

Abstract
The capacity of Staphylococcus aureus to form biofilms on host tissues and implanted medical devices is one of the major virulence traits underlying persistent and chronic infections. The matrix in which S. aureus cells are encased in a biofilm often consists of the polysaccharide intercellular adhesin (PIA) or poly-N-acetyl glucosamine (PNAG). However, surface proteins capable of promoting biofilm development in the absence of PIA/PNAG exopolysaccharide have been described. Here, we used two-dimensional nano-liquid chromatography and mass spectrometry to investigate the composition of a proteinaceous biofilm matrix and identified protein A (spa) as an essential component of the biofilm; protein A induced bacterial aggregation in liquid medium and biofilm formation under standing and flow conditions. Exogenous addition of synthetic protein A or supernatants containing secreted protein A to growth media induced biofilm development, indicating that protein A can promote biofilm development without being covalently anchored to the cell wall. Protein A-mediated biofilm formation was completely inhibited in a dose-dependent manner by addition of serum, purified immunoglobulin G, or anti-protein A-specific antibodies. A murine model of subcutaneous catheter infection unveiled a significant role for protein A in the development of biofilm-associated infections, as the amount of protein A-deficient bacteria recovered from the catheter was significantly lower than that of wild-type bacteria when both strains were used to coinfect the implanted medical device. Our results suggest a novel role for protein A complementary to its known capacity to interact with multiple immunologically important eukaryotic receptors.
Staphylococcus aureus is a gram-positive bacterium that lives as part of the normal microflora on the skin and mucous membranes of humans and animals. If S. aureus passes through the epithelial barrier and reaches internal organs, it can cause a variety of diseases, ranging from minor skin infections, such as furuncles or boils, to severe infections, such as bacteremia, pneumonia, osteomyelitis, or endocarditis. Despite the progress with antibiotics in the treatment of bacterial infections over the last 2 decades, the number of infections due to S. aureus has increased (11, 30). The infection rate has been correlated with an increase in the use of prosthetic and indwelling devices in modern medical practices (24, 26). S. aureus, as well as other coagulase-negative staphylococci, displays a strong capacity to irreversibly attach to the surface of implanted medical devices and forms multilayered communities of bacteria, known as biofilms, that grow embedded in a self-produced extracellular matrix (23). The biofilm formation process occurs in two steps: first, bacterial cells irreversibly attach to a surface, and second, they interact with each other and accumulate in multilayered cell clusters embedded in a self-produced extracellular matrix. Primary attachment is mediated by physico-chemical cell surface properties as well as specific factors that mediate the attachment to the host-derived extracellular matrix components that rapidly coat the biomaterial following insertion into the patient. Numerous proteins from the MSCRAMMs family (microbial surface components recognizing adhesive matrix molecules) are involved in the first step of S. aureus biofilm formation, such as clumping factors ClfA (37) and ClfB (41) and fibrinogen and fibronectin binding proteins (FnBPA and FnBPB) (25, 31). Once bacteria accumulate in multilayered cell clusters, most have no direct contact with the surface, and thus cell-to-cell interactions become essential for biofilm development and maintenance. An extracellular polysaccharide intercellular adhesin (PIA, or PNAG), produced by icaADBC operon-encoded enzymes, is currently the best-characterized element mediating intercellular interactions in vitro (8, 23, 34, 35, 38). Alternatively, a number of surface proteins can replace PIA/PNAG exopolysaccharide in promoting intercellular adhesion and biofilm development, including the surface protein Bap (9). All the tested staphylococcal isolates harboring the bap gene were shown to be strong biofilm producers, and inactivation of the icaADBC operon in bap-positive strains had no effect on in vitro biofilm formation (57). Remarkably, proteins homologous to Bap are involved in the biofilm formation process in diverse bacterial species (33). A second surface protein, SasG, as well as its homologous protein in Staphylococcus epidermidis, Aap, also mediates intercellular interactions and biofilm development in the absence of the ica operon (7, 51). More recently, two independent laboratories have shown that fibronectin binding proteins A and B (FnBPA and FnBPB) induce biofilm development of clinical isolates of S. aureus (45, 55). Finally, there is growing evidence that extracellular DNA, despite not being sufficient to replace PIA/PNAG exopolysaccharide, is an important S. aureus biofilm matrix component (50).
During the course of a systematic mutagenesis study of the 17 two-component systems of S. aureus that aimed to identify biofilm-negative regulators, we found that S. aureus agr arlRS double mutants developed an alternative, ica-independent biofilm in a chemically defined medium, Hussain-Hastings-White (HHW) medium (56). This study focused on the identification of the proteinaceous compound responsible for the biofilm developed by S. aureus agr arlRS mutants. Here, we show that S. aureus protein A is responsible for the aggregative phenotype and capacity for biofilm formation displayed by this strain. Furthermore, overproduction of protein A in wild-type S. aureus strains or addition of soluble protein A to bacterial growth medium induced aggregation and biofilm development, suggesting that protein A does not need to be covalently linked to the cell wall to promote multicellular behavior. Moreover, deletion of the spa gene significantly decreased the capacity of S. aureus to colonize subcutaneously implanted catheters. Our findings support a novel role for protein A in promoting multicellular behavior and suggest that protein A-mediated biofilm development may have a critical function during the infection process of S. aureus.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and plasmids.
The most relevant bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli XL1-Blue cells were grown in Luria-Bertani broth or on Luria-Bertani agar (Pronadisa, Madrid, Spain) with appropriate antibiotics. Staphylococcal strains were cultured using different media: trypticase soy agar (TSA), trypticase soy broth supplemented with glucose (0.25%, wt/vol) (TSBg), and chemically defined HHW modified (HHWm) medium. Lactococcus lactis strains were incubated in M17 medium (Pronadisa, Madrid, Spain). Media were supplemented with appropriate antibiotics at the following concentrations: erythromycin (Er), 20 μg ml−1, 1.5 μg ml−1, or 10 μg ml−1; ampicillin (Am), 100 μg ml−1; chloramphenicol (Cm), 20 μg ml−1; kanamycin (Km), 50 μg ml−1; tetracycline (Tet), 10 μg ml−1. When required, TSA was supplemented with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (Bioline, London, United Kingdom).
TABLE 1.
Strains and plasmids used in the study
| Strain or plasmid | Relevant characteristic(s) | Reference |
|---|---|---|
| S. aureus strains | ||
| ISP479r | ISP479c rsbU restored | 56 |
| ISP479r ΔarlRS | ISP479r ΔarlRS operon | 56 |
| ISP479r agr | ISP479r agr::tet | 56 |
| ISP479r agr Δspa | ISP479r agr::tet Δspa | This study |
| ISP479r agr ΔarlRS | ISP479r agr::tet ΔarlRS operon | 56 |
| ISP479r agr ΔarlRS srtA | ISP479r agr::tet ΔarlRS srtA::ermC | This study |
| ISP479r agr ΔarlRS Δspa | ISP479r agr::tet ΔarlRS Δspa | This study |
| ISP479r agr ΔarlRS ΔsdrD | ISP479r agr::tet ΔarlRS ΔsdrD | This study |
| ISP479r agr pCN40 | ISP479r agr transformed with pCN40 plasmid | This study |
| ISP479r agr ΔarlRS pCN40 | ISP479r agr ΔarlRS transformed with pCN40 plasmid | This study |
| ISP479r agr ΔarlRS Δspa pCN40 | ISP479r agr ΔarlRS Δspa transformed with pCN40 plasmid | This study |
| ISP479r agr ΔarlRS Δspa pCN40spa | ISP479r agr ΔarlRS Δspa complemented with spa gene | This study |
| Newman | 12 | |
| Newman srtA | Newman srtA::ermC | 36 |
| Newman spa | Newman spa::Km | 44 |
| Newman pCN40 | Newman transformed with pCN40 plasmid | This study |
| Newman pCN40spa | Newman complemented with spa gene | This study |
| Newman agr | Newman agr::tet | This study |
| Newman agr spa | Newman agr::tet spa::Km | This study |
| 12313 | Clinical strain isolate | 56 |
| Lactococcus lactis strains | ||
| MG1363 pKS80 | Plasmid-cured L. lactis strain complemented with pSK80 plasmid | 44 |
| MG1363 pKS80spa | Plasmid-cured L. lactis strain complemented with spa gene | 44 |
| Escherichia coli XL1-Blue | Used for cloning assays | |
| Plasmids | ||
| pMAD | E. coli-S. aureus shuttle vector with bgaB gene encoding a β-galactosidase; Apr/Err | 1 |
| pMAD spaAD | pMAD plasmid containing the mutant allele for deletion of spa gene | This study |
| pMAD sdrDAD | pMAD plasmid containing the mutant allele for deletion of sdrD gene | This study |
| pCN40 | E. coli-S. aureus shuttle vector with constitutive promoter PblaZ | 5 |
| pCN40spa | pCN40 plasmid containing spa gene | This study |
| pCN40spaΔLPXTG | pCN40 plasmid containing spa gene without charged tail and hydrophobic domain | This study |
DNA manipulations.
DNA plasmids were isolated from E. coli strains using the Qiagen plasmid mini prep kit (Bio-Rad Laboratories, Inc.) according to the manufacturer's protocol. Plasmids were transformed into staphylococci by electroporation, using a previously described protocol (9). Restriction enzymes were purchased from Takara Shuzo Co. Ltd. or New England Biolabs and used according to the manufacturers' instructions. Oligonucleotides were obtained from Thermo (Electron Corporation). The srtA gene was inactivated in S. aureus ISP479r agr by transferring srtA::ermC from S. aureus Newman srtA (36) by phage transduction using φ85 (42).
Allelic exchange of chromosomal genes.
To construct the deleted strains, we amplified by PCR two fragments of approximately 800 bp that flanked the left side (oligonucleotides A and B) and the right side (oligonucleotides C and D) of the sequence targeted for deletion (Table 2). The two obtained fragments were cloned in the pGEM-T Easy vector (Promega). Oligonucleotides B and C carry the same restriction site at the 3′ and 5′ ends, respectively, so that it is possible to fuse fragments AB and CD by ligation, creating the AD fragment. Besides, oligonucleotides A and D carry restriction sites, so that it is possible to fuse the AD fragment to the shuttle plasmid pMAD previously digested with the corresponding enzymes. The resulting plasmids were transformed into S. aureus by electroporation. pMAD contains a temperature-sensitive origin of replication and an erythromycin resistance gene (1). Homologous recombination experiments were performed as previously described (60). Erythromycin-sensitive white colonies, which no longer contained the pMAD plasmid, were tested by PCR using oligonucleotides E and F to confirm the gene replacement.
TABLE 2.
Oligonucleotides used in the study
| Name | Sequence |
|---|---|
| spa-A | 5′-GAATTCCAATTCTAGCTATTATCACTT-3′ |
| spa-B | 5′-TCTAGAATTAATACCCCCTGTATGTA-3′ |
| spa-C | 5′-TCTAGAAAACAAACAATACACAACGAT-3′ |
| spa-D | 5′-GGATCCTTAAAATGGAAAAGTGCAGG-3′ |
| spa-E | 5′-GATGATGTATACAATGTATTTC-3′ |
| spa-F | 5′-TGCGTCTCGATTTAATTGG-3′ |
| spa-G | 5′-GAATTCCACTTAAAACCATTTCCGAA-3′ |
| spa-H | 5′-GGATCCATCGTTGTGTATTGTTTGTTT-3′ |
| pCN40spa-1 | 5′-GGATCCAAGTTGTAAAACTTACATTTAAA-3′ |
| pCN40spa-2 | 5′-GAATTCATCGTTGTGTATTGTTTGTTT-3′ |
| pCN40spa-3 | 5′-GAATTCTTATGATAATCCACCAAATACAG-3′ |
| sdrD-A | 5′-GGATCCGTTACAGAAATGTAATTGTCTATG-3′ |
| sdrD-B | 5′-AGTTGCTGCATATCAACTTTATC-3′ |
| sdrD-C | 5′-CAGCTTATACCAGGTCCGTG-3′ |
| sdrD-D | 5′-CCATGGTGTTGAATCATTTTCTGTTGTCG-3′ |
| sdrD-E | 5′-GATTAACTTAACCAGGTCCATG-3′ |
| sdrD-F | 5′-CTTTAGCTTCTGGTTGTGAATC−3′ |
| arlRS-E | 5′-GGATCCTTGATTGATTGACGTCTCAGTCAT-3′ |
| arlRS-F | 5′-AAGCTTGTGAAATGTTTAAACTACGG-3′ |
| spa-Fw | 5′-GCAAACGGCACTACTGCTGA-3′ |
| spa-Rv | 5′-CACCAGTTTCTGGTAATGCTTGAG-3′ |
| gyr-Fw | 5′-TTATGGTGCTGGGCAAATACA-3′ |
| gyr-Rv | 5′-CACCATGTAAACCACCAGATA-3′ |
Complementation studies.
The spa gene was amplified with thermophylic DNA polymerase (Certamp long amplification kit; Biotools, Spain) from S. aureus strain ISP479r with primers pCN40spa-1 and pCN40spa-2 (Table 2). pCN40 is a staphyloccus-E. coli shuttle vector that harbors the constitutive PblaZ promoter (43). The PCR product was cloned into pCN40 (pCN40spa) so that it was expressed under the PblaZ promoter. This plasmid and the vector control were introduced in the different strains by phage transduction using φ85 (42). The spa gene lacking the carboxy-terminal region was amplified from strain ISP479r by PCR with primers pCN40spa-1 and pCN40spa-3 and it was cloned into pCN40 to create the pCN40spa ΔLPXTG plasmid. This plasmid was introduced in ISP479r agr ΔarlRS Δspa by phage transduction using φ85.
Surface-associated protein preparation.
Surface protein preparations were obtained from S. aureus cells under iso-osmotic conditions. Bacterial cells were recovered from 5-ml overnight cultures by centrifugation, washed twice in 1 ml of phosphate-buffered saline (PBS), and resuspended in 100 μl of iso-osmotic digestion buffer (PBS containing 26% [wt/vol] raffinose). After addition of 3 μl of a 1-mg/ml solution of lysostaphin (Sigma), the preparations were incubated with shaking at 37°C for 2 h. Protoplasts were sedimented by centrifugation at 8,000 × g for 30 min with slow deceleration, and the supernatant was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or stored at −20°C.
Biofilm formation assays.
A biofilm formation assay in microtiter wells was performed as described previously (28). Briefly, 5 μl of a culture of S. aureus grown overnight in HHWm or TSBg at 37°C was inoculated into the wells of microtiter plates (Iwaki) containing 195 μl of HHWm or TSBg (final dilution of the culture, 1:40). After 24 h of incubation at 37°C, the microplates were washed three times with 200 μl of H2O, dried in an inverted position, and stained with 100 μl of 0.25% crystal violet for 2 to 3 min at room temperature. Then, the microplates were rinsed again three times with H2O and dried, the dye was subsequently dissolved in 200 μl of ethanol-acetone (80:20), and the absorbance was measured at 595 nm. For the biofilm formation inhibition assay we used a nonspecific rabbit serum, purified immunoglobulin G (IgG [I5006]; Sigma), and anti-protein A-specific antiserum (P3775; Sigma) at concentrations ranging from 500 μg/ml to 3.6 μg/ml. Then, a biofilm formation assay was performed as described above. To analyze biofilm formation under flow conditions, we used 60-ml microfermentors (Pasteur Institute; (www.pasteur.fr/recherche/unites/Ggb/biofilmfermenter.html) with a continuous flow of 40 ml of chemically defined medium h−1 and constant aeration with sterile pressed air. Submerged Pyrex slides served as the growth substratum. Bacteria from an overnight culture grown at 37°C with aeration in chemically defined media were diluted to an optical density (OD) at 600 nm of 1. For the inoculations, the Pyrex slides were introduced in the bacterial solution for 2 min and placed back in the microfermentors. The microfermentors were cultivated for 24 h at 37°C. Biofilm development was recorded with a Nikon Coolpix 950 digital camera. To quantify the 24-h biofilm, bacteria that adhered to the surfaces of the Pyrex slides were resuspended in 10 ml of chemically defined medium. The OD of the suspension was measured at 650 nm. Biofilm formation assays in the presence of supernatants were performed as follows. Supernatants from ISP479r agr ΔarlRS (supernatant 1), ISP479r agr ΔarlRS Δspa complemented with pCN40spa (supernatant 2), ISP479r agr ΔarlRS Δspa (supernatant 3), and ISP479r agr ΔarlRS Δspa complemented with a truncated version of protein A that lacks the LPXTG domain (supernatant 4) were obtained after centrifugation of an overnight culture at 2,800 rpm for 20 min. Filtered supernatants were added 1:1 (vol/vol) to ISP479r agr ΔarlRS Δspa cultures. Soluble commercial protein A (P3838; Sigma) was added at a concentration of 10 μg/ml. Then, the biofilm formation assay was performed as previously described.
Cell aggregation assay.
Liquid cultures of S. aureus ISP479r agr ΔarlRS, ISP479r agr ΔarlRS Δspa, Newman pCN40, Newman pCN40spa, and Lactococcus lactis MG1363 pKS80 and MG1363 pKS80spa were separately grown overnight in HHWm, TSBg, or M17. Cultures were adjusted to an OD600 of 2.5. Cultures of 3 ml were incubated in 5-ml standing tubes at room temperature, and the OD600 of the upper part of the culture was measured throughout time. Assays were performed in triplicate.
Electrophoresis and Western blot analysis.
Electrophoretic separation of cell wall-associated protein preparations was carried out by SDS-PAGE. The acrylamide concentration was 12% in the resolving gel and 5% in the stacking gel. For Western blotting, proteins were transferred to Hybond-ECL nitrocellulose membranes (GE Healthcare) by electroblotting. The buffer used for transfer was 50 mM Tris (pH 8.3)-380 mM glycine-0.1% SDS-20% methanol. The blocking solution contained PBS, 5% milk powder, and 0.1% Tween 20. Probing was carried out only using a secondary antibody taking into account the property of protein A to bind the Fc region of the Igs. Nitrocellulose membranes were washed with PBS-Tween and then incubated for 1 h at room temperature with a 1:5,000 dilution of a peroxidase-conjugated AffiniPure rabbit anti-sheep IgG (Jackson InmunoResearch Laboratories, Inc.). The reaction was developed using the ECL Western blotting analysis system (GE Healthcare). Images were obtained with a Chemi-Doc apparatus (Bio-Rad).
Preparation of S. aureus strains for the two-dimensional nano-liquid chromatography and ion trap mass spectrometric (2DnLC-MS/MS) analysis.
Bacterial strains were grown overnight at 37°C in HHW growth medium. A 200-ml aliquot of fresh medium was then inoculated with a 1:10 dilution of the overnight culture and grown until an OD650 of 1 was reached. Bacteria were spun down by centrifugation (14,000 rpm, 10 min, 4°C) and washed twice with a 1/10 volume of the original culture in PBS. The bacteria were fixed for 1 h at 4°C in 20 ml of a solution containing 3% paraformaldehyde and then washed twice with ultrapure H2O. Fixed bacteria were resuspended in a 1/10 volume of the original culture of ammonium bicarbonate, 50 mM, and modified trypsin (sequencing grade; Promega, Madison, WI) was added at a final amount of 1 μg per sample. Digestion with trypsin was performed under shaking conditions for 1 h at 37°C. Digested peptides were separated from the bacterial cells by centrifugation (300,000 × g, 20 min, 30°C). The supernatant containing the peptide mixture was lyophilized and kept at −20°C.
2DnLC-MS/MS analysis.
The tryptic peptide mixtures (4 μg each) were injected onto a strong cationic exchange micro-precolumn (500 μm inner diameter [ID] by 15 mm; BioX-SCX; LC Packings, Amsterdam, The Netherlands) with a flow rate of 30 μl/min as a first-dimension separation. Peptides were eluted from the column as fractions by injecting three salt steps of increasing concentrations of ammonium acetate (10, 100, and 2,000 mM). Each of the three fractions together with the nonretained fraction was on-line injected onto a C18 reversed-phase microcolumn (300 μm ID by 5 mm; PepMap; LC Packings) to remove salts, and the peptides were analyzed in a continuous acetonitrile gradient consisting of 0 to 50% B in 45 min and 50 to 90% B in 1 min (B is 95% acetonitrile, 0.5% acetic acid in water) on a C18 reversed-phase self-packing nano-column (100 μm ID by 15 cm; Discovery BIO Wide pore; Supelco, Bellefonte, PA). A flow rate of 300 nl/min was used to elute peptides from the reversed-phase nanocolumn to a PicoTip emitter nano-spray needle (New Objective, Woburn, MA) for real-time ionization and peptide fragmentation on an Esquire HCT ion trap mass spectrometer (Bruker-Daltonics, Bremen, Germany). Every 1 s, the instrument cycled through acquisition of a full-scan mass spectrum and one MS/MS spectrum. A 4-Da window (precursor m/z ± 2), an MS/MS fragmentation amplitude of 0.80 V, and a dynamic exclusion time of 0.30 min were used for peptide fragmentation. 2DnLC was automatically performed on an advanced microcolumn switching device (Switchos; LC Packings) coupled to an autosampler (Famos; LC Packings) and a nano-gradient generator (Ultimate nano-HPLC; LC Packings). The software Hystar 2.3 was used to control the whole analytical process.
Database analysis.
MS/MS spectra were batch processed by using DataAnalysis 5.1 SR1 and MS BioTools 2.0 software packages and searched against the S. aureus protein databases using Mascot software (Matrix Science, London, United Kingdom).
Real-time quantitative PCR.
Total S. aureus RNA was prepared using the Fast RNA-Blue kit (Bio101) according to the manufacturer's instructions. Two micrograms of each RNA was subjected in triplicate to DNase I (Gibco-BRL) treatment for 30 min at 37°C. The enzyme was inactivated at 65°C in the presence of EDTA. To verify the absence of genomic DNA in every sample, the RNA triplicates were reverse transcribed in the presence and absence of Moloney murine leukemia virus reverse transcriptase (Gibco-BRL). All preparations were purified using CentriSep spin columns (Princeton Separations). A 1/20 fraction of each reaction mixture was used for real-time quantitative PCR using a LightCycler and the SYBR Green PCR master mix (Applied Biosystems). The spa transcripts were amplified using primers spa-FW and spa-RV (Table 2). The gyrB transcripts that are constitutively expressed were amplified as endogenous controls using primers gyr-FW and gyr-RV (63) (Table 2). To monitor the specificity, final PCR products were analyzed by melting curves and electrophoresis. Only samples with no gyrB amplification of the minus reverse transcriptase aliquot were considered in the study. The amount of spa transcript was expressed as the difference relative to the control gene (2−ΔCt, where ΔCt represents the difference in threshold cycle between the target and control genes).
Immunofluorescence microscopy.
Immunofluorescence microscopy analysis was performed as previously described (61). Overnight cultures of the different strains were diluted to an OD650 of 1 in HHW and 10 μl was loaded onto 0.1% poly-(l-lysine)-treated immunofluorescence microscope slides. Slides were washed three times with PBS between each step of this protocol. Cells were fixed with 3% paraformaldehyde for 10 min before quenching with 50 mM NH4Cl in PBS for 3 min. Slides were then saturated for 15 min with 0.5% bovine serum albumin in PBS before being incubated with a 1:200 dilution of a secondary anti-mouse IgG (whole molecule) antibody produced in rabbit (Sigma) conjugated to tetramethyl rhodamine isothiocyanate (TRITC). Finally, the slides were mounted with Vectashield HardSet mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories) and observed under an epifluorescence microscope with rhodamine and DAPI filters.
Experimental infection.
A mouse foreign body infection model was used to determine the role of protein A in the pathogenesis of S. aureus. A total of 60 adult male mice (Swiss-Albino) were used. A 1-cm segment of intravenous catheter (24 gauge; B. Braun) was aseptically implanted into the subcutaneous interscapular space. Each group of mice was coinoculated with 105 CFU of both S. aureus Newman and the spa mutant strain (spa::Km). The animals were euthanatized by cervical dislocation on days 3 or 7 postinfection. The catheter was aseptically removed, placed in a sterile tube containing 1 ml of TSBg, sonicated, and vortexed at high speed for 3 min. The CFU/catheter was determined by plating bacteria in either TSA or TSA-Km (50 μg/ml). An extra group of animals was inoculated with vehicle (PBS) and served as a negative control. The experiment was repeated twice.
Statistical analysis.
A two-tailed Student's test with Welch's correction was used to assess significant differences in bacterial recovery within groups in experimental infections. Differences were considered statistically significant when P was <0.05. The GraphPad 5.0 statistical package was used (GraphPad Software, San Diego, CA).
RESULTS
Non-gel analysis of the S. aureus cell wall proteome.
To identify the protein component responsible for the PIA/PNAG-independent biofilm development in agr arlRS mutants, we first generated a mutant of the sortase A gene (srtA) in ISP479r agr ΔarlRS strain. Sortase A was selected because it catalyzes the cell wall anchoring of the LPXTG family of surface proteins. Biofilm formation assays revealed that deletion of srtA completely inhibited the biofilm phenotype displayed by the agr ΔarlRS mutant, suggesting that an LPXTG protein was essential for the biofilm-forming capacity of these strains (data not shown). The cell wall proteomes of ISP479r agr and its corresponding isogenic arlRS mutant were then compared by 2DnLC-MS/MS. Fixed bacterial cells were digested with trypsin as described in Materials and Methods to yield a peptide mixture that was fractionated and analyzed by 2DnLC coupled to electrospray ionization and mass spectrometry (4). Two LPXTG proteins, protein A and SdrD, were detected in the cell wall proteome of the ISP479r agr ΔarlRS mutant (data not shown). In contrast to SdrD, protein A was also detected in the cell proteome of the ISP479r agr strain. To confirm that the levels of protein A were higher on the surface of ISP479r agr ΔarlRS compared to the wild-type ISP479r agr strain, the intensities of extracted ion chromatograms (EIC) were compared. As shown in Fig. 1, the intensity of the ions corresponding to the trypsin-digested peptides from protein A was fivefold higher in the ΔarlRS sample, confirming that protein A is present at higher levels in the arlRS mutant compared to the wild-type strain. In contrast, and as a control, analysis of the EIC corresponding to the tryptic peptides from the ClpB protein showed that the intensity of the signals was almost identical in both strains, indicating that the amount of ClpB on the surface of both bacteria is similar. Overall, these results demonstrate that the LPXTG proteins, protein A and SdrD, were present at higher levels on the cell surface of the S. aureus agr ΔarlRS double mutant than in the agr-deficient strain.
FIG. 1.

EICs of protein A and ClpB in S. aureus ISP479r agr and ISP479r agr ΔarlRS strains. On the left is the EIC from ions at m/z 783.38 (ion 1), 816.07 (ion 2), and 825.4 (ion 3) Da, corresponding to protein A-derived tryptic, triply charged peptides spanning sequences shown below. Ionic intensity from the three detected signals was increased fivefold in the ΔarlRS sample. On the right is the EIC for ions at m/z 794.37 (ion 4) and 796.37 (ion 5) Da, corresponding to doubly and triply charged tryptic peptides, respectively, from the ClpB protein, which is expressed at the same levels in both samples. Retention times for the ISP479r agr ΔarlRS sample have been shifted 1 minute to avoid chromatogram overlapping.
Deletion of the spa gene causes a decrease in the biofilm development promoted by the arlRS mutation.
We then asked whether either of the two LPXTG proteins whose levels are augmented in double agr arlRS mutant strains was responsible for the biofilm formation phenotype. We deleted the spa and sdrD genes in the ISP479r agr ΔarlRS strain. Biofilm assays in polystyrene petri dishes revealed that only deletion of the spa gene inhibited the biofilm formation capacity, whereas deletion of sdrD had no effect (Fig. 2A). This result indicated that protein A was responsible for the development of biofilm in agr ΔarlRS strains. To verify this finding, complementation studies were performed. When pCN40spa was introduced into ISP479r agr ΔarlRS Δspa, the biofilm phenotype was completely restored, confirming that inactivation of spa was responsible for the biofilm negative phenotype of this strain (Fig. 2B). Immunoblotting assays demonstrated that the levels of protein A were elevated in the ISP479r agr ΔarlRS strain compared to the corresponding single mutants and wild-type strain (Fig. 2C). To investigate whether this increase of protein A expression observed in the agr arlRS mutant was due to augmented expression of the spa gene, we employed real-time quantitative PCR. In agreement with previous results (16), the levels of spa gene expression were not affected by arlRS deletion (Fig. 2D). Taken together, these results indicated that protein A is an essential component of the biofilm produced by agr ΔarlRS strains and that these strains accumulate higher levels of protein A on their cell surface, promoting biofilm development in vitro.
FIG. 2.

Biofilm formation phenotype in polystyrene microtiter dishes in HHWm medium after 24 h of incubation. (A) S. aureus ISP479r, S. aureus ISP479r agr, S. aureus ISP479r agr Δspa, ISP479r agr ΔarlRS, ISP479r agr ΔarlRS Δspa, and ISP479r agr ΔarlRS ΔsdrD strains. (B) S. aureus ISP479r agr, ISP479r agr ΔarlRS, and ISP479r agr ΔarlRS Δspa strains with the pCN40 control plasmid and S. aureus ISP479r agr ΔarlRS Δspa strain complemented with the pCN40spa plasmid carrying the spa gene under the PblaZ promoter. HHWm was supplemented with 10 μg/ml of erythromycin as needed. (C) Western blot analysis for protein A expression of S. aureus ISP479r, ISP479r ΔarlRS, ISP479r agr, ISP479r agr ΔarlRS, and ISP479r agr ΔarlRS Δspa strains. The blot was probed with a 1:5,000 dilution of a peroxidase-conjugated AffiniPure rabbit anti-sheep IgG. (D) Real-time quantification of spa gene expression in S. aureus strains ISP479r, ISP479r ΔarlRS, ISP479r agr, and ISP479r agr ΔarlRS at mid-log exponential phase (OD650, 0.6).
Protein A mediates biofilm development in the absence of a functional agr system.
Recent studies have suggested a role for the agr system in S. aureus biofilm development, as agr mutants exhibit a high propensity to form biofilms, and cells dispersing from a biofilm have been observed to display an active agr system (3, 64). Thus, we wondered whether protein A might be responsible for the increased biofilm development displayed by agr mutants. As shown in Fig. 2A, disruption of the spa gene in the agr mutant completely inhibited biofilm development, suggesting that increased accumulation of protein A in the absence of an active agr system is responsible at least in some strains for the tendency of agr mutants to develop stronger biofilms.
Protein A induces aggregation and biofilm formation under flow cell conditions.
To further investigate whether the increased levels of protein A in agr ΔarlRS strains were able to induce other phenotypes associated with multicellular behavior, we monitored cell-to-cell interactions in an aggregation assay and biofilm formation capacity in a continuous flow culture on glass spatulas submerged in microfermentors (19). In contrast to protein A-deficient bacteria that remained in suspension, S. aureus agr ΔarlRS strongly aggregated and rapidly sedimented on the bottom of the tube (Fig. 3A). Accordingly, visualization of both macroscopic biofilm development in microfermentors and biofilm cell density on the removable glass spatulas inside the microfermentors showed that S. aureus agr ΔarlRS formed a robust biofilm after 24 h of incubation, in contrast to its corresponding spa mutant (Fig. 3B). Together, these results indicate that protein A promotes cell-to-cell interactions and biofilm formation under flow conditions.
FIG. 3.

(A) In vitro aggregation assay of S. aureus ISP479r agr ΔarlRS and ISP479r agr ΔarlRS Δspa strains. On the left side the aggregation phenotype observed in glass tubes is shown, and on the right side the quantification of the optical density at 650 nm at different times is displayed. Results of a representative experiment are shown. (B) Biofilm development of S. aureus ISP479r agr ΔarlRS and ISP479r agr ΔarlRS Δspa strains grown in microfermentors under continuous flow of HHWm for 24 h at 37°C. The microfermentor (left) and the glass slides where bacteria form the biofilm (right) are shown.
Expression of protein A leads to increased aggregation and biofilm formation in wild-type S. aureus strains.
We then asked whether deletion or overexpression of protein A might affect biofilm formation in wild-type S. aureus strains. We first inactivated spa in S. aureus Newman strain, which does not synthesize PIA/PNAG exopolysaccharide and displays a slight or null capacity to form a biofilm in microtiter plates in HHWm. As shown in Fig. 4, deletion of spa reduced the capacity of the strain to produce a biofilm in microfermentors, but it did not cause any noticeable effect on the capacity of the strain to produce a biofilm in microtiter plates. In agreement with previous results, mutation of agr bestowed the capacity to produce a biofilm in microtiter plate assay. Such increased biofilm formation capacity was completely inhibited when the spa gene was inactivated in the S. aureus Newman agr strain, indicating that protein A was required for increased biofilm development in the absence of agr.
FIG. 4.
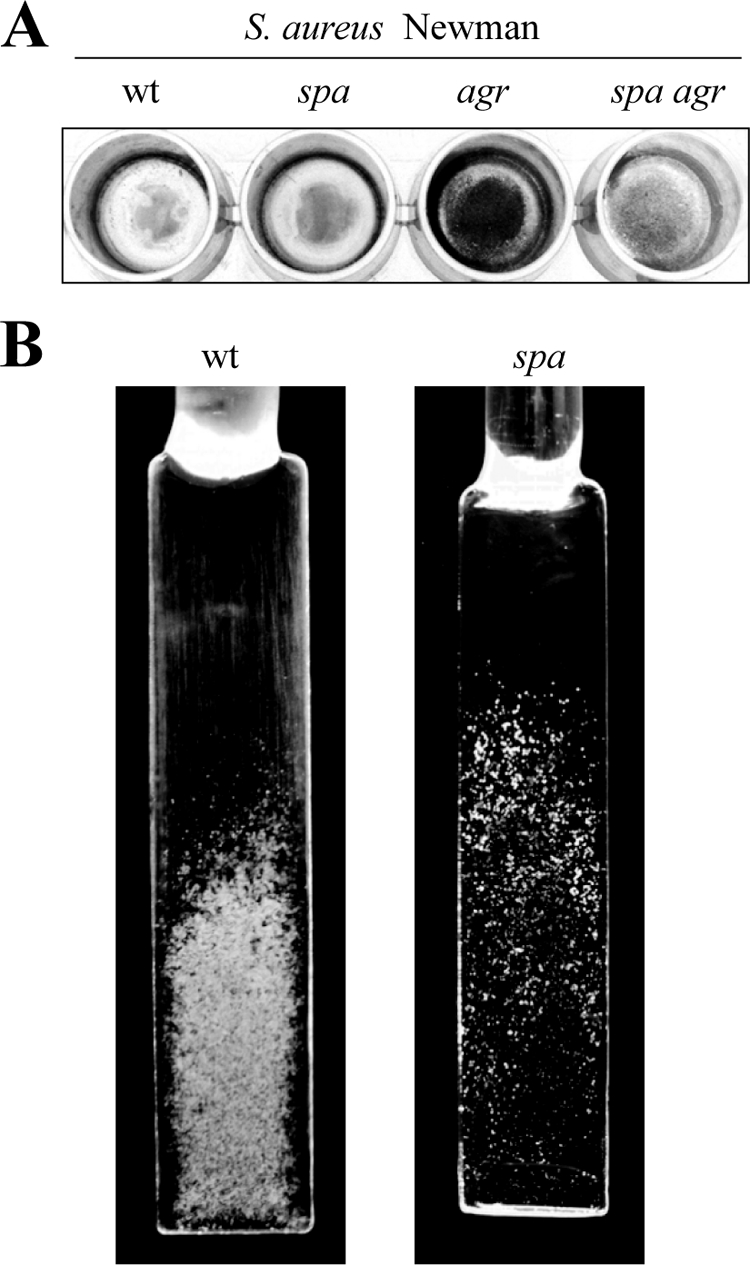
(A) Biofilm formation phenotype in polystyrene microtiter dishes with S. aureus Newman wild-type strain and its corresponding spa, agr, and spa agr mutant strains. (B) Biofilm development of S. aureus Newman wild-type strain and its corresponding spa mutant strain grown in continuous flow of HHWm for 24 h at 37°C. The glass slides where bacteria formed the biofilm are shown.
Then, we expressed protein A in S. aureus strain Newman from the multicopy plasmid pCN40. The results revealed that overexpression of protein A elicited aggregation in liquid culture and biofilm development in both polystyrene microtiter plates and microfermentors under flow conditions (Fig. 5A and B). To generalize the effects of protein A in multicellular behavior, we heterologously expressed protein A in Lactococcus lactis (Fig. 5C) and observed an increased aggregation phenotype indicative of bacterial clumping. Overall, these results demonstrate that production of protein A is sufficient to mediate biofilm development in wild-type S. aureus strains and cell-to-cell interactions in both wild-type S. aureus and L. lactis strains.
FIG. 5.
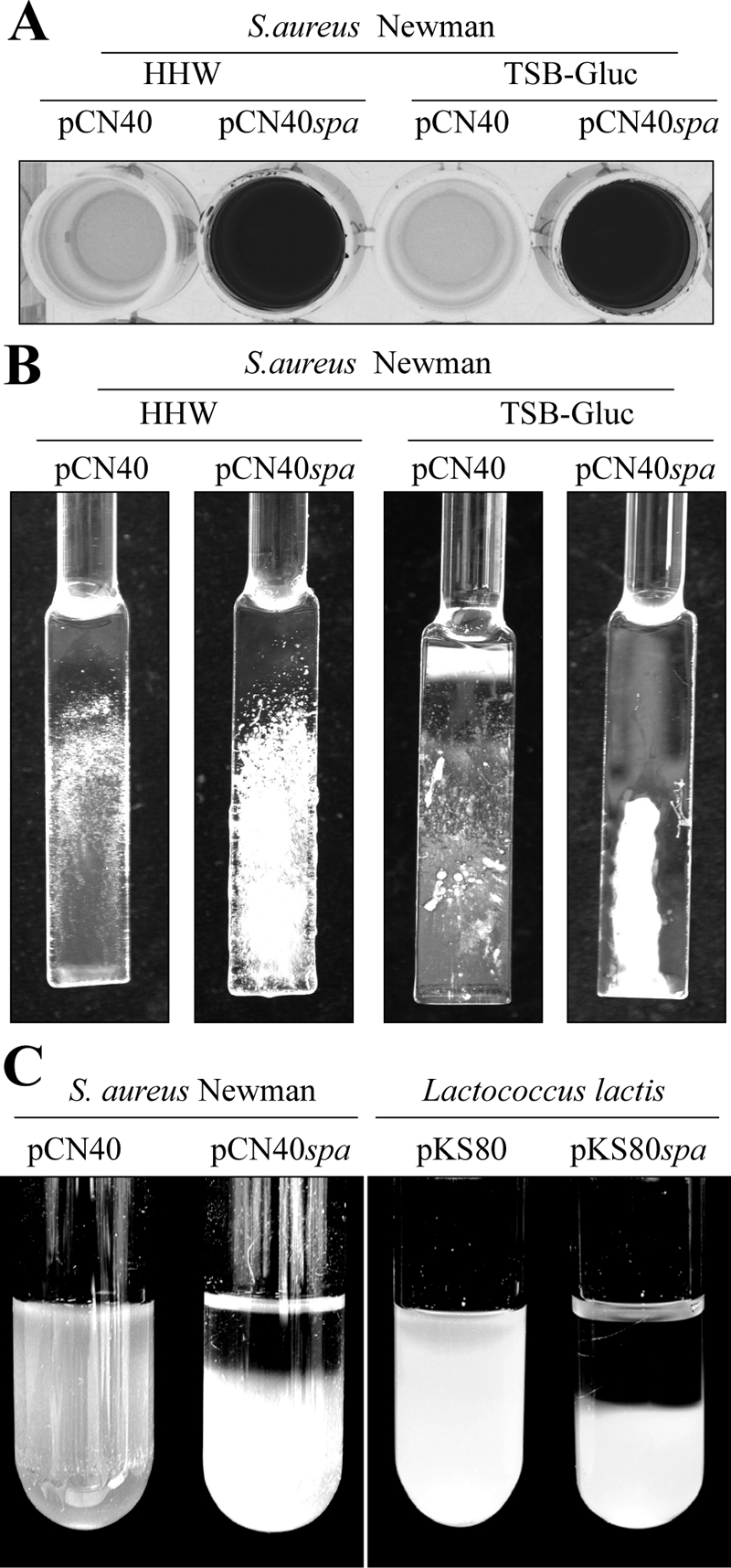
(A) Biofilm formation phenotype in polystyrene microtiter dishes with S. aureus Newman wild-type strain complemented with the pCN40 control plasmid and pCN40spa plasmid carrying the spa gene under the PblaZ promoter in HHWm and TSBg supplemented with 10 μg/ml of erythromycin. After 24 h of incubation, the microplates were washed and stained with crystal violet; the dye was dissolved in 200 μl of ethanol-acetone (80:20). (B) Biofilm development of S. aureus Newman wild-type strain with the pCN40 control plasmid and the pCN40spa plasmid expressing the spa gene under the PblaZ promoter grown in a continuous flow of HHWm or TSBg for 24 h at 37°C. The glass slides where bacteria formed the biofilm are shown. (C) In vitro aggregation assay in glass tubes. On the left side is S. aureus Newman wild-type strain with the pCN40 control plasmid and the pCN40spa plasmid carrying the spa gene under PblaZ promoter grown on HHWm supplemented with 10 μg/ml of erythromycin, and on the right side is Lactococcus lactis wild-type strain complemented with the pKS80 control plasmid and the pKS80spa plasmid grown on M17 supplemented with 10 μg/ml of erythromycin.
Antibodies inhibit protein A-induced biofilm formation.
Protein A directly reacts with the Fc portion of immunoglobulins (14). The IgG-binding regions of protein A consist of four or five domains, depending on the S. aureus strain (32). Based on this property, we reasoned that nonimmune binding of IgG to protein A could sterically interfere with the capacity of the protein to induce cell-cell interactions and biofilm formation. To test this hypothesis, we performed biofilm formation assays of S. aureus ISP479r agr ΔarlRS, S. aureus Newman pCN40spa, and S. aureus 12313 strains under the presence of increasing concentrations of a rabbit nonspecific serum, purified IgG, or polyclonal antibodies raised against protein A. The S. aureus 12313 strain was included as a control in the study, as its biofilm formation capacity depends on the production of the PIA/PNAG exopolysaccharide. We found that biofilm formation capacity of the ISP479r agr ΔarlRS and Newman pCN40spa strains was completely inhibited in a dose-dependent manner by nonspecific rabbit serum, purified IgG, and anti-protein A antibodies (Fig. 6). However, similar concentrations of antibodies did not affect the biofilm formation capacity of the clinical S. aureus 12313 strain. Taken together, these results indicate that interaction of antibodies with protein A sterically block the capacity of protein A to mediate biofilm development; this finding provides further evidence of the requirement for protein A in the biofilm development process.
FIG. 6.

Concentration-dependent inhibition of biofilm formation on microtiter plates. Increasing concentrations of unspecific rabbit antiserum, a purified IgG (Sigma), and specific anti-protein A antibodies (Sigma) were added to microtiter plates containing S. aureus ISP479r agr ΔarlRS strains, S. aureus Newman strain complemented with the pCN40spa plasmid carrying the spa gene under PblaZ promoter, and S. aureus strain 12313. The nonspecific rabbit antiserum, commercial purified IgG, and anti-protein A-specific serum were used at concentrations ranging from 500 μg/ml to 3.6 μg/ml. HHWm was supplemented with 10 μg/ml of erythromycin as needed. After 24 h of incubation, the microplates were washed and stained with crystal violet.
Extracellular protein A induces biofilm formation.
Protein A belongs to the LPXTG family of proteins characterized by its covalent linkage to the cell wall peptidoglycan. However, most S. aureus strains secrete significant levels of protein A (15 to 30%) into the cell culture (13). This prompted us to investigate whether protein A needs to be anchored to the cell wall to promote multicellular behavior or if secreted protein A might be functional. We first examined the levels of protein A in the supernatants of ISP479r agr, ISP479r agr ΔarlRS, ISP479r agr ΔarlRS Δspa, and ISP479r agr ΔarlRS Δspa pCN40spa. Significant levels of protein A in the supernatants of the two biofilm-positive strains, ISP479r agr ΔarlRS and ISP479r agr ΔarlRS Δspa pCN40spa, were confirmed (Fig. 7A). Supernatants of these bacterial strains, as well as pure commercial protein A and a supernatant containing a modified protein A lacking the LPXTG carboxy-terminal region, were added 1:1 (vol/vol) to ISP479r agr ΔarlRS Δspa cultures. As shown in Fig. 7B, addition of pure protein A or supernatants containing protein A induced biofilm formation in the ISP479r agr ΔarlRS Δspa strain, whereas protein A-deficient supernatants did not improve the biofilm formation capacity of this strain. Interestingly, the protein A derivative lacking the LPXTG domain retained the ability to induce biofilm formation. These results indicate that secreted protein A is fully functional in promoting biofilm development and suggest that protein A must interact with some compound of the bacterial surface to mediate biofilm development. To address this implication, we performed immunofluorescence microscopy analysis (Fig. 8). Protein A antiserum evenly bound to the cell surface of the ISP479r agr ΔarlRS Δspa strain when it was incubated with supernatants containing extracellular protein A, confirming that extracellular protein A interacts with the bacterial cell surface.
FIG. 7.

(A) Western Blot analysis for protein A expression of the supernatants of ISP479r agr, ISP479r agr ΔarlRS, ISP479r agr ΔarlRS Δspa complemented with the pCN40spa, and ISP479r agr ΔarlRS Δspa. The cultures were grown overnight at 37°C in HHWm, and cells were collected by centrifugation at 12,000 rpm for 2 min (the medium was supplemented with 10 μg/ml of erythromycin as needed). The supernatants were resolved by SDS-PAGE in a 12% polyacrylamide gel and then subjected to Western blot analysis. The blot was probed with a 1:5,000 dilution of a peroxidase-conjugated AffiniPure rabbit anti-sheep IgG. (B) Biofilm formation phenotype of the ISP479r agr ΔarlRS Δspa strain grown in the presence of supernatants obtained from ISP479r agr ΔarlRS (supernatant 1), ISP479r agr ΔarlRS Δspa complemented with the pCN40spa (supernatant 2), ISP479r agr ΔarlRS Δspa (supernatant 3), and ISP479r agr ΔarlRS Δspa complemented with a truncated version of protein A that lacks the LPXTG domain (supernatant 4). Additionally, ISP479r agr ΔarlRS Δspa was grown in the presence of pure commercial protein A (Sigma). The supernatants were added 1:1 to the culture medium, and the pure commercial protein A was added at a concentration of 10 μg/ml. After 24 h of incubation, the microplates were washed and stained with crystal violet. The dye was dissolved in 200 μl of ethanol-acetone (80:20).
FIG. 8.

Analysis of cell surface interactions of extracellular protein A by immunofluorescence microscopy. S. aureus ISP479r agr ΔarlRS Δspa cells were incubated with supernatants of ISP479r agr ΔarlRS (supernatant 1), ISP479r agr ΔarlRS Δspa pCN40spa (supernatant 2), and ISP479r agr ΔarlRS Δspa (supernatant 3), as well as pure protein A. The samples were washed twice with PBS and fixed with 3% paraformaldehyde in PBS before incubation with a 1:200 dilution of a secondary anti-mouse IgG (whole molecule)-TRITC rabbit antibody (Sigma). The slides were mounted with Vectashield HardSet mounting medium with DAPI (Vector Laboratories) (see Materials and Methods for details). The experiment was repeated twice.
Expression of protein A contributes to subcutaneous catheter infection.
To extend the relevance of our findings, we investigated the effects of protein A on the capacity of S. aureus to colonize a subcutaneous foreign catheter. A 1-cm segment of an intravenous catheter was aseptically implanted in the subscapular space of mice, and animals were coinfected with equal numbers of S. aureus Newman and the corresponding protein A-deficient strain. The ratio of protein A+ and protein A− clones recovered from infected catheters after 3 and 7 days was determined on agar plates with and without kanamycin. The results of two independent experiments are presented in Fig. 9. The number of protein A-positive colonies recovered from the catheters was approximately 10 times higher than protein A-negative colonies at day 3. Differences slightly increased at day 7 (P < 0.005, Student's t test). These data indicated that protein A significantly contributes to the development of biofilm-associated infections caused by S. aureus.
FIG. 9.

A 1-cm segment of an intravenous catheter was aseptically implanted into the subcutaneous interscapular spaces of adult male mice. Each group of five mice was coinoculated with 105 CFU of both S. aureus Newman and the spa mutant strain (spa::Km). The figure shows the recovery from the implanted subcutaneous catheters with S. aureus Newman and the Newman spa::Km mutant. The boxes represent the means and standard deviations of two independent experiments. Significant differences between the wild-type and mutant strains are represented by asterisks (P < 0.001) at day 3 and day 7 postinfection. Bacteria were not detectable in control animals at the end of the experimental period.
DISCUSSION
Protein A, the first surface protein of S. aureus to be identified, has served as a model for the characterization of the covalent anchorage of the LPXTG domain proteins by sortase A to the bacterial cell surface (40, 53, 54). Protein A is primarily known for its ability to bind to the Fc region of IgGs and inhibit opsonophagocytosis (14, 59). The consequence of this interaction is the coating of the bacterial surface with IgG molecules in the incorrect orientation to be recognized by the neutrophil Fc receptor (15). In addition, protein A can bind to the human von Willebrand factor, a large multimeric glycoprotein that mediates platelet adhesion at sites of endothelial damage to promote the appearance of blood clots (27, 46). Protein A also interacts with TNFR1 and stimulates an inflammatory response in airway epithelial cells (20, 21). In the present work, we have identified a novel function for protein A in the promotion of cell-to-cell interactions and biofilm formation by investigating the protein-dependent biofilm production of an agr arlRS double mutant (56). The regulation of protein A by the agr system, and specifically by its effector molecule, RNA III, has been well-characterized. Accumulation of RNA III during the postexponential phase of growth represses spa expression, not only at a transcriptional level but also by inhibiting its translation and inducing degradation of the stable spa mRNA by the double-strand-specific endoribonuclease III (RNase III) (6, 29, 43, 48). Thus, expression of protein A is induced at the exponential phase and repressed at the postexponential phase, when the expression of RNA III is maximal. In addition, arlRS has also been described as a repressor of spa expression, due to the fact that mutation in either of the two arlRS genes results in increased spa expression. The influence of arlRS on protein A expression requires the participation of agr or sarA regulators, as increased transcription was not observed in either an agr or sarA mutant strain (16). In agreement with these results, we did not observe significant upregulation of spa mRNA levels in agr arlRS mutants. However, the agr arlRS double mutant accumulates more protein A on the cell surface than the agr mutant, suggesting that arlRS either posttranscriptionally regulates spa mRNA stability or that decreased production of proteases due to the absence of arlRS indirectly provokes an increased accumulation of protein A. Whatever the molecular mechanism is, the final outcome of the double agr arlRS mutation is an increased accumulation of protein A on the bacterial surface. This accumulation together with the fact that most S. aureus strains are unable to produce PIA/PNAG exopolysaccharide in the HHWm synthetic medium has facilitated for us the observation that protein A can mediate biofilm formation. However, the effect of protein A on biofilm formation is not restricted to this double mutant. Thus, deletion of spa in wild-type strains reduces biofilm formation capacity in microfermentors, whereas it does not produce noticeable effects in microtiter plates, most likely because the amount of protein A under these conditions does not reach the threshold level required to promote cell-to-cell interactions and biofilm formation. In support of this hypothesis, we showed that overexpression of protein A or the increased accumulation of protein A in agr mutants induced biofilm development in microtiter plates. This last result correlates with previous data indicating that agr-deficient S. aureus mutants form more robust biofilms compared to wild-type strains (2, 3, 62, 64). Initially, it was proposed that the surfactant properties of δ-toxin encoded by RNA III limited the ability of the bacteria to form biofilms (62). More recently, it was shown that activation of the agr system through the addition of the AIP peptide at a physiological concentration represses biofilm development and triggers the detachment of established PIA/PNAG-independent biofilm matrix (3). The same study showed that the biofilm matrix produced by inactivation of the agr system was degraded by proteinase K and resistant to the treatment with dispersin B. Accordingly, agr-mediated biofilm detachment required the participation of extracellular proteases, as it did not occur in the presence of protease inhibitors or when the main protease encoding gene, aureolysin, was mutated (3). All these results strongly suggest that a proteinaceous compound is important for biofilm integrity and that agr activity promotes the detachment of a proteinaceous biofilm matrix. Our results support the idea that protein A is, at least in some strains, the proteinaceous compound responsible for biofilm development in S. aureus agr mutant strains.
Protein A-mediated aggregation and biofilm formation may result from homophilic interactions between two protein A molecules of neighboring cells. Such homophilic protein interactions leading to an autoaggregation phenotype have been described for protein H-mediated Streptococcus pyogenes cell aggregation (18), hemagglutinin-mediated cell aggregation in Bordetella pertussis (39), and antigen 43, which is involved in E. coli biofilm formation (10). Alternatively, protein A could mediate heterophilic interactions with other surface proteins or even with a nonproteinaceous cell wall structure. In support of the former hypothesis, expression of protein A in L. lactis also induced aggregation, likely via heterophilic interactions with a compound that must be present on the cell wall of both bacteria. However, expression of protein A in L. lactis only induced aggregation of bacterial cells and was not sufficient to induce the development of a mature biofilm on microtiter plates or on the glass slide in microfermenters, suggesting that either additional factors are required for biofilm development or that L. lactis is unable to produce biofilm under the experimental conditions tested.
The covalent attachment of protein A to the bacterial surface is not required for its ability to promote cell-to-cell interactions. Thus, secreted protein A from S. aureus strains or a protein A variant lacking the carboxy-terminal LPXTG domain is sufficient to induce biofilm development. In contrast to Aap- or SasG-mediated biofilm development that must undergo proteolytic processing to render the proteins active for cell-to-cell interactions (7, 51), protein A induces biofilm development in an agr arlRS mutant that produces low levels of proteases. Indeed, it appears that the decreased production of extracellular proteases is at least partially responsible for the increased accumulation of protein A in this mutant and thereby proteolytic processing seems to not be required to activate protein A for biofilm development.
Several studies have demonstrated the role of protein A in staphylococcal virulence. Protein A-defective mutants show reduced virulence in murine models of septic arthritis, septicemia, and skin abscesses, most likely due to the antiphagocytic effect of protein A bound to the Fc fractions of the IgGs (47, 49). Protein A has also been shown to trigger apoptosis of murine B cells that express antigen receptors with VH3-Fab analogues, which likely contributes to immunosuppression during S. aureus infection (22). In the present study, coinfection experiments using a subcutaneous foreign device murine infection model showed that protein A significantly contributed to the development of biofilm-related infections. As these experiments were performed with a wild-type Newman strain and its corresponding protein A-deficient mutant, these results imply the existence of physiological situations in which the levels of protein A are sufficient to contribute to biofilm development. We cannot exclude the possibility that the increased susceptibility of protein A-defective mutants to phagocytosis might be negatively affecting the survival of the bacteria in the implanted catheter. However, if this were the case, we would expect that the number of recovered protein A-deficient bacteria would decrease throughout the duration of the experiment. However, similar numbers of protein A-deficient bacteria were recuperated at days 3 and 7, suggesting that survival of the bacteria does not decrease during the infection period.
In summary, our results indicate that two of the consequences of inactivation of the agr system, namely, overexpression of protein A and induction of biofilm formation capacity, are related. Whether the common observed agr-negative variants during the course of the infection (17, 52, 58) may lead to the establishment of bacterial interactions and biofilm formation to evade the immune system is an interesting hypothesis that warrants further experiments.
Acknowledgments
Nekane Merino is a predoctoral fellow from the Basque Government, Spain. This work was supported by the BIO2005-08399 and ERA-Net pathogenomics GEN2006-27792-C2-1-E/PAT grants from the Spanish Ministerio de Educación y Ciencia and grant LSHM-CT-2006-019064 from the European Union.
Footnotes
Published ahead of print on 1 December 2008.
REFERENCES
- 1.Arnaud, M., A. Chastanet, and M. Débarbouillé. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 706887-6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beenken, K. E., P. M. Dunman, F. McAleese, D. Macapagal, E. Murphy, S. J. Projan, J. S. Blevins, and M. S. Smeltzer. 2004. Global gene expression in Staphylococcus aureus biofilms. J. Bacteriol. 1864665-4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boles, B. R., and A. R. Horswill. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 4e1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canas, B., D. Lopez-Ferrer, A. Ramos-Fernandez, E. Camafeita, and E. Calvo. 2006. Mass spectrometry technologies for proteomics. Brief Funct. Genomic Proteomic 4295-320. [DOI] [PubMed] [Google Scholar]
- 5.Charpentier, E., A. I. Anton, P. Barry, B. Alfonso, Y. Fang, and R. P. Novick. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 706076-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung, A. L., K. Eberhardt, and J. H. Heinrichs. 1997. Regulation of protein A synthesis by the sar and agr loci of Staphylococcus aureus. Infect. Immun. 652243-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrigan, R. M., D. Rigby, P. Handley, and T. J. Foster. 2007. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology 1532435-2446. [DOI] [PubMed] [Google Scholar]
- 8.Cramton, S. E., C. Gerke, N. F. Schnell, W. W. Nichols, and F. Gotz. 1999. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect. Immun. 675427-5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cucarella, C., C. Solano, J. Valle, B. Amorena, I. Lasa, and J. R. Penades. 2001. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 1832888-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danese, P. N., L. A. Pratt, S. L. Dove, and R. Kolter. 2000. The outer membrane protein, antigen 43, mediates cell-to-cell interactions within Escherichia coli biofilms. Mol. Microbiol. 37424-432. [DOI] [PubMed] [Google Scholar]
- 11.Donlan, R. M., and J. W. Costerton. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15167-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duthie, E. S., and L. L. Lorenz. 1952. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 695-107. [DOI] [PubMed] [Google Scholar]
- 13.Forsgren, A. 1970. Significance of protein A production by staphylococci. Infect. Immun. 2672-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsgren, A., and J. Sjoquist. 1966. “Protein A” from S. aureus. I. Pseudo-immune reaction with human gamma-globulin. J. Immunol. 97822-827. [PubMed] [Google Scholar]
- 15.Foster, T. J. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3948-958. [DOI] [PubMed] [Google Scholar]
- 16.Fournier, B., A. Klier, and G. Rapoport. 2001. The two-component system ArlS-ArlR is a regulator of virulence gene expression in Staphylococcus aureus. Mol. Microbiol. 41247-261. [DOI] [PubMed] [Google Scholar]
- 17.Fowler, V. G., Jr., G. Sakoulas, L. M. McIntyre, V. G. Meka, R. D. Arbeit, C. H. Cabell, M. E. Stryjewski, G. M. Eliopoulos, L. B. Reller, G. R. Corey, T. Jones, N. Lucindo, M. R. Yeaman, and A. S. Bayer. 2004. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J. Infect. Dis. 1901140-1149. [DOI] [PubMed] [Google Scholar]
- 18.Frick, I. M., M. Morgelin, and L. Bjorck. 2000. Virulent aggregates of Streptococcus pyogenes are generated by homophilic protein-protein interactions. Mol. Microbiol. 371232-1247. [DOI] [PubMed] [Google Scholar]
- 19.Ghigo, J. M. 2001. Natural conjugative plasmids induce bacterial biofilm development. Nature 412442-445. [DOI] [PubMed] [Google Scholar]
- 20.Gomez, M. I., A. Lee, B. Reddy, A. Muir, G. Soong, A. Pitt, A. Cheung, and A. Prince. 2004. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat. Med. 10842-848. [DOI] [PubMed] [Google Scholar]
- 21.Gomez, M. I., M. O'Seaghdha, M. Magargee, T. J. Foster, and A. S. Prince. 2006. Staphylococcus aureus protein A activates TNFR1 signaling through conserved IgG binding domains. J. Biol. Chem. 28120190-20196. [DOI] [PubMed] [Google Scholar]
- 22.Goodyear, C. S., and G. J. Silverman. 2004. Staphylococcal toxin induced preferential and prolonged in vivo deletion of innate-like B lymphocytes. Proc. Natl. Acad. Sci. USA 10111392-11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotz, F. 2002. Staphylococcus and biofilms. Mol. Microbiol. 431367-1378. [DOI] [PubMed] [Google Scholar]
- 24.Götz, F., and G. Peters. 2000. Colonization of medical devices by coagulase negative staphylococci, p. 41-54. In F. A. Waldvogel and A. L. Bisno (ed.), Infections associated with indwelling medical devices, 3rd ed. ASM Press, Washington, DC.
- 25.Greene, C., D. McDevitt, P. Francois, P. E. Vaudaux, D. P. Lew, and T. J. Foster. 1995. Adhesion properties of mutants of Staphylococcus aureus defective in fibronectin-binding proteins and studies on the expression of fnb genes. Mol. Microbiol. 171143-1152. [DOI] [PubMed] [Google Scholar]
- 26.Harris, L. G., and R. G. Richards. 2006. Staphylococci and implant surfaces: a review. Injury 37(Suppl. 2)S3-S14. [DOI] [PubMed] [Google Scholar]
- 27.Hartleib, J., N. Kohler, R. B. Dickinson, G. S. Chhatwal, J. J. Sixma, O. M. Hartford, T. J. Foster, G. Peters, B. E. Kehrel, and M. Herrmann. 2000. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood 962149-2156. [PubMed] [Google Scholar]
- 28.Heilmann, C., O. Schweitzer, C. Gerke, N. Vanittanakom, D. Mack, and F. Gotz. 1996. Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol. Microbiol. 201083-1091. [DOI] [PubMed] [Google Scholar]
- 29.Huntzinger, E., S. Boisset, C. Saveanu, Y. Benito, T. Geissmann, A. Namane, G. Lina, J. Etienne, B. Ehresmann, C. Ehresmann, A. Jacquier, F. Vandenesch, and P. Romby. 2005. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 24824-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jass, J., S. Surman, and J. T. Walker (ed.). 2003. Medical biofilms, p. 51-72. John Wiley & Sons Ltd., Sussex, England.
- 31.Jonsson, K., C. Signas, H. P. Muller, and M. Lindberg. 1991. Two different genes encode fibronectin binding proteins in Staphylococcus aureus. The complete nucleotide sequence and characterization of the second gene. Eur. J. Biochem. 2021041-1048. [DOI] [PubMed] [Google Scholar]
- 32.Kreiswirth, B. N., S. Lofdahl, M. J. Betley, M. O'Reilly, P. M. Schlievert, M. S. Bergdoll, and R. P. Novick. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305709-712. [DOI] [PubMed] [Google Scholar]
- 33.Lasa, I., and J. R. Penades. 2006. Bap: a family of surface proteins involved in biofilm formation. Res. Microbiol. 15799-107. [DOI] [PubMed] [Google Scholar]
- 34.Mack, D., W. Fischer, A. Krokotsch, K. Leopold, R. Hartmann, H. Egge, and R. Laufs. 1996. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear β-1,6-linked glucosaminoglycan: purification and structural analysis. J. Bacteriol. 178175-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maira-Litran, T., A. Kropec, C. Abeygunawardana, J. Joyce, G. Mark III, D. A. Goldmann, and G. B. Pier. 2002. Immunochemical properties of the staphylococcal poly-N-acetylglucosamine surface polysaccharide. Infect. Immun. 704433-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mazmanian, S. K., G. Liu, E. R. Jensen, E. Lenoy, and O. Schneewind. 2000. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA 975510-5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDevitt, D., T. Nanavaty, K. House-Pompeo, E. Bell, N. Turner, L. McIntire, T. Foster, and M. Hook. 1997. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur. J. Biochem. 247416-424. [DOI] [PubMed] [Google Scholar]
- 38.McKenney, D., J. Hubner, E. Muller, Y. Wang, D. A. Goldmann, and G. B. Pier. 1998. The ica locus of Staphylococcus epidermidis encodes production of the capsular polysaccharide/adhesin. Infect. Immun. 664711-4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menozzi, F. D., P. E. Boucher, G. Riveau, C. Gantiez, and C. Locht. 1994. Surface-associated filamentous hemagglutinin induces autoagglutination of Bordetella pertussis. Infect. Immun. 624261-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Navarre, W. W., and O. Schneewind. 1994. Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. Mol. Microbiol. 14115-121. [DOI] [PubMed] [Google Scholar]
- 41.Ni Eidhin, D., S. Perkins, P. Francois, P. Vaudaux, M. Hook, and T. J. Foster. 1998. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 30245-257. [DOI] [PubMed] [Google Scholar]
- 42.Novick, R. P. 1991. Genetic systems in staphylococci. Methods Enzymol. 204587-636. [DOI] [PubMed] [Google Scholar]
- 43.Novick, R. P., H. F. Ross, S. J. Projan, J. Kornblum, B. Kreiswirth, and S. Moghazeh. 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 123967-3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Brien, L., S. W. Kerrigan, G. Kaw, M. Hogan, J. Penades, D. Litt, D. J. Fitzgerald, T. J. Foster, and D. Cox. 2002. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol. Microbiol. 441033-1044. [DOI] [PubMed] [Google Scholar]
- 45.O'Neill, E., C. Pozzi, P. Houston, H. Humphreys, D. A. Robinson, A. Loughman, T. J. Foster, and J. P. O'Gara. 2008. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 1903835-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Seaghdha, M., C. J. van Schooten, S. W. Kerrigan, J. Emsley, G. J. Silverman, D. Cox, P. J. Lenting, and T. J. Foster. 2006. Staphylococcus aureus protein A binding to von Willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J. 2734831-4841. [DOI] [PubMed] [Google Scholar]
- 47.Palmqvist, N., T. Foster, A. Tarkowski, and E. Josefsson. 2002. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb. Pathog. 33239-249. [DOI] [PubMed] [Google Scholar]
- 48.Patel, A. H., J. Kornblum, B. Kreiswirth, R. Novick, and T. J. Foster. 1992. Regulation of the protein A-encoding gene in Staphylococcus aureus. Gene 11425-34. [DOI] [PubMed] [Google Scholar]
- 49.Patel, A. H., P. Nowlan, E. D. Weavers, and T. Foster. 1987. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 553103-3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rice, K. C., E. E. Mann, J. L. Endres, E. C. Weiss, J. E. Cassat, M. S. Smeltzer, and K. W. Bayles. 2007. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 1048113-8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohde, H., C. Burdelski, K. Bartscht, M. Hussain, F. Buck, M. A. Horstkotte, J. K. Knobloch, C. Heilmann, M. Herrmann, and D. Mack. 2005. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol. Microbiol. 551883-1895. [DOI] [PubMed] [Google Scholar]
- 52.Sakoulas, G., G. M. Eliopoulos, R. C. Moellering, Jr., C. Wennersten, L. Venkataraman, R. P. Novick, and H. S. Gold. 2002. Accessory gene regulator (agr) locus in geographically diverse Staphylococcus aureus isolates with reduced susceptibility to vancomycin. Antimicrob. Agents Chemother. 461492-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneewind, O., A. Fowler, and K. F. Faull. 1995. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 268103-106. [DOI] [PubMed] [Google Scholar]
- 54.Schneewind, O., P. Model, and V. A. Fischetti. 1992. Sorting of protein A to the staphylococcal cell wall. Cell 70267-281. [DOI] [PubMed] [Google Scholar]
- 55.Shanks, R. M., M. A. Meehl, K. M. Brothers, R. M. Martinez, N. P. Donegan, M. L. Graber, A. L. Cheung, and G. A. O'Toole. 2008. Genetic evidence for an alternative citrate-dependent biofilm formation pathway of Staphylococcus aureus dependent on fibronectin binding proteins and the GraRS two-component regulatory system. Infect. Immun. 762469-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toledo-Arana, A., N. Merino, M. Vergara-Irigaray, M. Debarbouille, J. R. Penades, and I. Lasa. 2005. Staphylococcus aureus develops an alternative, ica-independent biofilm in the absence of the arlRS two-component system. J. Bacteriol. 1875318-5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tormo, M. A., E. Knecht, F. Gotz, I. Lasa, and J. R. Penades. 2005. Bap-dependent biofilm formation by pathogenic species of Staphylococcus: evidence of horizontal gene transfer? Microbiology 1512465-2475. [DOI] [PubMed] [Google Scholar]
- 58.Traber, K. E., E. Lee, S. Benson, R. Corrigan, M. Cantera, B. Shopsin, and R. P. Novick. 2008. agr function in clinical Staphylococcus aureus isolates. Microbiology 1542265-2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uhlen, M., B. Guss, B. Nilsson, F. Gotz, and M. Lindberg. 1984. Expression of the gene encoding protein A in Staphylococcus aureus and coagulase-negative staphylococci. J. Bacteriol. 159713-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valle, J., A. Toledo-Arana, C. Berasain, J. M. Ghigo, B. Amorena, J. R. Penades, and I. Lasa. 2003. SarA and not σB is essential for biofilm development by Staphylococcus aureus. Mol. Microbiol. 481075-1087. [DOI] [PubMed] [Google Scholar]
- 61.Vergara-Irigaray, M., T. Maira-Litran, N. Merino, G. B. Pier, J. R. Penades, and I. Lasa. 2008. Wall teichoic acids are dispensable for anchoring the PNAG exopolysaccharide to the Staphylococcus aureus cell surface. Microbiology 154865-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vuong, C., H. L. Saenz, F. Gotz, and M. Otto. 2000. Impact of the agr quorum-sensing system on adherence to polystyrene in Staphylococcus aureus. J. Infect. Dis. 1821688-1693. [DOI] [PubMed] [Google Scholar]
- 63.Wolz, C., C. Goerke, R. Landmann, W. Zimmerli, and U. Fluckiger. 2002. Transcription of clumping factor A in attached and unattached Staphylococcus aureus in vitro and during device-related infection. Infect. Immun. 702758-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yarwood, J. M., D. J. Bartels, E. M. Volper, and E. P. Greenberg. 2004. Quorum sensing in Staphylococcus aureus biofilms. J. Bacteriol. 1861838-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]