. 2003. Increased production of matrix metalloproteinases 1 and 3 by smooth muscle cells upon infection with Chlamydia pneumoniae. FEMS Immunol. Med. Microbiol. 38159-164. [DOI] [PubMed] [Google Scholar]
- 36.Rödel, J., M. Woytas, A. Groh, K.-H. Schmidt, M. Hartmann, M. Lehmann, and E. Straube. 2000. Production of basic fibroblast growth factor and interleukin 6 by human smooth muscle cells following infection with Chlamydia pneumoniae. Infect. Immun. 683635-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rolfs, A., I. Schuller, U. Finckh, and I. Weber-Rolfs. 1992. PCR: clinical diagnostics and research. Springer-Verlag, Berlin, Germany.
- 38.Ross, R. 1999. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 340115-126. [DOI] [PubMed] [Google Scholar]
- 39.Rothfuchs, A. G., C. Trumstedt, F. Mattei, G. Schiavoni, Å. Hidmark, H. Wigzell, and M. E. Rottenberg. 2006. STAT1 regulates IFN-αβ- and IFN-γ-dependent control of infection with Chlamydia pneumoniae by nonhemopoietic cells. J. Immunol. 1766982-6990. [DOI] [PubMed] [Google Scholar]
- 40.Rupp, J., D. Droemann, T. Goldmann, P. Zabel, W. Solbach, E. Vollmer, D. Branscheid, K. Dalhoff, and M. Maass. 2004. Alveolar epithelial cells type II are major target cells for C. pneumoniae in chronic but not in acute respiratory infection. FEMS Immunol. Med. Microbiol. 41197-203. [DOI] [PubMed] [Google Scholar]
- 41.Saikku, P., M. Leinonen, K. Mattila, M. R. Ekman, M. S. Nieminen, P. H. Makela, J. K. Huttonen, and V. Valtonen. 1988. Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 2983-986. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt, R., V. Redecke, Y. Breitfeld, N. Wanita, T. Miethke, S. Massberg, S. Fischel, F.-J. Neumann, A. Schömig, and A. E. May. 2006. EMMPRIN (CD 147) is a central activator of extracellular matrix degradation by Chlamydia pneumoniae-infected monocytes. Implications for plaque rupture. Thromb. Haemost. 95151-158. [PubMed] [Google Scholar]
- 43.Schmidt, S. M., C. E. Müller, and S. K. W. Wiersbitzky. 2005. Inverse association between Chlamydia pneumoniae respiratory tract infection and initiation of asthma or allergic rhinitis in children. Pediatr. Allergy Immunol. 16137-144. [DOI] [PubMed] [Google Scholar]
- 44.Shor, A., J. I. Phillips, G. Ong, B. J. Thomas, and D. Taylor-Robinson. 1998. Chlamydia pneumoniae in atheroma: consideration of criteria for causality. J. Clin. Pathol. 51812-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subbarayan, V., A. L. Sabichi, N. Llansa, S. M. Lippman, and D. G. Menter. 2001. Differential expression of cyclooxygenase-2 and its regulation by tumor necrosis factor-a in normal and malignant prostate cells. Cancer Res. 612720-2726. [PubMed] [Google Scholar]
- 46.Tanabe, J., A. Izawa, N. Takemi, Y. Miyauchi, Y. Torii, H. Tsuchiyama, T. Suzuki, S. Sone, and K. Ando. 2007. Interferon-beta reduces the mouse liver fibrosis induced by repeated administration of concanavalin A via the direct and indirect effects. Immunology 122562-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Theegarten, D., G. Mogilevski, O. Anhenn, G. Stamatis, R. Jaeschock, and K. Morgenroth. 2000. The role of Chlamydia in the pathogenesis of pulmonary emphysema: electron microscopy and immunofluorescence reveal corresponding findings as in atherosclerosis. Virchows Arch. 437190-193. [DOI] [PubMed] [Google Scholar]
- 48.Vignola, A. M., F. Mirabella, G. Costanzo, R. Di Giorni, M. Cjomarkaj, V. Bellia, and G. Monsignore. 2003. Airway remodeling in asthma. Chest 123417S-422S. [DOI] [PubMed] [Google Scholar]
- 49.Vignola, A. M., P. Chanez, G. Chiappara, A. Merendino, E. Pace, A. Rizzo, A. M. la Rocca, V. Bellia, G. Bonsignore, and J. Bousquet. 1997. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 156591-599. [DOI] [PubMed] [Google Scholar]
- 50.Von Hertzen, L., H. Alakärppä, R. Koskinen, K. Liippo, H. M. Surcel, M. Leinonen, and P. Saikku. 1997. Chlamydia pneumoniae infection in patients with chronic obstructive airway disease. Epidemiol. Infect. 118155-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Lampe, B., B. Barthel, S. E. Coupland, E.-O. Riecken, and S. Rockewicz. 2000. Differential expression of matrix metalloproteinases and their tissue inhibitors in colon mucosa of patients with inflammatory bowel disease. Gut 4763-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Webley, W. C., P. S. Salva, C. Andrzejewski, F. Cirino, C. A. West, Y. Tilahun, and E. S. Stuart. 2005. The bronchial lavage of pediatric patients with asthma contains infectious Chlamydia. Am. J. Respir. Crit. Care Med. 1711083-1088. [DOI] [PubMed] [Google Scholar]
- 53.Williams, D. M., B. G. Grubbs, S. Park-Snyder, R. G. Rank, and L. F. Bonewald. 1996. Activation of latent transforming growth factor beta during Chlamydia trachomatis-induced murine pneumonia. Res. Microbiol. 147251-262. [DOI] [PubMed] [Google Scholar]
- 54.Xue, M. L., H. Zhu, A. Thakur, and M. Willcox. 2002. 1-alpha,25-Dihydroxyvitamin D3 inhibits proinflammatory cytokine and chemokine expression in human corneal epithelial cells colonized with Pseudomonas aeruginosa. Immunol. Cell Biol. 80340-345. [DOI] [PubMed] [Google Scholar]